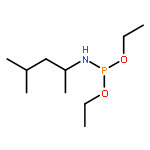
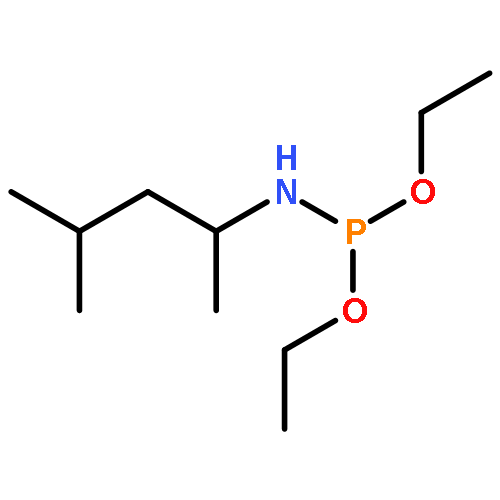
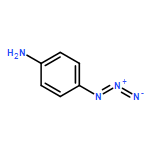
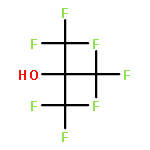
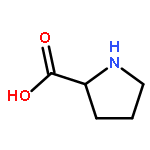
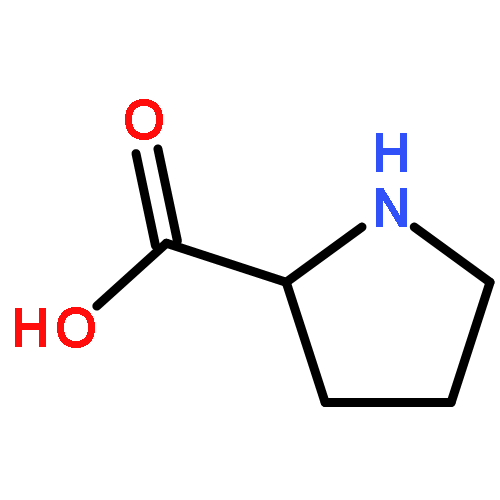
Co-reporter:Caitlin M. Tressler;Neal J. Zondlo
Biochemistry February 28, 2017 Volume 56(Issue 8) pp:1062-1074
Publication Date(Web):February 6, 2017
DOI:10.1021/acs.biochem.6b01020
Highly fluorinated amino acids can stabilize proteins and complexes with proteins, via enhanced hydrophobicity, and provide novel methods for identification of specific molecular events in complex solutions, via selective detection by 19F NMR and the absence of native 19F signals in biological contexts. However, the potential applications of 19F NMR in probing biological processes are limited both by the strong propensities of most highly fluorinated amino acids for the extended conformation and by the relatively modest sensitivity of NMR spectroscopy, which typically constrains measurements to mid-micromolar concentrations. Herein, we demonstrate that perfluoro-tert-butyl homoserine exhibits a propensity for compact conformations, including α-helix and polyproline helix (PPII), that is similar to that of methionine. Perfluoro-tert-butyl homoserine has nine equivalent fluorines that do not couple to any other nuclei, resulting in a sharp singlet that can be sensitively detected rapidly at low micromolar concentrations. Perfluoro-tert-butyl homoserine was incorporated at sites of leucine residues within the α-helical LXXLL short linear motif of estrogen receptor (ER) coactivator peptides. A peptide containing perfluoro-tert-butyl homoserine at position i + 3 of the ER coactivator LXXLL motif exhibited a Kd of 2.2 μM for the estradiol-bound estrogen receptor, similar to that of the native ligand. 19F NMR spectroscopy demonstrated the sensitive detection (5 μM concentration, 128 scans) of binding of the peptide to the ER and of inhibition of protein–protein interaction by the native ligand or by the ER antagonist tamoxifen. These results suggest diverse potential applications of perfluoro-tert-butyl homoserine in probing protein function and protein–protein interfaces in complex solutions.
Co-reporter:Christina R. Forbes, Sudipta K. Sinha, Himal K. Ganguly, Shi Bai, Glenn P. A. Yap, Sandeep Patel, and Neal J. Zondlo
Journal of the American Chemical Society 2017 Volume 139(Issue 5) pp:1842-1855
Publication Date(Web):January 12, 2017
DOI:10.1021/jacs.6b08415
Thiols can engage favorably with aromatic rings in S–H/π interactions, within abiological systems and within proteins. However, the underlying bases for S–H/π interactions are not well understood. The crystal structure of Boc-l-4-thiolphenylalanine tert-butyl ester revealed crystal organization centered on the interaction of the thiol S–H with the aromatic ring of an adjacent molecule, with a through-space Hthiol···Caromatic distance of 2.71 Å, below the 2.90 Å sum of the van der Waals radii of H and C. The nature of this interaction was further examined by DFT calculations, IR spectroscopy, solid-state NMR spectroscopy, and analysis of the Cambridge Structural Database. The S–H/π interaction was found to be driven significantly by favorable molecular orbital interactions, between an aromatic π donor orbital and the S–H σ* acceptor orbital (a π → σ* interaction). For comparison, a structural analysis of O–H/π interactions and of cation/π interactions of alkali metal cations with aromatic rings was conducted. Na+ and K+ exhibit a significant preference for the centroid of the aromatic ring and distances near the sum of the van der Waals and ionic radii, as expected for predominantly electrostatic interactions. Li+ deviates substantially from Na+ and K+. The S–H/π interaction differs from classical cation/π interactions by the preferential alignment of the S–H σ* toward the ring carbons and an aromatic π orbital rather than toward the aromatic centroid. These results describe a potentially broadly applicable approach to understanding the interactions of weakly polar bonds with π systems.
Co-reporter:Caitlin M. Tressler and Neal J. Zondlo
Organic Letters 2016 Volume 18(Issue 24) pp:6240-6243
Publication Date(Web):December 6, 2016
DOI:10.1021/acs.orglett.6b02858
A practical synthesis of the novel highly fluorinated amino acid Fmoc-perfluoro-tert-butyl tyrosine was developed. The sequence proceeds in two steps from commercially available Fmoc-4-NH2-phenylalanine via diazotization followed by diazonium coupling reaction with perfluoro-tert-butanol. In peptides, perfluoro-tert-butyl tyrosine was detected in 30 s by NMR spectroscopy at 500 nM peptide concentration due to nine chemically equivalent fluorines that are a sharp singlet by 19F NMR. Perfluoro-tert-butyl ether has an estimated σp Hammett substituent constant of +0.30.
Co-reporter:Christina R. Forbes, Anil K. Pandey, Himal K. Ganguly, Glenn P. A. Yap and Neal J. Zondlo
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 7) pp:2327-2346
Publication Date(Web):19 Jan 2016
DOI:10.1039/C5OB02473K
Bioorthogonal reactions allow the introduction of new functionalities into peptides, proteins, and other biological molecules. The most readily accessible amino acids for bioorthogonal reactions have modest conformational preferences or bases for molecular interactions. Herein we describe the synthesis of 4 novel amino acids containing functional groups for bioorthogonal reactions. (2S,4R)- and (2S,4S)-iodophenyl ethers of hydroxyproline are capable of modification via rapid, specific Suzuki and Sonogashira reactions in water. The synthesis of these amino acids, as Boc-, Fmoc- and free amino acids, was achieved through succinct sequences. These amino acids exhibit well-defined conformational preferences, with the 4S-iodophenyl hydroxyproline crystallographically exhibiting β-turn (ϕ, ψ ∼ –80°, 0°) or relatively extended (ϕ, ψ ∼ –80°, +170°) conformations, while the 4R-diastereomer prefers a more compact conformation (ϕ ∼ –60°). The aryloxyproline diastereomers present the aryl groups in a highly divergent manner, suggesting their stereospecific use in molecular design, medicinal chemistry, and catalysis. Thus, the 4R- and 4S-iodophenyl hydroxyprolines can be differentially applied in distinct structural contexts. The pentynoate ester of 4R-hydroxyproline introduces an alkyne functional group within an amino acid that prefers compact conformations. The propargyl thioether of 4-thiolphenylalanine was synthesized via copper-mediated cross-coupling reaction of thioacetic acid with protected 4-iodophenylalanine, followed by thiolysis and alkylation. This amino acid combines an alkyne functional group with an aromatic amino acid and the ability to tune aromatic and side chain properties via sulfur oxidation. These amino acids provide novel loci for peptide functionalization, with greater control of conformation possible than with other amino acids containing these functional groups.
Co-reporter:Michael A. Brister ; Anil K. Pandey ; Agata A. Bielska ;Neal J. Zondlo
Journal of the American Chemical Society 2014 Volume 136(Issue 10) pp:3803-3816
Publication Date(Web):February 21, 2014
DOI:10.1021/ja407156m
Phosphorylation and OGlcNAcylation are dynamic intracellular protein post-translational modifications that frequently are alternatively observed on the same serine and threonine residues. Phosphorylation and OGlcNAcylation commonly occur in natively disordered regions of proteins, and often have opposing functional effects. In the microtubule-associated protein tau, hyperphosphorylation is associated with protein misfolding and aggregation as the neurofibrillary tangles of Alzheimer’s disease, whereas OGlcNAcylation stabilizes the soluble form of tau. A series of peptides derived from the proline-rich domain (residues 174–251) of tau was synthesized, with free Ser/Thr hydroxyls, phosphorylated Ser/Thr (pSer/pThr), OGlcNAcylated Ser/Thr, and diethylphosphorylated Ser/Thr. Phosphorylation and OGlcNAcylation were found by CD and NMR to have opposing structural effects on polyproline helix (PPII) formation, with phosphorylation favoring PPII, OGlcNAcylation opposing PPII, and the free hydroxyls intermediate in structure, and with phosphorylation structural effects greater than OGlcNAcylation. For tau196–209, phosphorylation and OGlcNAcylation had similar structural effects, opposing a nascent α-helix. Phosphomimic Glu exhibited PPII-favoring structural effects. Structural changes due to Thr phosphorylation were greater than those of Ser phosphorylation or Glu, with particular conformational restriction as the dianion, with mean 3JαN = 3.5 Hz (pThr) versus 5.4 Hz (pSer), compared to 7.2, 6.8, and 6.2 Hz for Thr, Ser, and Glu, respectively, values that correlate with the backbone torsion angle ϕ. Dianionic phosphothreonine induced strong phosphothreonine amide protection and downfield amide chemical shifts (δmean = 9.63 ppm), consistent with formation of a stable phosphate-amide hydrogen bond. These data suggest potentially greater structural importance of threonine phosphorylation than serine phosphorylation due to larger induced structural effects.
Co-reporter:Anil K. Pandey, Krista M. Thomas, Christina R. Forbes, and Neal J. Zondlo
Biochemistry 2014 Volume 53(Issue 32) pp:5307-5314
Publication Date(Web):July 30, 2014
DOI:10.1021/bi500696k
Aromatic rings exhibit defined interactions via the unique aromatic π face. Aromatic amino acids interact favorably with proline residues via both the hydrophobic effect and aromatic–proline interactions, C−H/π interactions between the aromatic π face and proline ring C–H bonds. The canonical aromatic amino acids Trp, Tyr, and Phe strongly disfavor a polyproline helix (PPII) when they are present in proline-rich sequences because of the large populations of cis amide bonds induced by favorable aromatic–proline interactions (aromatic–cis-proline and proline–cis-proline–aromatic interactions). We demonstrate the ability to tune polyproline helix conformation and cis–trans isomerism in proline-rich sequences using aromatic electronic effects. Electron-rich aromatic residues strongly disfavor polyproline helix and exhibit large populations of cis amide bonds, while electron-poor aromatic residues exhibit small populations of cis amide bonds and favor polyproline helix. 4-Aminophenylalanine is a pH-dependent electronic switch of polyproline helix, with cis amide bonds favored as the electron-donating amine, but trans amide bonds and polyproline helix preferred as the electron-withdrawing ammonium. Peptides with block proline–aromatic PPXPPXPPXPP sequences exhibited electronically switchable pH-dependent structures. Electron-poor aromatic amino acids provide special capabilities to integrate aromatic residues into polyproline helices and to serve as the basis of aromatic electronic switches to change structure.
Co-reporter:Michael B. Elbaum and Neal J. Zondlo
Biochemistry 2014 Volume 53(Issue 14) pp:
Publication Date(Web):March 18, 2014
DOI:10.1021/bi500117c
OGlcNAcylation and phosphorylation are the major competing intracellular post-translational modifications of serine and threonine residues. The structural effects of both post-translational modifications on serine and threonine were examined within Baldwin model α-helical peptides (Ac-AKAAAAKAAAAKAAGY-NH2 or Ac-YGAKAAAAKAAAAKAA-NH2). At the N-terminus of an α-helix, both phosphorylation and OGlcNAcylation stabilized the α-helix relative to the free hydroxyls, with a larger induced structure for phosphorylation than for OGlcNAcylation, for the dianionic phosphate than for the monoanionic phosphate, and for modifications on threonine than for modifications on serine. Both phosphoserine and phosphothreonine resulted in peptides more α-helical than alanine at the N-terminus, with dianionic phosphothreonine the most α-helix-stabilizing residue here. In contrast, in the interior of the α-helix, both post-translational modifications were destabilizing with respect to the α-helix, with the greatest destabilization seen for threonine OGlcNAcylation at residue 5 and threonine phosphorylation at residue 10, with peptides containing either post-translational modification existing as random coils. At the C-terminus, both OGlcNAcylation and phosphorylation were destabilizing with respect to the α-helix, though the induced structural changes were less than in the interior of the α-helix. In general, the structural effects of modifications on threonine were greater than the effects on serine, because of both the lower α-helical propensity of Thr and the more defined induced structures upon modification of threonine than serine, suggesting threonine residues are particularly important loci for structural effects of post-translational modifications. The effects of serine and threonine post-translational modifications are analogous to the effects of proline on α-helices, with the effects of phosphothreonine being greater than those of proline throughout the α-helix. These results provide a basis for understanding the context-dependent structural effects of these competing protein post-translational modifications.
Co-reporter:Caitlin M. Tressler and Neal J. Zondlo
The Journal of Organic Chemistry 2014 Volume 79(Issue 12) pp:5880-5886
Publication Date(Web):May 28, 2014
DOI:10.1021/jo5008674
(2S,4R)- and (2S,4S)-perfluoro-tert-butyl 4-hydroxyproline were synthesized (as Fmoc-, Boc-, and free amino acids) in 2–5 steps. The key step of each synthesis was a Mitsunobu reaction with perfluoro-tert-butanol, which incorporated a perfluoro-tert-butyl group, with nine chemically equivalent fluorines. Both amino acids were incorporated in model α-helical and polyproline helix peptides. Each amino acid exhibited distinct conformational preferences, with (2S,4R)-perfluoro-tert-butyl 4-hydroxyproline promoting polyproline helix. Peptides containing these amino acids were sensitively detected by 19F NMR, suggesting their use in probes and medicinal chemistry.
Co-reporter:Anil K. Pandey, Glenn P. A. Yap, and Neal J. Zondlo
The Journal of Organic Chemistry 2014 Volume 79(Issue 9) pp:4174-4179
Publication Date(Web):April 10, 2014
DOI:10.1021/jo500367d
(2S,4R)-4-Hydroxyproline(4-nitrobenzoate) was synthesized. The crystal structure revealed an exo ring pucker, with the nitrobenzoate pseudoaxial on the pyrrolidine envelope and antiperiplanar to Cβ and Cδ C–H bonds. The unit cell exhibited variation in Cδ–H/Cγ–O and Cβ–H/Cγ–O torsion angles, with a 15° increase in torsion angle (148° to 163°) observed to result in a 0.018 Å decrease in Cδ–H/Cγ–O bond length, consistent with favorable σC–H → σ*C–O hyperconjugative interactions increasing with greater orbital overlap.
Co-reporter:Anil K. Pandey ; Devan Naduthambi ; Krista M. Thomas ;Neal J. Zondlo
Journal of the American Chemical Society 2013 Volume 135(Issue 11) pp:4333-4363
Publication Date(Web):February 12, 2013
DOI:10.1021/ja3109664
Functionalized proline residues have diverse applications. Herein we describe a practical approach, proline editing, for the synthesis of peptides with stereospecifically modified proline residues. Peptides are synthesized by standard solid-phase peptide synthesis to incorporate Fmoc-hydroxyproline (4R-Hyp). In an automated manner, the Hyp hydroxyl is protected and the remainder of the peptide synthesized. After peptide synthesis, the Hyp protecting group is orthogonally removed and Hyp selectively modified to generate substituted proline amino acids, with the peptide main chain functioning to “protect” the proline amino and carboxyl groups. In a model tetrapeptide (Ac-TYPN-NH2), 4R-Hyp was stereospecifically converted to 122 different 4-substituted prolyl amino acids, with 4R or 4S stereochemistry, via Mitsunobu, oxidation, reduction, acylation, and substitution reactions. 4-Substituted prolines synthesized via proline editing include incorporated structured amino acid mimetics (Cys, Asp/Glu, Phe, Lys, Arg, pSer/pThr), recognition motifs (biotin, RGD), electron-withdrawing groups to induce stereoelectronic effects (fluoro, nitrobenzoate), handles for heteronuclear NMR (19F:fluoro; pentafluorophenyl or perfluoro-tert-butyl ether; 4,4-difluoro; 77SePh) and other spectroscopies (fluorescence, IR: cyanophenyl ether), leaving groups (sulfonate, halide, NHS, bromoacetate), and other reactive handles (amine, thiol, thioester, ketone, hydroxylamine, maleimide, acrylate, azide, alkene, alkyne, aryl halide, tetrazine, 1,2-aminothiol). Proline editing provides access to these proline derivatives with no solution-phase synthesis. All peptides were analyzed by NMR to identify stereoelectronic and steric effects on conformation. Proline derivatives were synthesized to permit bioorthogonal conjugation reactions, including azide–alkyne, tetrazine-trans-cyclooctene, oxime, reductive amination, native chemical ligation, Suzuki, Sonogashira, cross-metathesis, and Diels–Alder reactions. These proline derivatives allowed three parallel bioorthogonal reactions to be conducted in one solution.
Co-reporter:Devan Naduthambi, Santosh Bhor, Michael B. Elbaum, and Neal J. Zondlo
Organic Letters 2013 Volume 15(Issue 18) pp:4892-4895
Publication Date(Web):September 9, 2013
DOI:10.1021/ol402334j
α-Helices are ubiquitous protein recognition elements that bind diverse biomolecular targets. The synthesis of a small molecule scaffold to present the side chains of an α-helix is described. The 1,3,5,7-tetrasubstituted 1,2,3,4-tetrahydronaphthalene scaffold, providing mimicry of the i, i+3, and i+4 positions of an α-helix, was synthesized using a novel MgBr2-catalyzed Friedel–Crafts epoxide cycloalkylation as the key step. Each position may be differentiated via O-alkylation after scaffold synthesis, generating a diversity-oriented approach to readily synthesize proteomimetics for different targets.
Co-reporter:Christina R. Forbes and Neal J. Zondlo
Organic Letters 2012 Volume 14(Issue 2) pp:464-467
Publication Date(Web):January 6, 2012
DOI:10.1021/ol202947f
Aryl thiolates have unique reactive, redox, electronic, and spectroscopic properties. A practical approach to synthesize peptides containing thiophenylalanine has been developed via a novel Cu(I)-mediated cross-coupling reaction between thiolacetic acid and iodophenylalanine-containing peptides in the solid phase. This approach is compatible with all canonical proteinogenic functional groups, providing general access to aryl thiolates in peptides. Peptides containing thiophenylalanine (pKa 6.4) were readily elaborated to contain methyl, allyl, and nitrobenzyl thioethers, disulfides, sulfoxides, sulfones, or sulfonates.
Co-reporter:Alaina M. Brown and Neal J. Zondlo
Biochemistry 2012 Volume 51(Issue 25) pp:
Publication Date(Web):June 5, 2012
DOI:10.1021/bi3002924
Type II polyproline helices (PPII) are a fundamental secondary structure of proteins, common in globular and nonglobular regions and important in cellular signaling. We developed a propensity scale for PPII using a host–guest system with sequence Ac-GPPXPPGY-NH2, where X represents any amino acid. We found that proline has the highest PPII propensity, but most other amino acids display significant PPII propensities. The PPII propensity of leucine was the highest of all propensities of non-proline residues. Alanine and residues with linear side chains displayed the next highest PPII propensities. Three classes of residues displayed lower PPII propensities: β-branched amino acids (Thr, Val, and Ile), short amino acids with polar side chains (Asn, protonated Asp, Ser, Thr, and Cys), and aromatic amino acids (Phe, Tyr, and Trp). tert-Leucine particularly disfavored PPII. The basis of the low PPII propensities of aromatic amino acids in this context was significant cis–trans isomerism, with proline-rich peptides containing aromatic residues exhibiting 45–60% cis amide bonds, due to Pro–cis-Pro–aromatic and aromatic–cis-Pro amide bonds.
Co-reporter:Dr. Shalini Balakrishnan;Michael J. Scheuermann; Dr. Neal J. Zondlo
ChemBioChem 2012 Volume 13( Issue 2) pp:259-270
Publication Date(Web):
DOI:10.1002/cbic.201100638
Abstract
Arginine residues are broadly employed for specific biomolecular recognition, including in protein–protein, protein–DNA, and protein–RNA interactions. Arginine recognition commonly exploits the potential for bidentate electrostatic and hydrogen-bonding interactions. However, in arginine residues, the guanidinium functional group is located at the terminus of a flexible hydrocarbon side chain, which lacks the functionality to contribute to specific arginine-mediated recognition and may entropically disfavor binding. In order to enhance the potential for specificity and affinity in arginine-mediated molecular recognition, we have developed an approach to the synthesis of peptides that incorporates an α-guanidino acid as a novel arginine mimetic. α-Guanidino acids, derived from α-amino acids, with guanidinylation of the amino group, were incorporated stereospecifically into peptides on solid phase via coupling of an Fmoc amino acid to diaminopropionic acid (Dap), Fmoc deprotection, guanidinylation of the amine on solid phase, and deprotection, generating a peptide containing an α-functionalized arginine mimetic. This approach was examined by incorporating arginine mimetics into ligands for the Src, Grb, and Crk SH3 domains at the site of the key recognition arginine. Protein binding was examined for peptides containing guanidino acids derived from Gly, L-Val, L-Phe, L-Trp, D-Val, D-Phe, and D-Trp. We demonstrate that paralogue specificity and target site affinity may be modulated with the use of α-guanidino acid-derived arginine mimetics, generating peptides that exhibit enhanced Src specificity by selection against Grb and peptides that reverse the specificity of the native peptide ligand, with enhancements in Src target specificity of up to 15-fold (1.6 kcal mol−1).
Co-reporter:Susan Carr Zondlo ; Feng Gao ;Neal J. Zondlo
Journal of the American Chemical Society 2010 Volume 132(Issue 16) pp:5619-5621
Publication Date(Web):April 2, 2010
DOI:10.1021/ja100862u
Tyrosine kinases are critical mediators of intracellular signaling and of intracellular responses to extracellular signaling. Changes in tyrosine kinase activity are implicated in numerous human diseases, including cancers, diabetes, and pathogen infectivity. To address questions in tyrosine phosphorylation, we have designed a protein tyrosine kinase-inducible domain, a small, genetically encodable protein motif whose structure is dependent on its tyrosine phosphorylation state. Tyrosine kinase-inducible domain peptides are based on EF-hand loops in which a structurally critical Glu12 residue is replaced by tyrosine at residue 11 or at residue 15 of the protein. Tyrosine kinase-inducible domain peptides bind terbium(III) in a phosphorylation-dependent manner, showing strong terbium luminescence when phosphorylated but weak terbium luminescence when not phosphorylated. Lanthanide binding was confirmed by NMR. A tyrosine kinase-inducible domain peptide, pKID-Abl, was designed to incorporate a recognition sequence of the Abl kinase. Incubation of pKID-Abl with Abl kinase resulted in a large increase in terbium luminescence. This increase in luminescence was abolished when pKID-Abl and Abl kinase were incubated with the Abl kinase inhibitor Gleevec. In addition, incubation of phosphorylated pKID-Abl with the tyrosine phosphatase YOP resulted in a large reduction in terbium luminescence. pKID-Abl was employed as a fluorescent sensor of Abl tyrosine kinase activity in HeLa cell extracts, exhibiting low luminescence with extracts from serum-starved cells and increased luminescence using extracts from EGF-treated cells. These results indicate that tyrosine kinase-inducible domains may be used as sensors of tyrosine kinase and tyrosine phosphatase activity and in the detection of tyrosine kinase inhibitors.
Co-reporter:Christopher W. am Ende;Hai Yun Meng;Mao Ye Dr.;Anil K. Pey ;Neal J. Zondlo Dr.
ChemBioChem 2010 Volume 11( Issue 12) pp:1738-1747
Publication Date(Web):
DOI:10.1002/cbic.201000056
Abstract
Lanthanides have interesting chemical properties; these include luminescent, magnetic, and catalytic functions. Toward the development of proteins incorporating novel functions, we have designed a new lanthanide-binding motif, lanthanide fingers. These were designed based on the Zif268 zinc finger, which exhibits a ββα structural motif. Lanthanide fingers utilize an Asp2Glu2 metal-coordination environment to bind lanthanides through a tetracarboxylate peptide ligand. The iterative design of a general lanthanide-binding peptide incorporated the following key elements: 1) residues with high α-helix and β-sheet propensities in the respective secondary structures; 2) an optimized big box α-helix N-cap; 3) a Schellman α-helix C-cap motif; and 4) an optional D-Pro-Ser type II’ β-turn in the β-hairpin. The peptides were characterized for lanthanide binding by circular dichroism (CD), NMR, and fluorescence spectroscopy. In all instances, stabilization of the peptide secondary structures resulted in an increase in metal affinity. The optimized protein design was a 25-residue peptide that was a general lanthanide-binding motif; this binds all lanthanides examined in a competitive aqueous environment, with a dissociation constant of 9.3 μM for binding Er3+. CD spectra of the peptide-lanthanide complexes are similar to those of zinc fingers and other ββα proteins. Metal binding involves residues from the N-terminal β-hairpin and the C terminal α-helical segments of the peptide. NMR data indicated that metal binding induced a global change in the peptide structure. The D-Pro-Ser type II’ β-turn motif could be replaced by Thr–Ile to generate genetically encodable lanthanide fingers. Replacement of the central Phe with Trp generated genetically encodable lanthanide fingers that exhibited terbium luminescence greater than that of an EF-hand peptide.
Co-reporter:Christina R. Forbes, Anil K. Pandey, Himal K. Ganguly, Glenn P. A. Yap and Neal J. Zondlo
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 7) pp:NaN2346-2346
Publication Date(Web):2016/01/19
DOI:10.1039/C5OB02473K
Bioorthogonal reactions allow the introduction of new functionalities into peptides, proteins, and other biological molecules. The most readily accessible amino acids for bioorthogonal reactions have modest conformational preferences or bases for molecular interactions. Herein we describe the synthesis of 4 novel amino acids containing functional groups for bioorthogonal reactions. (2S,4R)- and (2S,4S)-iodophenyl ethers of hydroxyproline are capable of modification via rapid, specific Suzuki and Sonogashira reactions in water. The synthesis of these amino acids, as Boc-, Fmoc- and free amino acids, was achieved through succinct sequences. These amino acids exhibit well-defined conformational preferences, with the 4S-iodophenyl hydroxyproline crystallographically exhibiting β-turn (ϕ, ψ ∼ –80°, 0°) or relatively extended (ϕ, ψ ∼ –80°, +170°) conformations, while the 4R-diastereomer prefers a more compact conformation (ϕ ∼ –60°). The aryloxyproline diastereomers present the aryl groups in a highly divergent manner, suggesting their stereospecific use in molecular design, medicinal chemistry, and catalysis. Thus, the 4R- and 4S-iodophenyl hydroxyprolines can be differentially applied in distinct structural contexts. The pentynoate ester of 4R-hydroxyproline introduces an alkyne functional group within an amino acid that prefers compact conformations. The propargyl thioether of 4-thiolphenylalanine was synthesized via copper-mediated cross-coupling reaction of thioacetic acid with protected 4-iodophenylalanine, followed by thiolysis and alkylation. This amino acid combines an alkyne functional group with an aromatic amino acid and the ability to tune aromatic and side chain properties via sulfur oxidation. These amino acids provide novel loci for peptide functionalization, with greater control of conformation possible than with other amino acids containing these functional groups.

![(3S,4S,5S,6S)-6-[4-(2-aminoethyl)-2-hydroxy-phenoxy]-3,4,5-trihydroxy-tetrahydropyran-2-carboxylic acid](http://img.cochemist.com/ccimg/36000/35954-65-5.png)
![(3S,4S,5S,6S)-6-[4-(2-aminoethyl)-2-hydroxy-phenoxy]-3,4,5-trihydroxy-tetrahydropyran-2-carboxylic acid](http://img.cochemist.com/ccimg/36000/35954-65-5_b.png)
![L-Threonine, N-[(9H-fluoren-9-ylmethoxy)carbonyl]-O-[3,4,6-tri-O-acetyl-2-(acetylamino)-2-deoxy-β-D-glucopyranosyl]-](/data/chemimg/533500/160168-40-1.png)
![L-Threonine, N-[(9H-fluoren-9-ylmethoxy)carbonyl]-O-[3,4,6-tri-O-acetyl-2-(acetylamino)-2-deoxy-β-D-glucopyranosyl]-](/data/chemimg/533500/160168-40-1_b.png)
![L-Serine,N-[(9H-fluoren-9-ylmethoxy)carbonyl]-O-[3,4,6-tri-O-acetyl-2-(acetylamino)-2-deoxy-b-D-glucopyranosyl]-](http://img.cochemist.com/ccimg/160100/160067-63-0.png)
![L-Serine,N-[(9H-fluoren-9-ylmethoxy)carbonyl]-O-[3,4,6-tri-O-acetyl-2-(acetylamino)-2-deoxy-b-D-glucopyranosyl]-](http://img.cochemist.com/ccimg/160100/160067-63-0_b.png)
![L-Phenylalanine, N-[(1,1-dimethylethoxy)carbonyl]-4-iodo-, 1,1-dimethylethyl ester](/data/chemimg/1792000/217326-89-1.png)
![L-Phenylalanine, N-[(1,1-dimethylethoxy)carbonyl]-4-iodo-, 1,1-dimethylethyl ester](/data/chemimg/1792000/217326-89-1_b.png)


![L-PHENYLALANINE, N-[(1,1-DIMETHYLETHOXY)CARBONYL]-4-MERCAPTO-](http://img.cochemist.com/ccimg/88200/88170-93-8.png)
![L-PHENYLALANINE, N-[(1,1-DIMETHYLETHOXY)CARBONYL]-4-MERCAPTO-](http://img.cochemist.com/ccimg/88200/88170-93-8_b.png)