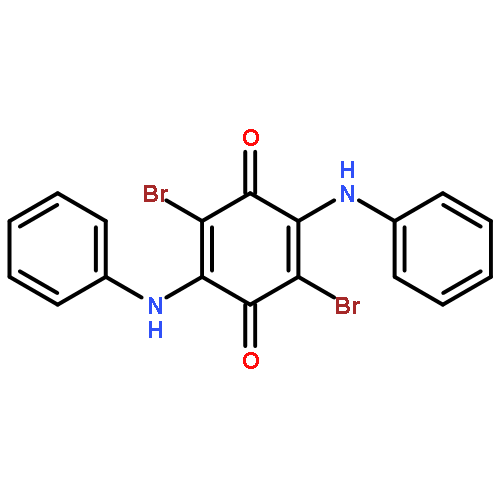
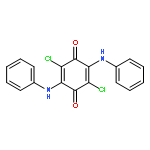
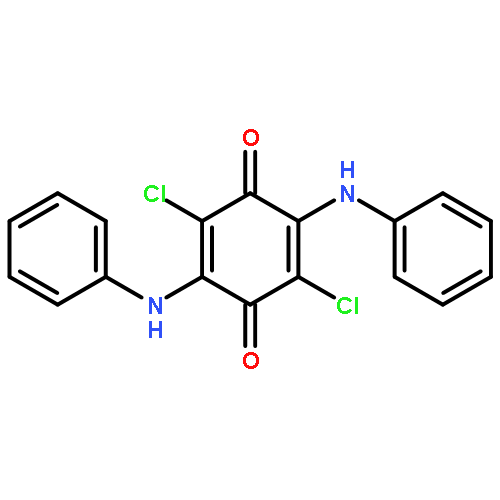
Co-reporter:Agnes E. Thorarinsdottir, Kang Du, James H. P. Collins, and T. David Harris
Journal of the American Chemical Society November 8, 2017 Volume 139(Issue 44) pp:15836-15836
Publication Date(Web):October 13, 2017
DOI:10.1021/jacs.7b08574
We report a Co2-based magnetic resonance (MR) probe that enables the ratiometric quantitation and imaging of pH through chemical exchange saturation transfer (CEST). This approach is illustrated in a series of air- and water-stable CoII2 complexes featuring CEST-active tetra(carboxamide) and/or hydroxyl-substituted bisphosphonate ligands. For the complex bearing both ligands, variable-pH CEST and NMR analyses reveal highly shifted carboxamide and hydroxyl peaks with intensities that increase and decrease with increasing pH, respectively. The ratios of CEST peak intensities at 104 and 64 ppm are correlated with solution pH in the physiological range 6.5–7.6 to construct a linear calibration curve of log(CEST104 ppm/CEST64 ppm) versus pH, which exhibits a remarkably high pH sensitivity of 0.99(7) pH unit–1 at 37 °C. In contrast, the analogous CoII2 complex with a CEST-inactive bisphosphonate ligand exhibits no such pH response, confirming that the pH sensitivity stems from the integration of amide and hydroxyl CEST effects that show base- and acid-catalyzed proton exchange, respectively. Importantly, the pH calibration curve is independent of the probe concentration and is identical in aqueous buffer and fetal bovine serum. Furthermore, phantom images reveal analogous linear pH behavior. The CoII2 probe is stable toward millimolar concentrations of H2PO4–/HPO42–, CO32–, SO42–, CH3COO–, and Ca2+ ions, and more than 50% of melanoma cells remain viable in the presence of millimolar concentrations of the complex. The stability of the probe in physiological environments suggests that it may be suitable for in vivo studies. Together, these results highlight the ability of dinuclear transition metal PARACEST probes to provide a concentration-independent measure of pH, and they provide a potential design strategy toward the development of MR probes for ratiometric pH imaging.
Co-reporter:Jordan A. DeGayner, Ie-Rang Jeon, Lei Sun, Mircea Dincă, and T. David Harris
Journal of the American Chemical Society March 22, 2017 Volume 139(Issue 11) pp:4175-4175
Publication Date(Web):February 23, 2017
DOI:10.1021/jacs.7b00705
We report the magnetism and conductivity for a redox pair of iron-quinoid metal–organic frameworks (MOFs). The oxidized compound, (Me2NH2)2[Fe2L3]·2H2O·6DMF (LH2 = 2,5-dichloro-3,6-dihydroxo-1,4-benzoquinone) was previously shown to magnetically order below 80 K in its solvated form, with the ordering temperature decreasing to 26 K upon desolvation. Here, we demonstrate this compound to exhibit electrical conductivity values up to σ = 1.4(7) × 10–2 S/cm (Ea = 0.26(1) cm–1) and 1.0(3) × 10–3 S/cm (Ea = 0.19(1) cm–1) in its solvated and desolvated forms, respectively. Upon soaking in a DMF solution of Cp2Co, the compound undergoes a single-crystal-to-single-crystal one-electron reduction to give (Cp2Co)1.43(Me2NH2)1.57[Fe2L3]·4.9DMF. Structural and spectroscopic analysis confirms this reduction to be ligand-based, and as such the trianionic framework is formulated as [FeIII2(L3–•)3]3–. Magnetic measurements for this reduced compound reveal the presence of dominant intralayer metal–organic radical coupling to give a magnetically ordered phase below Tc = 105 K, one of the highest reported ordering temperatures for a MOF. This high ordering temperature is significantly increased relative to the oxidized compound, and stems from the overall increase in coupling strength afforded by an additional organic radical. In line with the high critical temperature, the new MOF exhibits magnetic hysteresis up to 100 K, as revealed by variable-field measurements. Finally, this compound is electrically conductive, with values up to σ = 5.1(3) × 10–4 S/cm with Ea = 0.34(1) eV. Taken together, these results demonstrate the unique ability of metal-quinoid MOFs to simultaneously exhibit both high magnetic ordering temperatures and high electrical conductivity.
Co-reporter:Lujia Liu, T. David Harris
Inorganica Chimica Acta 2017 Volume 460(Volume 460) pp:
Publication Date(Web):24 April 2017
DOI:10.1016/j.ica.2016.08.038
•A zinc 2,5-diiminobenzoquinoid chain compound is structurally characterized.•Preservation of the imino groups is confirmed by X-ray crystallography and several spectroscopic measurements.•The compound exhibits an electronic conductivity value of σ = 1.076(5) × 10−12 S·cm−1.The incorporation of amino-substituted benzoquinoid bridging ligands into extended solids may provide a route to access light-weight metal-organic solids with high-temperature magnetic behavior and electronic conductivity. Reaction of Zn(NO3)2·6H2O with 2,5-diamino-3,6-dibromo-1,4-benzoquinone (BrLH2) in DMF at 130 °C gave a black crystalline solid of the compound Zn(BrL)(DMF), which features a structure comprised of one-dimensional chains made up of alternating ZnII ions and BrL2− bridging ligands. The preservation of NH groups on BrL2− was confirmed by X-ray crystallography, a suite of spectroscopic measurements, and elemental analysis. To our knowledge, this chain compound represents the first structurally-characterized solid that contains amino-substituted benzoquinoid bridging ligands. Finally, an ambient temperature electronic conductivity value of σ = 1.076(5) × 10−12 S·cm−1 was measured for the chain compound.The incorporation of amino-substituted benzoquinoid bridging ligands into extended solids may provide a route to access light-weight metal-organic solids with high-temperature magnetic behavior and electronic conductivity. Reaction of Zn(NO3)2·6H2O with 2,5-diamino-3,6-dibromo-1,4-benzoquinone (BrLH2) in DMF at 130 °C gave black block-shaped single crystals of the compound Zn(BrL)(DMF), which features a structure comprised of one-dimensional chains made up of alternating ZnII ions and BrL2− bridging ligands. The preservation of NH groups on BrL2− was confirmed by X-ray crystallography, a suite of spectroscopic measurements, and elemental analysis. To our knowledge, this chain compound represents the first structurally-characterized solid that contains amino-substituted benzoquinoid bridging ligands. Finally, an ambient temperature electronic conductivity value of σ = 1.076(5) × 10−12 S·cm−1 was measured for the chain compound.Download high-res image (54KB)Download full-size image
Co-reporter:Kang Du;Emily A. Waters
Chemical Science (2010-Present) 2017 vol. 8(Issue 6) pp:4424-4430
Publication Date(Web):2017/05/30
DOI:10.1039/C7SC00562H
We demonstrate the ability of a molecular Fe2 complex to enable magnetic resonance (MR)-based ratiometric quantitation of redox status, namely through redox-dependent paramagnetic chemical exchange saturation transfer (PARACEST). Metalation of a tetra(carboxamide) ligand with FeII and/or FeIII in the presence of etidronate ion affords analogous FeII2, FeIIFeIII, and FeIII2 complexes. Both FeII2 and FeIIFeIII complexes give highly-shifted, sharp, and non-overlapping NMR spectra, with multiple resonances for each complex corresponding to exchangeable carboxamide protons. These protons can be selectively irradiated to give CEST peaks at 74 and 83 ppm vs. H2O for the FeIIFeIII complex and at 29, 40 and 68 ppm for the FeII2 complex. The CEST spectra obtained from a series of samples containing mixtures of FeII2 and FeIIFeIII are correlated with independently-determined open-circuit potentials to construct a Nernstian calibration curve of potential vs. CEST peak intensity ratio. In addition, averaged intensities of phantom images collected on a 9.4 T MRI scanner show analogous Nernstian behavior. Finally, both the FeII2 and FeIIFeIII forms of the complex are stable to millimolar concentrations of H2PO4−/HPO42−, CO32−, SO42−, CH3COO−, and Ca2+ ions, and the FeIII2 form is air-stable in aqueous buffer and shows >80% viability in melanoma cells at millimolar concentration. The stability suggests the possible application of this or related complexes for in vivo studies. To our knowledge, this concentration-independent method based on a single Fe2 probe provides the first example of MR-based ratiometric quantitation of redox environment.
Co-reporter:Alexandra I. Gaudette;Agnes E. Thorarinsdottir
Chemical Communications 2017 vol. 53(Issue 96) pp:12962-12965
Publication Date(Web):2017/11/30
DOI:10.1039/C7CC08158H
An FeII complex that features a pH-dependent spin state population, by virtue of a variable ligand protonation state, is described. This behavior leads to a highly pH-dependent 19F NMR chemical shift with a sensitivity of 13.9(5) ppm per pH unit at 37 °C, thereby demonstrating the potential utility of the complex as a 19F chemical shift-based pH sensor.
Co-reporter:Kang Du
Journal of the American Chemical Society 2016 Volume 138(Issue 25) pp:7804-7807
Publication Date(Web):June 8, 2016
DOI:10.1021/jacs.6b03060
The ability of magnetic exchange coupling to enable observation of paramagnetic chemical exchange saturation transfer (PARACEST) in transition metal ions with long electronic relaxation times (τs) is demonstrated. Metalation of the dinucleating, tetra(carboxamide) ligand HL with Cu2+ in the presence of pyrophosphate (P2O7)4– affords the complex [LCuII2(P2O7)]−. Solution-phase variable-temperature magnetic susceptibility data reveal weak ferromagnetic superexchange coupling between the two S = 1/2 CuII centers, with a coupling constant of J = +2.69(5) cm–1, to give an S = 1 ground state. This coupling results in a sharpened NMR line width relative to a GaCu analogue, indicative of a shortening of τs. Presaturation of the amide protons in the Cu2 complex at 37 °C leads to a 14% intensity decrease in the bulk water 1H NMR signal through the CEST effect. Conversely, no CEST effect is observed in the GaCu complex. These results provide the first example of a Cu-based PARACEST magnetic resonance contrast agent and demonstrate the potential to expand the metal ion toolbox for PARACEST agents through introduction of magnetic exchange coupling.
Co-reporter:Ie-Rang Jeon; Lei Sun; Bogdan Negru; Richard P. Van Duyne; Mircea Dincă
Journal of the American Chemical Society 2016 Volume 138(Issue 20) pp:6583-6590
Publication Date(Web):April 26, 2016
DOI:10.1021/jacs.6b02485
We demonstrate that incorporation of a redox-active benzoquinoid ligand into a one-dimensional chain compound can give rise to a material that exhibits simultaneous solid-state redox switching of optical, magnetic, and electronic properties. Metalation of the ligand 4,5-bis(pyridine-2-carboxamido)-1,2-catechol (N,OLH4) with MnIII affords the chain compound Mn(N,OL)(DMSO). Structural and spectroscopic analysis of this compound show the presence of MnII centers bridged by N,OL2– ligands, resulting partially from a spontaneous ligand-to-metal electron transfer. Upon soaking in a solution of the reductant Cp2Co, Mn(N,OL)(DMSO) undergoes a ligand-centered solid-state reduction to [Mn(N,OL)]−, as revealed by a suite of techniques, including Raman and X-ray absorption spectroscopy. The ligand-based reduction engenders a dramatic modulation of the physical properties of the chain compound. An electrochromic response, evidenced by a color change from dark green to dark purple is accompanied by a nearly 40-fold increase in magnetic coupling strength, from J = −0.38(1) to −15.6(2) cm–1, and a 10,000-fold increase in electronic conductivity, from σ = 2.33(1) × 10–12 S/cm (Ea = 0.64(1) eV) to 8.61(1) × 10–8 S/cm (Ea = 0.39(1) eV). Importantly, the chemical reduction is reversible: treatment of the reduced compound with [Cp2Fe]+ regenerates the oxidized chain. Taken together, these results highlight the ability of benzoquinoid ligands to facilitate solid-state ligand-based redox reactions in nonporous coordination solids, giving rise to reversible switching of optical properties, magnetic exchange interactions, and electronic conductivity.
Co-reporter:M. L. Kelty, W. Morris, A. T. Gallagher, J. S. Anderson, K. A. Brown, C. A. Mirkin and T. D. Harris
Chemical Communications 2016 vol. 52(Issue 50) pp:7854-7857
Publication Date(Web):01 Jun 2016
DOI:10.1039/C6CC03264H
We describe and employ a high-throughput screening method to accelerate the synthesis and identification of pure-phase, nanocrystalline metal–organic frameworks (MOFs). We demonstrate the efficacy of this method through its application to a series of porphyrinic zirconium MOFs, resulting in the isolation of MOF-525, MOF-545, and PCN-223 on the nanoscale.
Co-reporter:Ie-Rang Jeon and T. David Harris
Chemical Communications 2016 vol. 52(Issue 5) pp:1006-1008
Publication Date(Web):11 Nov 2015
DOI:10.1039/C5CC08482B
The asymmetric redox-active ligand 4,5-bis(pyridine-2-carboxamido)-1,2-catechol (N,OLH4) is prepared and metalated to afford the hexanuclear complex [Mn6(N,OL)6]6−. Structural analysis and magnetic measurements reveal this complex to feature MnII ions bridged by N,OL3−˙ radicals, which are antiferromagnetically coupled to give an S = 12 ground state.
Co-reporter:Ie-Rang Jeon; Bogdan Negru; Richard P. Van Duyne
Journal of the American Chemical Society 2015 Volume 137(Issue 50) pp:15699-15702
Publication Date(Web):November 17, 2015
DOI:10.1021/jacs.5b10382
The incorporation of tetraoxolene radical bridging ligands into a microporous magnetic solid is demonstrated. Metalation of the redox-active bridging ligand 2,5-dichloro-3,6-dihydroxy-1,4-benzoquinone (LH2) with FeII affords the solid (Me2NH2)2[Fe2L3]·2H2O·6DMF. Analysis of X-ray diffraction, Raman spectra, and Mössbauer spectra confirm the presence of FeIII centers with mixed-valence ligands of the form (L3)8– that result from a spontaneous electron transfer from FeII to L2–. Upon removal of DMF and H2O solvent molecules, the compound undergoes a slight structural distortion to give the desolvated phase (Me2NH2)2[Fe2L3], and a fit to N2 adsorption data of this activated compound gives a BET surface area of 885(105) m2/g. Dc magnetic susceptibility measurements reveal a spontaneous magnetization below 80 and 26 K for the solvated and the activated solids, respectively, with magnetic hysteresis up to 60 and 20 K. These results highlight the ability of redox-active tetraoxolene ligands to support the formation of a microporous magnet and provide the first example of a structurally characterized extended solid that contains tetraoxolene radical ligands.
Co-reporter:Alexandra I. Gaudette; Ie-Rang Jeon; John S. Anderson; Fernande Grandjean; Gary J. Long
Journal of the American Chemical Society 2015 Volume 137(Issue 39) pp:12617-12626
Publication Date(Web):September 16, 2015
DOI:10.1021/jacs.5b07251
The ability of a benzoquinonoid bridging ligand to mediate double-exchange coupling in a mixed-valence Fe2 complex is demonstrated. Metalation of the bridging ligand 2,5-di(2,6-dimethylanilino)-3,6-dibromo-1,4-benzoquinone (LH2) with FeII in the presence of the capping ligand tris((6-methyl-2-pyridyl)methyl)amine (Me3TPyA) affords the dinuclear complex [(Me3TPyA)2FeII2(L)]2+. The dc magnetic measurements, in conjunction with X-ray diffraction and Mössbauer spectroscopy, reveal the presence of weak ferromagnetic superexchange coupling between FeII centers through the diamagnetic bridging ligand to give an S = 4 ground state. The ac magnetic susceptibility measurements, collected in a small dc field, show this complex to behave as a single-molecule magnet with a relaxation barrier of Ueff = 14(1) cm–1. The slow magnetic relaxation in the FeII2 complex can be switched off through one-electron oxidation to the mixed-valence congener [(Me3TPyA)2Fe2(L)]3+, where X-ray diffraction and Mössbauer spectroscopy indicate a metal-centered oxidation. The dc magnetic measurements show an S = 9/2 ground state for the mixed-valence complex, stemming from strong ferromagnetic exchange coupling that is best described considering electron hopping through a double-exchange coupling mechanism, with a double-exchange parameter of B = 69(4) cm–1. In accordance with double-exchange, an intense feature is observed in the near-infrared region and is assigned as an intervalence charge-transfer band. The rate of intervalence electron hopping is comparable to that of the Mössbauer time scale, such that variable-temperature Mössbauer spectra reveal a thermally activated transition from a valence-trapped to detrapped state and provide an activation energy for electron hopping of 63(8) cm–1. These results demonstrate the ability of quinonoid ligands to mediate electron hopping between high-spin metal centers, by providing the first example of an Fe complex that exhibits double-exchange through an organic bridging ligand and the largest metal–metal separation yet observed in any metal complex with double-exchange coupling.
Co-reporter:Jordan A. DeGayner, Ie-Rang Jeon and T. David Harris
Chemical Science 2015 vol. 6(Issue 11) pp:6639-6648
Publication Date(Web):18 Aug 2015
DOI:10.1039/C5SC02725J
The ability of tetraazalene radical bridging ligands to mediate exceptionally strong magnetic exchange coupling across a range of transition metal complexes is demonstrated. The redox-active bridging ligand N,N′,N′′,N′′′-tetra(2-methylphenyl)-2,5-diamino-1,4-diiminobenzoquinone (NMePhLH2) was metalated to give the series of dinuclear complexes [(TPyA)2M2(NMePhL2−)]2+ (TPyA = tris(2-pyridylmethyl)amine, M = MnII, FeII, CoII). Variable-temperature dc magnetic susceptibility data for these complexes reveal the presence of weak superexchange interactions between metal centers, and fits to the data provide coupling constants of J = −1.64(1) and −2.16(2) cm−1 for M = MnII and FeII, respectively. One-electron reduction of the complexes affords the reduced analogues [(TPyA)2M2(NMePhL3−˙)]+. Following a slightly different synthetic procedure, the related complex [(TPyA)2CrIII2(NMePhL3−˙)]3+ was obtained. X-ray diffraction, cyclic voltammetry, and Mössbauer spectroscopy indicate the presence of radical NMePhL3−˙ bridging ligands in these complexes. Variable-temperature dc magnetic susceptibility data of the radical-bridged species reveal the presence of strong magnetic interactions between metal centers and ligand radicals, with simulations to data providing exchange constants of J = −626(7), −157(7), −307(9), and −396(16) cm−1 for M = CrIII, MnII, FeII, and CoII, respectively. Moreover, the strength of magnetic exchange in the radical-bridged complexes increases linearly with decreasing M–L bond distance in the oxidized analogues. Finally, ac magnetic susceptibility measurements reveal that [(TPyA)2Fe2(NMePhL3−˙)]+ behaves as a single-molecule magnet with a relaxation barrier of Ueff = 52(1) cm−1. These results highlight the ability of redox-active tetraazalene bridging ligands to enable dramatic enhancement of magnetic exchange coupling upon redox chemistry and provide a rare opportunity to examine metal–radical coupling trends across a transmetallic series of complexes.
Co-reporter:Jesse G. Park; Ie-Rang Jeon
Inorganic Chemistry 2015 Volume 54(Issue 1) pp:359-369
Publication Date(Web):December 12, 2014
DOI:10.1021/ic5025586
A series of four isostructural FeII2 complexes, [(TPyA)2Fe2(XL)]2+ (TPyA = tris(2-pyridylmethyl)amine; XL2– = doubly deprotonated form of 3,6-disubstituted-2,5-dianilino-1,4-benzoquinone; X = H, Br, Cl, and F), were synthesized to enable a systematic study of electronic effects on spin crossover behavior. Comparison of X-ray diffraction data for these complexes reveals the sole presence of high-spin FeII at 225 K and mixtures of high-spin and low-spin FeII at 100 K, which is indicative of incomplete spin crossover. In addition, crystal packing diagrams show that these complexes are well-isolated from one another in the solid state, owing primarily to the presence of bulky tetra(aryl)borate counteranions, such that spin crossover is likely not significantly affected by intermolecular interactions. Variable-temperature dc magnetic susceptibility data confirm the structural observations and reveal that 54(1), 56(1), 62(1), and 84(1)% of FeII centers remain high-spin even below 65 K. Moreover, fits to magnetic data provide crossover temperatures of T1/2 = 160(1), 124(1), 121(1), and 110(1) K for X = H, Br, Cl, and F, respectively, along with enthalpies of ΔH = 11.4(3), 8.5(3), 8.3(3), and 7.5(2) kJ/mol, respectively. These parameters decrease with increasing electronegativity of X and thus increasing electron-withdrawing character of XL2–, suggesting that the observed trends originate primarily from inductive effects of X. Moreover, when plotted as a function of the Pauling electronegativity of X, both T1/2 and ΔH undergo a linear decrease. Further analyses of the low-temperature magnetic data and variable-temperature Mössbauer spectroscopy suggest that the incomplete spin crossover behavior in [(TPyA)2Fe2(XL)]2+ is best described as a transition from purely [FeHS-FeHS] (HS = high-spin) complexes at high temperature to a mixture of [FeHS-FeHS] and [FeHS-FeLS] (LS = low-spin) complexes at low temperature, with the number of [FeHS-FeHS] species increasing with decreasing electron-withdrawing character of XL2–.
Co-reporter:John S. Anderson ; Audrey T. Gallagher ; Jarad A. Mason
Journal of the American Chemical Society 2014 Volume 136(Issue 47) pp:16489-16492
Publication Date(Web):November 7, 2014
DOI:10.1021/ja5103103
The porphyrinic metal–organic framework (MOF) PCN-224 is metalated with FeII to yield a 4-coordinate ferrous heme-containing compound. The heme center binds O2 at −78 °C to give a 5-coordinate heme-O2 complex. For the first time, this elusive species is structurally characterized, revealing an FeIII center coordinated to superoxide via an end-on, η1 linkage. Mössbauer spectroscopy supports the structural observations and indicates the presence of a low-spin electronic configuration for FeIII. Finally, variable-temperature O2 adsorption data enable quantification of the Fe–O2 interaction, providing a binding enthalpy of −34(4) kJ/mol. This value is nearly half of that observed for comparable 6-coordinate, imidazole-bound heme-O2 complexes, a difference that further illustrates the importance of axial ligands in biological heme-mediated O2 transport and storage. These results demonstrate the ability of a MOF, by virtue of its rigid solid-state structure, to enable isolation and thorough characterization of a species that can only be observed transiently in molecular form.
Co-reporter:Ie-Rang Jeon, Jesse G. Park, Chad R. Haney and T. David Harris
Chemical Science 2014 vol. 5(Issue 6) pp:2461-2465
Publication Date(Web):04 Apr 2014
DOI:10.1039/C4SC00396A
We demonstrate the potential utility of spin crossover iron(II) complexes as temperature-responsive paramagnetic chemical exchange saturation transfer (PARACEST) contrast agents in magnetic resonance imaging (MRI) thermometry. This approach is illustrated in the two molecular complexes [Fe(3-bpp)2]2+ (3-bpp = 2,6-di(pyrazol-3-yl)pyridine) and [(Me2NPY5Me2)Fe(H2O)]2+ (Me2NPY5Me2 = 4-dimethylamino-2,6-bis(1,1-bis(2-pyridyl)ethyl)pyridine). Variable-temperature magnetic susceptibility data collected for aqueous solutions of these complexes reveal that they exhibit spin crossover behaviour in H2O over the temperature range 20–60 °C. Selective presaturation of pyrazolyl and coordinated water protons in these complexes, respectively, leads to a significant decrease in the NMR signal intensity of bulk water protons through CEST. The corresponding Z-spectra reveal a strong linear temperature dependence of chemical shift of those protons, 0.23(1) ppm °C−1 and 1.02(1) ppm °C−1, respectively, arising from thermal conversion between low-spin S = 0 and high-spin S = 2 iron(II), representing 23- and 100-fold higher sensitivity than that afforded by conventional proton resonance frequency shift thermometry. Finally, temperature maps generated for an aqueous solution containing [(Me2NPY5Me2)Fe(H2O)]2+ show excellent agreement with independently measured temperatures of the solution.
Co-reporter:Ie-Rang Jeon ; Jesse G. Park ; Dianne J. Xiao
Journal of the American Chemical Society () pp:
Publication Date(Web):October 28, 2013
DOI:10.1021/ja409927v
One-electron reduction of the complex [(TPyA)2FeII2(NPhL2–)]2+ (TPyA = tris(2-pyridylmethyl)amine, NPhLH2 = azophenine = N,N′,N″,N‴-tetraphenyl-2,5-diamino-1,4-diiminobenzoquinone) affords the complex [(TPyA)2FeII2(NPhL3–•)]+. X-ray diffraction and Mössbauer spectroscopy confirm that the reduction occurs on NPhL2– to give an S = 1/2 radical bridging ligand. Dc magnetic susceptibility measurements demonstrate the presence of extremely strong direct antiferromagnetic exchange between S = 2 FeII centers and NPhL3–• in the reduced complex, giving an S = 7/2 ground state with an estimated coupling constant magnitude of |J| ≥ 900 cm–1. Mössbauer spectroscopy and ac magnetic susceptibility reveal that this complex behaves as a single-molecule magnet with a spin relaxation barrier of Ueff = 50(1) cm–1. To our knowledge, this complex exhibits by far the strongest magnetic exchange coupling ever to be observed in a single-molecule magnet.
Co-reporter:Kang Du, Emily A. Waters and T. David Harris
Chemical Science (2010-Present) 2017 - vol. 8(Issue 6) pp:NaN4430-4430
Publication Date(Web):2017/04/19
DOI:10.1039/C7SC00562H
We demonstrate the ability of a molecular Fe2 complex to enable magnetic resonance (MR)-based ratiometric quantitation of redox status, namely through redox-dependent paramagnetic chemical exchange saturation transfer (PARACEST). Metalation of a tetra(carboxamide) ligand with FeII and/or FeIII in the presence of etidronate ion affords analogous FeII2, FeIIFeIII, and FeIII2 complexes. Both FeII2 and FeIIFeIII complexes give highly-shifted, sharp, and non-overlapping NMR spectra, with multiple resonances for each complex corresponding to exchangeable carboxamide protons. These protons can be selectively irradiated to give CEST peaks at 74 and 83 ppm vs. H2O for the FeIIFeIII complex and at 29, 40 and 68 ppm for the FeII2 complex. The CEST spectra obtained from a series of samples containing mixtures of FeII2 and FeIIFeIII are correlated with independently-determined open-circuit potentials to construct a Nernstian calibration curve of potential vs. CEST peak intensity ratio. In addition, averaged intensities of phantom images collected on a 9.4 T MRI scanner show analogous Nernstian behavior. Finally, both the FeII2 and FeIIFeIII forms of the complex are stable to millimolar concentrations of H2PO4−/HPO42−, CO32−, SO42−, CH3COO−, and Ca2+ ions, and the FeIII2 form is air-stable in aqueous buffer and shows >80% viability in melanoma cells at millimolar concentration. The stability suggests the possible application of this or related complexes for in vivo studies. To our knowledge, this concentration-independent method based on a single Fe2 probe provides the first example of MR-based ratiometric quantitation of redox environment.
Co-reporter:Ie-Rang Jeon and T. David Harris
Chemical Communications 2016 - vol. 52(Issue 5) pp:NaN1008-1008
Publication Date(Web):2015/11/11
DOI:10.1039/C5CC08482B
The asymmetric redox-active ligand 4,5-bis(pyridine-2-carboxamido)-1,2-catechol (N,OLH4) is prepared and metalated to afford the hexanuclear complex [Mn6(N,OL)6]6−. Structural analysis and magnetic measurements reveal this complex to feature MnII ions bridged by N,OL3−˙ radicals, which are antiferromagnetically coupled to give an S = 12 ground state.
Co-reporter:Jordan A. DeGayner, Ie-Rang Jeon and T. David Harris
Chemical Science (2010-Present) 2015 - vol. 6(Issue 11) pp:NaN6648-6648
Publication Date(Web):2015/08/18
DOI:10.1039/C5SC02725J
The ability of tetraazalene radical bridging ligands to mediate exceptionally strong magnetic exchange coupling across a range of transition metal complexes is demonstrated. The redox-active bridging ligand N,N′,N′′,N′′′-tetra(2-methylphenyl)-2,5-diamino-1,4-diiminobenzoquinone (NMePhLH2) was metalated to give the series of dinuclear complexes [(TPyA)2M2(NMePhL2−)]2+ (TPyA = tris(2-pyridylmethyl)amine, M = MnII, FeII, CoII). Variable-temperature dc magnetic susceptibility data for these complexes reveal the presence of weak superexchange interactions between metal centers, and fits to the data provide coupling constants of J = −1.64(1) and −2.16(2) cm−1 for M = MnII and FeII, respectively. One-electron reduction of the complexes affords the reduced analogues [(TPyA)2M2(NMePhL3−˙)]+. Following a slightly different synthetic procedure, the related complex [(TPyA)2CrIII2(NMePhL3−˙)]3+ was obtained. X-ray diffraction, cyclic voltammetry, and Mössbauer spectroscopy indicate the presence of radical NMePhL3−˙ bridging ligands in these complexes. Variable-temperature dc magnetic susceptibility data of the radical-bridged species reveal the presence of strong magnetic interactions between metal centers and ligand radicals, with simulations to data providing exchange constants of J = −626(7), −157(7), −307(9), and −396(16) cm−1 for M = CrIII, MnII, FeII, and CoII, respectively. Moreover, the strength of magnetic exchange in the radical-bridged complexes increases linearly with decreasing M–L bond distance in the oxidized analogues. Finally, ac magnetic susceptibility measurements reveal that [(TPyA)2Fe2(NMePhL3−˙)]+ behaves as a single-molecule magnet with a relaxation barrier of Ueff = 52(1) cm−1. These results highlight the ability of redox-active tetraazalene bridging ligands to enable dramatic enhancement of magnetic exchange coupling upon redox chemistry and provide a rare opportunity to examine metal–radical coupling trends across a transmetallic series of complexes.
Co-reporter:M. L. Kelty, W. Morris, A. T. Gallagher, J. S. Anderson, K. A. Brown, C. A. Mirkin and T. D. Harris
Chemical Communications 2016 - vol. 52(Issue 50) pp:NaN7857-7857
Publication Date(Web):2016/06/01
DOI:10.1039/C6CC03264H
We describe and employ a high-throughput screening method to accelerate the synthesis and identification of pure-phase, nanocrystalline metal–organic frameworks (MOFs). We demonstrate the efficacy of this method through its application to a series of porphyrinic zirconium MOFs, resulting in the isolation of MOF-525, MOF-545, and PCN-223 on the nanoscale.
Co-reporter:Ie-Rang Jeon, Jesse G. Park, Chad R. Haney and T. David Harris
Chemical Science (2010-Present) 2014 - vol. 5(Issue 6) pp:NaN2465-2465
Publication Date(Web):2014/04/04
DOI:10.1039/C4SC00396A
We demonstrate the potential utility of spin crossover iron(II) complexes as temperature-responsive paramagnetic chemical exchange saturation transfer (PARACEST) contrast agents in magnetic resonance imaging (MRI) thermometry. This approach is illustrated in the two molecular complexes [Fe(3-bpp)2]2+ (3-bpp = 2,6-di(pyrazol-3-yl)pyridine) and [(Me2NPY5Me2)Fe(H2O)]2+ (Me2NPY5Me2 = 4-dimethylamino-2,6-bis(1,1-bis(2-pyridyl)ethyl)pyridine). Variable-temperature magnetic susceptibility data collected for aqueous solutions of these complexes reveal that they exhibit spin crossover behaviour in H2O over the temperature range 20–60 °C. Selective presaturation of pyrazolyl and coordinated water protons in these complexes, respectively, leads to a significant decrease in the NMR signal intensity of bulk water protons through CEST. The corresponding Z-spectra reveal a strong linear temperature dependence of chemical shift of those protons, 0.23(1) ppm °C−1 and 1.02(1) ppm °C−1, respectively, arising from thermal conversion between low-spin S = 0 and high-spin S = 2 iron(II), representing 23- and 100-fold higher sensitivity than that afforded by conventional proton resonance frequency shift thermometry. Finally, temperature maps generated for an aqueous solution containing [(Me2NPY5Me2)Fe(H2O)]2+ show excellent agreement with independently measured temperatures of the solution.
.jpg)


![2-Pyridinemethanamine, 6-methyl-N,N-bis[(6-methyl-2-pyridinyl)methyl]-](http://img.cochemist.com/ccimg/25600/25599-08-0.png)
![2-Pyridinemethanamine, 6-methyl-N,N-bis[(6-methyl-2-pyridinyl)methyl]-](http://img.cochemist.com/ccimg/25600/25599-08-0_b.png)