Co-reporter:Jonathan A. Dines, Charles M. Marson
Tetrahedron 2016 Volume 72(Issue 52) pp:8584-8592
Publication Date(Web):29 December 2016
DOI:10.1016/j.tet.2016.11.039
Alkylation of malonamic esters provides a direct approach to derivatives of suberoylanilide hydroxamic acid (SAHA) that are branched at the amide carbon atom, a location pivotal for enhancing biological and therapeutic activity. Alkylations use NaH in THF followed by addition of the ester of 6-bromohexanoic acid; no protection of the amidic NH group is necessary. By this means, carboxylic acid, ester, amide, hydroxymethyl and 2-benzimidazolyl branching units have been appended to the SAHA backbone. Routes to vary one of the branching units at a time have been developed.
Co-reporter:Charles M. Marson, Kin Cheung Yau
Tetrahedron 2015 Volume 71(Issue 39) pp:7459-7469
Publication Date(Web):30 September 2015
DOI:10.1016/j.tet.2015.06.052
A flexible route to piperidine-2,4-diones variously substituted at the 6-, 5,6- and 2,6-positions, both with and without 1-substitution, is described; no N-protective group is required. A related regioselective Dieckmann cyclisation is also described that uses Davies' α-methylbenzylamine auxiliary and affords 6-substituted piperidine-2,4-diones enantioselectively.
Co-reporter:Charles M. Marson ; Christopher J. Matthews ; Elena Yiannaki ; Stephen J. Atkinson ; Peter E. Soden ; Lena Shukla ; Nermina Lamadema ;N. Shaun B. Thomas
Journal of Medicinal Chemistry 2013 Volume 56(Issue 15) pp:6156-6174
Publication Date(Web):July 6, 2013
DOI:10.1021/jm400634n
The synthesis of a novel series of potent chiral inhibitors of histone deacetylase (HDAC) is described that contain a heterocyclic capping group and a N-(2-aminophenyl)benzamide unit that binds in the active site. In vitro assays for the inhibition of HDAC1, HDAC2, HDAC3-NCoR1, and HDAC8 by the N-(2-aminophenyl)benzamide 24a gave respective IC50 values of 930, 85, 12, and 4100 nM, exhibiting class I selectivity and potent inhibition of HDAC3-NCoR1. Both imidazolinone and thiazoline rings are shown to be effective replacements for the pyrimidine ring present in many other 2-(aminophenyl)benzamides previously reported, an example of each ring system at 1 μM causing an increase in histone H3K9 acetylation in the human cell lines Jurkat and HeLa and an increase in cell death consistent with induction of apoptosis. Inhibition of the growth of MCF-7, A549, DU145, and HCT116 cell lines by 24a was observed, with respective IC50 values of 5.4, 5.8, 6.4, and 2.2 mM.
Co-reporter:Patrizia Ferretti, Kin Pong U, Barbora Vagaska, Rohan Merchant, Christopher J. Matthews and Charles M. Marson
MedChemComm 2013 vol. 4(Issue 7) pp:1109-1113
Publication Date(Web):30 May 2013
DOI:10.1039/C3MD00091E
The synthesis and biological properties of a structurally novel, potent and non-peptidic inhibitor of peptidylarginine deiminase are described. The novel drug-like PAD inhibitor contains a 3,5-dihydroimidazol-4-one ring that replaces the acyclic guanidine-binding unit present in arginine residues. This new drug-like PAD inhibitor was effective at 100 nM or below and could have relevance to diseases in which PAD expression is up-regulated, including rheumatoid arthritis, cancer, multiple sclerosis, and neural injury.
Co-reporter:Mehrnoosh Ostovar, Charles M. Marson
Tetrahedron 2013 69(32) pp: 6639-6647
Publication Date(Web):
DOI:10.1016/j.tet.2013.05.125
Co-reporter:Charles M. Marson
Chemical Society Reviews 2012 vol. 41(Issue 23) pp:7712-7722
Publication Date(Web):24 Aug 2012
DOI:10.1039/C2CS35183H
Reactions in which several components are combined in sequence, and without isolation of intermediates, are greatly sought because of the inherent molecular diversity, efficiency, and atom-economy. However, organocatalytic reactions, employing an organic catalyst to assemble products of high enantiomeric excess (a single optical isomer), are also cutting-edge methodology. This tutorial review covers the overlap of these two areas, outling the structural diversity and stereocontrol arising from one-pot combinations of at least three components, powerful approaches with great potential that minimise formation of by-products and operating costs.
Co-reporter:Charles M. Marson
Chemical Reviews 2011 Volume 111(Issue 11) pp:7121
Publication Date(Web):August 15, 2011
DOI:10.1021/cr900166w
Co-reporter:E. S. K. Assem;S. Mann;B. Y. C. Wan;C. M. Marson
Inflammation Research 2010 Volume 59( Issue 2 Supplement) pp:235-237
Publication Date(Web):2010 March
DOI:10.1007/s00011-009-0137-0
The aim of this study was to investigate the ability of (a) antioxidants, some related to α-lipoic acid (LA), (b) histone deacetylase (HDAC) inhibitors, and (c) hybrid compounds possessing both α-lipoic acid-derived antioxidant properties and HDAC inhibitory activity to inhibit guinea pig smooth muscle contraction.Guinea pig isolated tracheal rings (GPTR) were prepared and their isometric tension measured using a transducer. Histamine, carbachol and 5-hydroxytryptamine (5-HT) served as agonists. Tests with antigen (ovalbumin) used GPTR from sensitised guinea pigs or rings from non-sensitised animals that had been incubated for at least 2 h with diluted serum from sensitised animals. All antioxidants tested showed a relaxant effect on resting tension and on tension induced by histamine or carbachol, with EC50(s) of 0.2–5.0 mM and a rank order of potency: LA derivatives > glutathione (GSH) > ascorbic acid (AA). However, low concentrations (<50 μM) of GSH, AA and LA potentiated histamine-induced contractions. The benzamide HDAC inhibitor MGCD0103 inhibited mast cell activation and GPTR contraction produced by antigen and certain agonists, although a 2–6 h pre-incubation was required for those effects to be apparent. Two LA–benzamide HDAC hybrid compounds, UCL M084 and UCL M109 inhibited GPTR contraction after 30 min pre-incubation; however, even after long pre-incubation (up to 6 h) those hybrid compounds showed less potent inhibition of agonist-induced contraction than did MGCD0103.The results showed that GSH more potently inhibited contraction induced by histamine than that induced by 5-HT or carbachol, whereas LA, and especially UCL M084 and UCL M109, more potently blocked contraction induced by carbachol and 5-HT than that induced by histamine. For GSH, and possibly also for LA-type compounds, the inhibition of agonist-induced tracheal smooth muscle contractions may be due to NO formation. This study did not detect a synergistic relaxant effect in two compounds incorporating the structural union of a benzamide HDAC inhibitor terminus with a LA-derived antioxidant moiety.
Co-reporter:B. Y. C. Wan;S. Mann;E. S. K. Assem;C. M. Marson
Inflammation Research 2010 Volume 59( Issue 2 Supplement) pp:231-233
Publication Date(Web):2010 March
DOI:10.1007/s00011-009-0136-1
The effects of the endogenous antioxidant α-lipoic acid on guinea pig colon smooth muscle contraction (Gpcc) induced by hydrogen peroxide were examined. Having previously shown that the histone deacetylase (HDAC) benzamide inhibitor MGCD0103 inhibits guinea-pig smooth muscle contraction, as do various sulfur-containing antioxidants, we asked whether hybrid compounds possessing both α-lipoic acid-derived antioxidant properties and HDAC inhibitory activity could inhibit Gpcc.Guinea pig colon (Gpc) was incubated at 37°C with Krebs buffer; the four stimulants—hydrogen peroxide, carbachol, histamine, and sodium fluoride—were added independently. The response to each stimulant alone was compared with that in the presence of each of the test compounds: MGCD0103, α-lipoic acid, and two of their hybrids, UCL M084 and UCL M109.NaF (10 mM), carbachol (0.05 μM), histamine (0.1 μM), and hydrogen peroxide (1 μM) produced Gpcc of about 50–60% above basal level. With the exception of MGCD0103 against hydrogen peroxide, all four test compounds at 1 μM—MGCD0103, α-lipoic acid, UCL M084, and UCL M109—produced a significant inhibition of 35–60% of Gpcc induced by hydrogen peroxide, NaF, and carbachol, although none reduced histamine or ovalbumin-induced Gpcc. Benzalkonium chloride (Bcl), a G-protein inhibitor, reduced the hydrogen peroxide-induced Gpcc by 35%.Contraction by stimulants used to induce Gpcc is known to involve G-proteins. All four test compounds—MGCD0103, α-lipoic acid and two of their hybrids, UCL M084 and UCL M109—reduced Gpcc induced by NaF and carbachol, suggesting that G-protein pathway involvement is relevant to the action of the test compounds, as is also indicated by the Bcl-induced inhibition of hydrogen peroxide-induced contractions. Additionally, α-lipoic acid and the two hybrids showed >30% inhibition of hydrogen peroxide-induced contractions, consistent with the antioxidant properties of the 1,2-dithiolane ring.
Co-reporter:Charles M. Marson, Esra Edaan, James M. Morrell, Simon J. Coles, Michael B. Hursthouse and David T. Davies
Chemical Communications 2007 (Issue 24) pp:2494-2496
Publication Date(Web):26 Mar 2007
DOI:10.1039/B701548H
3(2H)-Furanones can be prepared by a catalytic asymmetric protocol from enynones, which, if electron-rich, require only one reagent and involve two reactions in a single operation—a domino process.
Co-reporter:Charles M. Marson, Thevaki Mahadevan, Jon Dines, Stéphane Sengmany, James M. Morrell, John P. Alao, Simon P. Joel, David M. Vigushin, R. Charles Coombes
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 1) pp:136-141
Publication Date(Web):1 January 2007
DOI:10.1016/j.bmcl.2006.09.085
Syntheses of aryloxyalkanoic acid hydroxyamides are described, all of which are potent inhibitors of histone deacetylase, some being more potent in vitro than trichostatin A (IC50 = 3 nM). Variation of the substituents on the benzene ring as well as fusion of a second ring have marked effects on potency, in vitro IC50 values down to 1 nM being obtained.Aryl ether inhibitors of histone deacetylase are described.
Co-reporter:Charles M. Marson and Mona Saadi
Organic & Biomolecular Chemistry 2006 vol. 4(Issue 21) pp:3892-3893
Publication Date(Web):02 Oct 2006
DOI:10.1039/B613791C
A protocol for the construction of poly-oxazoles with consecutive 2,4′-linkages is described, and has afforded an efficient route to a penta-oxazole which demarcates a route to telomestatin and related macrocyclic poly-oxazole systems.
Co-reporter:Charles M. Marson, Robert C. Melling, Simon J. Coles, Michael B. Hursthouse
Tetrahedron: Asymmetry 2005 Volume 16(Issue 16) pp:2799-2809
Publication Date(Web):22 August 2005
DOI:10.1016/j.tetasy.2005.07.024
Enantiopure 1,1′-ethylenedipyrrolidines possessing 3,4-disubstitution have been prepared from esters of l-(+)-tartaric acid. Although diacylation routes via the diacetoxypyrrolidin-2,5-diones were problematic, N,N-dialkylation protocols proved to be reliable and led to the synthesis of (3S,3S′,4S,4S′)-1,1′-ethylenedipyrrolidine-3,3′,4,4′-tetraol, (3R,3′S,4R,4′S)-3,4-diamino-1,1′-ethylenedipyrrolidine-3′,4′-diol and (3R,3′R,4S,4′S)-3,3′-diamino-1,1′-ethylenedipyrrolidine-4,4′-diol. The tetraol possesses a crystal structure that exhibits an unusual zig-zag intermolecular pattern comprising a network of hydrogen bonds involving the terminal hydroxyl groups and a nitrogen atom of one of the pyrrolidine rings.(3S,3′S′,4S,4′S′)-3,3′,4,4′-Tetrakis(methoxymethyloxy)-1,1′-ethylenedipyrrolidineC18H36N2O8Ee = 100%[α]D30=-10.6 (c 0.15, CHCl3)Source of chirality: asymmetric synthesisAbsolute configuration: (3S,3′S′,4S,4′S′)(3S,3S′,4S,4S′)-1,1′-Ethylenedipyrrolidine-3,3′,4,4′-tetraolC10H20N2O4Ee = 100%[α]D30=+15.4 (c 0.35, CH3OH)Source of chirality: asymmetric synthesisAbsolute configuration: (3S,3′S′,4S,4′S′)(3R,3′S,4R,4′S)-3,4-Diazido-3′,4′-bis(methoxymethyloxy)-1,1′-ethylenedipyrrolidineC14H26N8O4Ee = 100%[α]D34=-6.0 (c 0.2, CHCl3)Source of chirality: asymmetric synthesisAbsolute configuration: (3S,3′S′,4S,4′S′)(3R,3′S,4R,4′S)-3,4-Diamino-3′,4′-bis(methoxymethyloxy)-1,1′-ethylenedipyrrolidineC10H30N4O4Ee = 100%[α]D33=-9.7 (c 1.1, CHCl3)Source of chirality: asymmetric synthesisAbsolute configuration: (3S,3′S′,4S,4′S′)(2S,3R)-3-Azido-1,4-bis(methanesulfonyloxy)butane-2-olC6H13N3O7S2Ee = 100%[α]D30=-35.4 (c 0.6, CHCl3)Source of chirality: asymmetric synthesisAbsolute configuration: (2S,3R)(3R,3′R,4S,4′S)-3,3′-Diazido-1,1′-ethylenedipyrrolidine-4,4′-diolC10H18N8O2Ee = 100%[α]D28=+44.7 (c 1.2, CH3OH)Source of chirality: asymmetric synthesisAbsolute configuration: (3R,3′R,4S,4′S)(3R,3′R,4S,4′S)-3,3′-Diamino-1,1′-ethylenedipyrrolidine-4,4′-diolC10H22N4O2Ee = 100%[α]D32=+36.3 (c 1.3, CH3OH)Source of chirality: asymmetric synthesisAbsolute configuration: (3R,3′R,4S,4′S)
Co-reporter:Charles M. Marson, Nawal Serradji, Alphonso S. Rioja, Sebastien P. Gastaud, John P. Alao, R.Charles Coombes, David M. Vigushin
Bioorganic & Medicinal Chemistry Letters 2004 Volume 14(Issue 10) pp:2477-2481
Publication Date(Web):17 May 2004
DOI:10.1016/j.bmcl.2004.03.012
Syntheses of (2E,4E)-5-arylpenta-2,4-dienoic acid hydroxyamides are described, some of which are potent inhibitors of histone deacetylase, a double bond conferring more than a 10-fold increase in potency compared with the triple bond analogue oxamflatin. Variation of substituents on the aromatic ring has a marked effect on potency, in vitro IC50 values down to 50 nM being obtained.Synthesis and evaluation of unsaturated inhibitors of histone deacetylase are described. Inhibition of histone deacetylase IC50=49 nM.
Co-reporter:Charles M. Marson, Alphonso S. Rioja, Greg Brooke, R.Charles Coombes, David M. Vigushin
Bioorganic & Medicinal Chemistry Letters 2002 Volume 12(Issue 2) pp:255-259
Publication Date(Web):21 January 2002
DOI:10.1016/S0960-894X(01)00718-1
Cyclic acid anhydrides possessing a lipid chain have been shown to be a new class of non-peptidic inhibitors of geranylgeranyl protein-transferase type I (GGPTase-I).Cyclic anhydride inhibitors selective for GGPTase-I are described.
Co-reporter:Charles M. Marson; Christopher J. Matthews; Stephen J. Atkinson; Nermina Lamadema;N. Shaun B. Thomas
Journal of Medicinal Chemistry () pp:
Publication Date(Web):August 19, 2015
DOI:10.1021/acs.jmedchem.5b00545
A novel series of potent chiral inhibitors of histone deacetylase (HDAC) is described that contains an oxazoline capping group and a N-(2-aminophenyl)-benzamide unit. Among several new inhibitors of this type exhibiting Class I selectivity and potent inhibition of HDAC3-NCoR2, in vitro assays for the inhibition of HDAC1, HDAC2, and HDAC3-NCoR2 by N-(2-aminophenyl)-benzamide 15k gave respective IC50 values of 80, 110, and 6 nM. Weak inhibition of all other HDAC isoforms (HDAC4, 5, 6, 7, and 9: IC50 > 100 000 nM; HDAC8: IC50 = 25 000 nM; HDAC10: IC50 > 4000 nM; HDAC11: IC50 > 2000 nM) confirmed the Class I selectivity of 15k. 2-Aminoimidazolinyl, 2-thioimidazolinyl, and 2-aminooxazolinyl units were shown to be effective replacements for the pyrimidine ring present in many other 2-(aminophenyl)-benzamides previously reported, but the 2-aminooxazolinyl unit was the most potent in inhibiting HDAC3-NCoR2. Many of the new HDAC inhibitors showed higher solubilities and lower binding to human serum albumin than that of Mocetinostat. Increases in histone H3K9 acetylation in the human cell lines U937 and PC-3 was observed for all three oxazolinyl inhibitors evaluated; those HDAC inhibitors also lowered cyclin E expression in U937 cells but not in PC-3 cells, indicating underlying differences in the mechanisms of action of the inhibitors on those two cell lines.
Co-reporter:Charles M. Marson, Esra Edaan, James M. Morrell, Simon J. Coles, Michael B. Hursthouse and David T. Davies
Chemical Communications 2007(Issue 24) pp:NaN2496-2496
Publication Date(Web):2007/03/26
DOI:10.1039/B701548H
3(2H)-Furanones can be prepared by a catalytic asymmetric protocol from enynones, which, if electron-rich, require only one reagent and involve two reactions in a single operation—a domino process.
Co-reporter:Charles M. Marson
Chemical Society Reviews 2012 - vol. 41(Issue 23) pp:NaN7722-7722
Publication Date(Web):2012/08/24
DOI:10.1039/C2CS35183H
Reactions in which several components are combined in sequence, and without isolation of intermediates, are greatly sought because of the inherent molecular diversity, efficiency, and atom-economy. However, organocatalytic reactions, employing an organic catalyst to assemble products of high enantiomeric excess (a single optical isomer), are also cutting-edge methodology. This tutorial review covers the overlap of these two areas, outling the structural diversity and stereocontrol arising from one-pot combinations of at least three components, powerful approaches with great potential that minimise formation of by-products and operating costs.









![3,4-Pyrrolidinediol, 1-[1,1'-biphenyl]-2-yl-, (3S,4S)-](http://img.cochemist.com/ccimg/880500/880497-58-5.png)
![3,4-Pyrrolidinediol, 1-[1,1'-biphenyl]-2-yl-, (3S,4S)-](http://img.cochemist.com/ccimg/880500/880497-58-5_b.png)






![Methanesulfonamide, N,N'-[(3R,4R)-1-phenyl-3,4-pyrrolidinediyl]bis-](http://img.cochemist.com/ccimg/880500/880497-78-9.png)
![Methanesulfonamide, N,N'-[(3R,4R)-1-phenyl-3,4-pyrrolidinediyl]bis-](http://img.cochemist.com/ccimg/880500/880497-78-9_b.png)























![Cyclooctanone, 2-[(1R)-1-hydroxyethyl]-2-phenyl-, (2R)-rel-](http://img.cochemist.com/ccimg/477900/477883-05-9.png)
![Cyclooctanone, 2-[(1R)-1-hydroxyethyl]-2-phenyl-, (2R)-rel-](http://img.cochemist.com/ccimg/477900/477883-05-9_b.png)
![Cycloheptanone, 2-[(1R)-1-hydroxyethyl]-2-phenyl-, (2S)-rel-](http://img.cochemist.com/ccimg/477900/477883-01-5.png)
![Cycloheptanone, 2-[(1R)-1-hydroxyethyl]-2-phenyl-, (2S)-rel-](http://img.cochemist.com/ccimg/477900/477883-01-5_b.png)
![Hexanamide, N-hydroxy-6-[4-[methyl(4-pyridinylmethyl)amino]phenoxy]-](http://img.cochemist.com/ccimg/926900/926893-11-0.png)
![Hexanamide, N-hydroxy-6-[4-[methyl(4-pyridinylmethyl)amino]phenoxy]-](http://img.cochemist.com/ccimg/926900/926893-11-0_b.png)
![Hexanamide, 6-[4-(dimethylamino)phenoxy]-N-hydroxy-](http://img.cochemist.com/ccimg/926900/926893-08-5.png)
![Hexanamide, 6-[4-(dimethylamino)phenoxy]-N-hydroxy-](http://img.cochemist.com/ccimg/926900/926893-08-5_b.png)






![Heptanamide, N-hydroxy-7-[4-[[2-(1H-indol-3-yl)ethyl]amino]phenoxy]-](http://img.cochemist.com/ccimg/926900/926893-19-8.png)
![Heptanamide, N-hydroxy-7-[4-[[2-(1H-indol-3-yl)ethyl]amino]phenoxy]-](http://img.cochemist.com/ccimg/926900/926893-19-8_b.png)
![Hexanoic acid, 6-[4-(dimethylamino)phenoxy]-, ethyl ester](http://img.cochemist.com/ccimg/926900/926892-97-9.png)
![Hexanoic acid, 6-[4-(dimethylamino)phenoxy]-, ethyl ester](http://img.cochemist.com/ccimg/926900/926892-97-9_b.png)



![Cycloheptanone, 2-[(1R)-1-hydroxyethyl]-2-methyl-, (2R)-rel-](http://img.cochemist.com/ccimg/477900/477883-03-7.png)
![Cycloheptanone, 2-[(1R)-1-hydroxyethyl]-2-methyl-, (2R)-rel-](http://img.cochemist.com/ccimg/477900/477883-03-7_b.png)
![Cyclooctanol, 1-[(2R,3S)-2,3-dimethyloxiranyl]-, rel-](http://img.cochemist.com/ccimg/477900/477882-99-8.png)
![Cyclooctanol, 1-[(2R,3S)-2,3-dimethyloxiranyl]-, rel-](http://img.cochemist.com/ccimg/477900/477882-99-8_b.png)
![Cycloheptanol, 1-[(2R,3R)-3-methyl-2-phenyloxiranyl]-, rel-](http://img.cochemist.com/ccimg/477900/477882-97-6.png)
![Cycloheptanol, 1-[(2R,3R)-3-methyl-2-phenyloxiranyl]-, rel-](http://img.cochemist.com/ccimg/477900/477882-97-6_b.png)
![Cyclohexanol, 1-[(2R,3S)-2,3-dimethyloxiranyl]-, rel-](http://img.cochemist.com/ccimg/477900/477882-95-4.png)
![Cyclohexanol, 1-[(2R,3S)-2,3-dimethyloxiranyl]-, rel-](http://img.cochemist.com/ccimg/477900/477882-95-4_b.png)








![Cyclononanone, 2-[(1R)-1-hydroxyethyl]-2-methyl-, (2R)-rel-](http://img.cochemist.com/ccimg/477900/477883-07-1.png)
![Cyclononanone, 2-[(1R)-1-hydroxyethyl]-2-methyl-, (2R)-rel-](http://img.cochemist.com/ccimg/477900/477883-07-1_b.png)
![Cyclohexanol, 1-[(2R,3S)-3-methyl-2-phenyloxiranyl]-, rel-](http://img.cochemist.com/ccimg/477900/477882-93-2.png)
![Cyclohexanol, 1-[(2R,3S)-3-methyl-2-phenyloxiranyl]-, rel-](http://img.cochemist.com/ccimg/477900/477882-93-2_b.png)
![CYCLONONANONE, 2-[(1R)-1-HYDROXYETHYL]-2-METHYL-, (2S)-](http://img.cochemist.com/ccimg/477900/477883-08-2.png)
![CYCLONONANONE, 2-[(1R)-1-HYDROXYETHYL]-2-METHYL-, (2S)-](http://img.cochemist.com/ccimg/477900/477883-08-2_b.png)
![CYCLOOCTANOL, 1-[(2R,3R)-2,3-DIMETHYLOXIRANYL]-](http://img.cochemist.com/ccimg/477900/477883-00-4.png)
![CYCLOOCTANOL, 1-[(2R,3R)-2,3-DIMETHYLOXIRANYL]-](http://img.cochemist.com/ccimg/477900/477883-00-4_b.png)












![Ethanol, 2,2'-[(1R,2R)-1,2-cyclohexanediyldiimino]bis-](http://img.cochemist.com/ccimg/200100/200008-68-0.png)
![Ethanol, 2,2'-[(1R,2R)-1,2-cyclohexanediyldiimino]bis-](http://img.cochemist.com/ccimg/200100/200008-68-0_b.png)












![PYRROLIDINE, 1-[1,1'-BIPHENYL]-2-YL-3,4-BIS(PHENYLMETHOXY)-, (3S,4S)-](http://img.cochemist.com/ccimg/880500/880497-57-4.png)
![PYRROLIDINE, 1-[1,1'-BIPHENYL]-2-YL-3,4-BIS(PHENYLMETHOXY)-, (3S,4S)-](http://img.cochemist.com/ccimg/880500/880497-57-4_b.png)




![PYRROLIDINE, 3,4-DIAZIDO-1-[1,1'-BIPHENYL]-2-YL-, (3R,4R)-](http://img.cochemist.com/ccimg/880500/880497-70-1.png)
![PYRROLIDINE, 3,4-DIAZIDO-1-[1,1'-BIPHENYL]-2-YL-, (3R,4R)-](http://img.cochemist.com/ccimg/880500/880497-70-1_b.png)




![BUTANAMIDE, N-[(1R,2R)-2-AMINOCYCLOHEXYL]-2-HYDROXY-3-METHYL-, (2R)-](http://img.cochemist.com/ccimg/847400/847340-81-2.png)
![BUTANAMIDE, N-[(1R,2R)-2-AMINOCYCLOHEXYL]-2-HYDROXY-3-METHYL-, (2R)-](http://img.cochemist.com/ccimg/847400/847340-81-2_b.png)





![ACETAMIDE, 2-AMINO-N-[1-(1,3-DIOXAN-2-YL)ETHYL]-N-METHOXY-](http://img.cochemist.com/ccimg/817600/817556-42-6.png)
![ACETAMIDE, 2-AMINO-N-[1-(1,3-DIOXAN-2-YL)ETHYL]-N-METHOXY-](http://img.cochemist.com/ccimg/817600/817556-42-6_b.png)














![3,4-PYRROLIDINEDIAMINE, 1-[1,1'-BIPHENYL]-2-YL-, (3R,4R)-](http://img.cochemist.com/ccimg/880500/880497-76-7.png)
![3,4-PYRROLIDINEDIAMINE, 1-[1,1'-BIPHENYL]-2-YL-, (3R,4R)-](http://img.cochemist.com/ccimg/880500/880497-76-7_b.png)




![Arsonium, [(2E)-4-ethoxy-4-oxo-2-butenyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/97900/97856-60-5.png)
![Arsonium, [(2E)-4-ethoxy-4-oxo-2-butenyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/97900/97856-60-5_b.png)




![2,4-Pentadienoic acid, 5-[4-(dimethylamino)phenyl]-4-methyl-, ethylester, (2E,4E)-](http://img.cochemist.com/ccimg/711100/711014-70-9.png)
![2,4-Pentadienoic acid, 5-[4-(dimethylamino)phenyl]-4-methyl-, ethylester, (2E,4E)-](http://img.cochemist.com/ccimg/711100/711014-70-9_b.png)




![2,4-Hexadienoic acid, 6-[[4-(dimethylamino)phenyl]thio]-, methyl ester,(2E,4E)-](http://img.cochemist.com/ccimg/698400/698387-79-0.png)
![2,4-Hexadienoic acid, 6-[[4-(dimethylamino)phenyl]thio]-, methyl ester,(2E,4E)-](http://img.cochemist.com/ccimg/698400/698387-79-0_b.png)
![2,4-Heptadienoic acid, 6-[[4-(dimethylamino)phenyl]thio]-4-methyl-,ethyl ester, (2E,4E)-](http://img.cochemist.com/ccimg/698400/698387-93-8.png)
![2,4-Heptadienoic acid, 6-[[4-(dimethylamino)phenyl]thio]-4-methyl-,ethyl ester, (2E,4E)-](http://img.cochemist.com/ccimg/698400/698387-93-8_b.png)
![Hexanamide, 6-[[4-(dimethylamino)phenyl]sulfinyl]-N-hydroxy-](http://img.cochemist.com/ccimg/698400/698352-65-7.png)
![Hexanamide, 6-[[4-(dimethylamino)phenyl]sulfinyl]-N-hydroxy-](http://img.cochemist.com/ccimg/698400/698352-65-7_b.png)
![Hexanamide, 6-[(4-chlorophenyl)thio]-N-hydroxy-](http://img.cochemist.com/ccimg/698400/698352-39-5.png)
![Hexanamide, 6-[(4-chlorophenyl)thio]-N-hydroxy-](http://img.cochemist.com/ccimg/698400/698352-39-5_b.png)
![Hexanoic acid, 6-[[4-(dimethylamino)phenyl]thio]-, methyl ester](http://img.cochemist.com/ccimg/698400/698352-26-0.png)
![Hexanoic acid, 6-[[4-(dimethylamino)phenyl]thio]-, methyl ester](http://img.cochemist.com/ccimg/698400/698352-26-0_b.png)







![(3S,10S,13S,17E)-17-hydrazinylidene-10,13-dimethyl-1,2,3,4,5,6,7,8,9,11,12,14,15,16-tetradecahydrocyclopenta[a]phenanthren-3-ol](http://img.cochemist.com/ccimg/10500/10481-80-8.png)
![(3S,10S,13S,17E)-17-hydrazinylidene-10,13-dimethyl-1,2,3,4,5,6,7,8,9,11,12,14,15,16-tetradecahydrocyclopenta[a]phenanthren-3-ol](http://img.cochemist.com/ccimg/10500/10481-80-8_b.png)


![2-Propenal, 3-[4-(dimethylamino)phenyl]-2-methyl-, (2E)-](http://img.cochemist.com/ccimg/87300/87234-33-1.png)
![2-Propenal, 3-[4-(dimethylamino)phenyl]-2-methyl-, (2E)-](http://img.cochemist.com/ccimg/87300/87234-33-1_b.png)




![2,4-Pentadienoic acid, 5-[4-[[(4-methoxyphenyl)sulfonyl]amino]phenyl]-,ethyl ester, (2E,4E)-](http://img.cochemist.com/ccimg/711100/711014-68-5.png)
![2,4-Pentadienoic acid, 5-[4-[[(4-methoxyphenyl)sulfonyl]amino]phenyl]-,ethyl ester, (2E,4E)-](http://img.cochemist.com/ccimg/711100/711014-68-5_b.png)




![2,4-Pentadienamide,N-hydroxy-5-[4-[[(4-methoxyphenyl)sulfonyl]amino]phenyl]-4-methyl-,(2E,4E)-](http://img.cochemist.com/ccimg/711100/711014-80-1.png)
![2,4-Pentadienamide,N-hydroxy-5-[4-[[(4-methoxyphenyl)sulfonyl]amino]phenyl]-4-methyl-,(2E,4E)-](http://img.cochemist.com/ccimg/711100/711014-80-1_b.png)
![2,4-Pentadienoic acid,5-[4-[[(4-chlorophenyl)sulfonyl]amino]phenyl]-4-methyl-, ethyl ester,(2E,4E)-](http://img.cochemist.com/ccimg/711100/711014-76-5.png)
![2,4-Pentadienoic acid,5-[4-[[(4-chlorophenyl)sulfonyl]amino]phenyl]-4-methyl-, ethyl ester,(2E,4E)-](http://img.cochemist.com/ccimg/711100/711014-76-5_b.png)




![Hexanamide, 6-[(4-chlorophenyl)sulfinyl]-N-hydroxy-](http://img.cochemist.com/ccimg/698400/698352-60-2.png)
![Hexanamide, 6-[(4-chlorophenyl)sulfinyl]-N-hydroxy-](http://img.cochemist.com/ccimg/698400/698352-60-2_b.png)
![2,4-Hexadienoic acid, 6-[(4-chlorophenyl)thio]-, methyl ester, (2E,4E)-](http://img.cochemist.com/ccimg/698400/698387-78-9.png)
![2,4-Hexadienoic acid, 6-[(4-chlorophenyl)thio]-, methyl ester, (2E,4E)-](http://img.cochemist.com/ccimg/698400/698387-78-9_b.png)





![2,4-Pentadienamide,N-hydroxy-5-[4-[[(4-methoxyphenyl)sulfonyl]amino]phenyl]-, (2E,4E)-](http://img.cochemist.com/ccimg/711100/711014-66-3.png)
![2,4-Pentadienamide,N-hydroxy-5-[4-[[(4-methoxyphenyl)sulfonyl]amino]phenyl]-, (2E,4E)-](http://img.cochemist.com/ccimg/711100/711014-66-3_b.png)
![2,4-Pentadienamide,N-hydroxy-4-methyl-5-[4-[(phenylsulfonyl)amino]phenyl]-, (2E,4E)-](http://img.cochemist.com/ccimg/711100/711014-78-7.png)
![2,4-Pentadienamide,N-hydroxy-4-methyl-5-[4-[(phenylsulfonyl)amino]phenyl]-, (2E,4E)-](http://img.cochemist.com/ccimg/711100/711014-78-7_b.png)








![Benzenemethanol, 3-[[4-(dimethylamino)phenyl]thio]-](http://img.cochemist.com/ccimg/698400/698352-89-5.png)
![Benzenemethanol, 3-[[4-(dimethylamino)phenyl]thio]-](http://img.cochemist.com/ccimg/698400/698352-89-5_b.png)
![Benzo[b]thiophene-2-methanol, 5-(dimethylamino)-](http://img.cochemist.com/ccimg/698400/698352-77-1.png)
![Benzo[b]thiophene-2-methanol, 5-(dimethylamino)-](http://img.cochemist.com/ccimg/698400/698352-77-1_b.png)
![(3S,8R,9S,10R,13S,14S)-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15-decahydr o-1H-cyclopenta[a]phenanthren-3-ol](http://img.cochemist.com/ccimg/1300/1224-94-8.png)
![(3S,8R,9S,10R,13S,14S)-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15-decahydr o-1H-cyclopenta[a]phenanthren-3-ol](http://img.cochemist.com/ccimg/1300/1224-94-8_b.png)
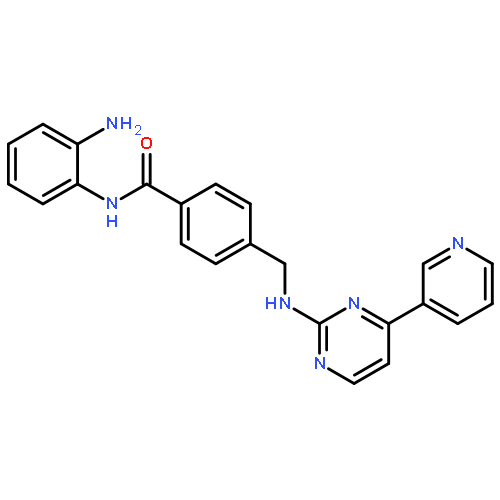
![4-({[4-(Pyridin-3-yl)pyrimidin-2-yl]amino}methyl)benzoic acid](http://img.cochemist.com/ccimg/849300/849235-68-3.png)
![4-({[4-(Pyridin-3-yl)pyrimidin-2-yl]amino}methyl)benzoic acid](http://img.cochemist.com/ccimg/849300/849235-68-3_b.png)





![2,4-Hexadienamide, N-hydroxy-6-[(4-methoxyphenyl)sulfinyl]-, (2E,4E)-](http://img.cochemist.com/ccimg/698400/698387-87-0.png)
![2,4-Hexadienamide, N-hydroxy-6-[(4-methoxyphenyl)sulfinyl]-, (2E,4E)-](http://img.cochemist.com/ccimg/698400/698387-87-0_b.png)
![2,4-Hexadienamide, 6-[(4-chlorophenyl)sulfinyl]-N-hydroxy-, (2E,4E)-](http://img.cochemist.com/ccimg/698400/698387-86-9.png)
![2,4-Hexadienamide, 6-[(4-chlorophenyl)sulfinyl]-N-hydroxy-, (2E,4E)-](http://img.cochemist.com/ccimg/698400/698387-86-9_b.png)
![2,4-Hexadienoic acid, 6-[(4-chlorophenyl)sulfinyl]-, methyl ester,(2E,4E)-](http://img.cochemist.com/ccimg/698400/698387-83-6.png)
![2,4-Hexadienoic acid, 6-[(4-chlorophenyl)sulfinyl]-, methyl ester,(2E,4E)-](http://img.cochemist.com/ccimg/698400/698387-83-6_b.png)
![Benzoic acid, 3-[[4-(dimethylamino)phenyl]thio]-, methyl ester](http://img.cochemist.com/ccimg/698400/698352-87-3.png)
![Benzoic acid, 3-[[4-(dimethylamino)phenyl]thio]-, methyl ester](http://img.cochemist.com/ccimg/698400/698352-87-3_b.png)
![Benzoic acid, 3-[(4-nitrophenyl)thio]-, methyl ester](http://img.cochemist.com/ccimg/698400/698352-85-1.png)
![Benzoic acid, 3-[(4-nitrophenyl)thio]-, methyl ester](http://img.cochemist.com/ccimg/698400/698352-85-1_b.png)

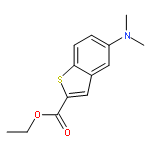

![Heptanamide, 7-[(4-chlorophenyl)sulfinyl]-N-hydroxy-](http://img.cochemist.com/ccimg/698400/698352-63-5.png)
![Heptanamide, 7-[(4-chlorophenyl)sulfinyl]-N-hydroxy-](http://img.cochemist.com/ccimg/698400/698352-63-5_b.png)
![Hexanoic acid, 6-[[4-(dimethylamino)phenyl]sulfinyl]-, methyl ester](http://img.cochemist.com/ccimg/698400/698352-51-1.png)
![Hexanoic acid, 6-[[4-(dimethylamino)phenyl]sulfinyl]-, methyl ester](http://img.cochemist.com/ccimg/698400/698352-51-1_b.png)
![Heptanoic acid, 7-[(4-chlorophenyl)sulfinyl]-, ethyl ester](http://img.cochemist.com/ccimg/698400/698352-49-7.png)
![Heptanoic acid, 7-[(4-chlorophenyl)sulfinyl]-, ethyl ester](http://img.cochemist.com/ccimg/698400/698352-49-7_b.png)
![Hexanoic acid, 6-[(4-chlorophenyl)sulfinyl]-, methyl ester](http://img.cochemist.com/ccimg/698400/698352-47-5.png)
![Hexanoic acid, 6-[(4-chlorophenyl)sulfinyl]-, methyl ester](http://img.cochemist.com/ccimg/698400/698352-47-5_b.png)
![Hexanamide,6-[[4-[[(4-chlorophenyl)sulfonyl]amino]phenyl]thio]-N-hydroxy-](http://img.cochemist.com/ccimg/698400/698352-43-1.png)
![Hexanamide,6-[[4-[[(4-chlorophenyl)sulfonyl]amino]phenyl]thio]-N-hydroxy-](http://img.cochemist.com/ccimg/698400/698352-43-1_b.png)
![Heptanoic acid, 7-[(4-chlorophenyl)thio]-, ethyl ester](http://img.cochemist.com/ccimg/698400/698352-21-5.png)
![Heptanoic acid, 7-[(4-chlorophenyl)thio]-, ethyl ester](http://img.cochemist.com/ccimg/698400/698352-21-5_b.png)
![Hexanoic acid, 6-[(4-chlorophenyl)thio]-, methyl ester](http://img.cochemist.com/ccimg/698400/698352-19-1.png)
![Hexanoic acid, 6-[(4-chlorophenyl)thio]-, methyl ester](http://img.cochemist.com/ccimg/698400/698352-19-1_b.png)
![2,4-Pentadienamide, 5-[4-(dimethylamino)phenyl]-N-hydroxy-, (2E,4E)-](http://img.cochemist.com/ccimg/674800/674767-42-1.png)
![2,4-Pentadienamide, 5-[4-(dimethylamino)phenyl]-N-hydroxy-, (2E,4E)-](http://img.cochemist.com/ccimg/674800/674767-42-1_b.png)
![Hexanoic acid, 6-[(4-aminophenyl)thio]-, methyl ester](http://img.cochemist.com/ccimg/125200/125173-94-6.png)
![Hexanoic acid, 6-[(4-aminophenyl)thio]-, methyl ester](http://img.cochemist.com/ccimg/125200/125173-94-6_b.png)
























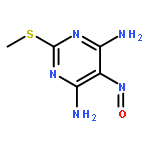

![2-Penten-1-ol, 4-[[4-(dimethylamino)phenyl]thio]-2-methyl-, (2E)-](http://img.cochemist.com/ccimg/875800/875737-08-9.png)
![2-Penten-1-ol, 4-[[4-(dimethylamino)phenyl]thio]-2-methyl-, (2E)-](http://img.cochemist.com/ccimg/875800/875737-08-9_b.png)
![2-Pentenoic acid, 4-[[4-(dimethylamino)phenyl]thio]-2-methyl-, methylester, (2E)-](http://img.cochemist.com/ccimg/875800/875737-07-8.png)
![2-Pentenoic acid, 4-[[4-(dimethylamino)phenyl]thio]-2-methyl-, methylester, (2E)-](http://img.cochemist.com/ccimg/875800/875737-07-8_b.png)








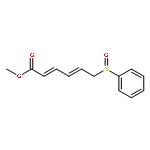



![2,4-Pentadienoic acid, 5-[4-(dimethylamino)phenyl]-, ethyl ester, (2E,4E)-](/data/chemimg/1148200/16913-49-8.png)
![2,4-Pentadienoic acid, 5-[4-(dimethylamino)phenyl]-, ethyl ester, (2E,4E)-](/data/chemimg/1148200/16913-49-8_b.png)







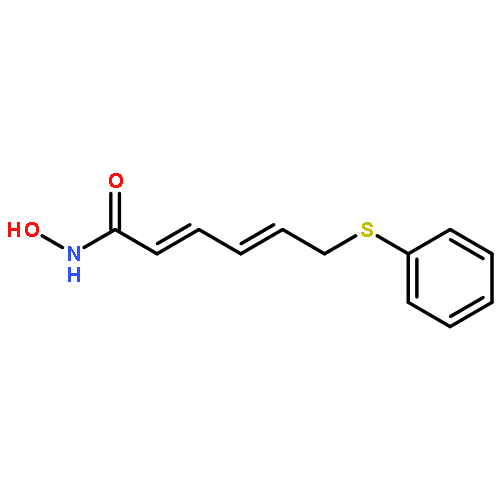

































![Methyl 4-({[4-(pyridin-3-yl)pyrimidin-2-yl]amino}methyl)benzoate](http://img.cochemist.com/ccimg/849300/849235-67-2.png)
![Methyl 4-({[4-(pyridin-3-yl)pyrimidin-2-yl]amino}methyl)benzoate](http://img.cochemist.com/ccimg/849300/849235-67-2_b.png)
![[2-[[4-(Aminomethyl)benzoyl]amino]phenyl]-carbamic Acid tert-Butyl Ester](http://img.cochemist.com/ccimg/209800/209784-85-0.png)
![[2-[[4-(Aminomethyl)benzoyl]amino]phenyl]-carbamic Acid tert-Butyl Ester](http://img.cochemist.com/ccimg/209800/209784-85-0_b.png)
![N-(2-Aminophenyl)-4-[[[(4S)-4,5-dihydro-4-phenyl-2-thiazolyl]amino]methyl]-benzamide](http://img.cochemist.com/ccimg/1448400/1448350-50-2.png)
![N-(2-Aminophenyl)-4-[[[(4S)-4,5-dihydro-4-phenyl-2-thiazolyl]amino]methyl]-benzamide](http://img.cochemist.com/ccimg/1448400/1448350-50-2_b.png)



![Carbamic acid,[2-(hydroxyamino)-2-oxoethyl]-, 1,1-dimethylethyl ester (9CI)](http://img.cochemist.com/ccimg/156800/156706-50-2.png)
![Carbamic acid,[2-(hydroxyamino)-2-oxoethyl]-, 1,1-dimethylethyl ester (9CI)](http://img.cochemist.com/ccimg/156800/156706-50-2_b.png)


















































![Benzene, 1-methoxy-4-[(4-pentynyloxy)methyl]-](http://img.cochemist.com/ccimg/197300/197219-01-5.png)
![Benzene, 1-methoxy-4-[(4-pentynyloxy)methyl]-](http://img.cochemist.com/ccimg/197300/197219-01-5_b.png)
![3-Nitro-[1,1'-biphenyl]-4-amine](http://img.cochemist.com/ccimg/4100/4085-18-1.png)
![3-Nitro-[1,1'-biphenyl]-4-amine](http://img.cochemist.com/ccimg/4100/4085-18-1_b.png)











![(1S,4AR,5R,8AS)-1,4A-DIMETHYL-6-METHYLENE-5-[(1E)-3-OXO-1-BUTEN-1-YL]DECAHYDRO-1-NAPHTHALENECARBOXYLIC ACID](http://img.cochemist.com/ccimg/103200/103123-51-9.png)
![(1S,4AR,5R,8AS)-1,4A-DIMETHYL-6-METHYLENE-5-[(1E)-3-OXO-1-BUTEN-1-YL]DECAHYDRO-1-NAPHTHALENECARBOXYLIC ACID](http://img.cochemist.com/ccimg/103200/103123-51-9_b.png)