Co-reporter:Andrew W. Baggett and Shih-Yuan Liu
Journal of the American Chemical Society October 25, 2017 Volume 139(Issue 42) pp:15259-15259
Publication Date(Web):October 2, 2017
DOI:10.1021/jacs.7b09491
Upon reaction with either molecular oxygen or di-tert-butylperoxide in the presence of a simple copper(I) salt and an alcohol, a range of 1,2-azaborines readily exchange B-alkyl or B-aryl moieties for B-alkoxide fragments. This transformation allows alkyl and aryl groups to serve for the first time as removable protecting groups for the boron position of 1,2-azaborines during reactions that are not compatible with the easily modifiable B-alkoxide moiety. This reaction can be applied to synthesize a previously inaccessible BN isostere of ethylbenzene, a compound of interest in biomedical research. A sequence of epoxide ring opening using N-deprotonated 1,2-azaborines followed by an intramolecular version of the boron deprotection reaction can be applied to access the first examples of BN isosteres of dihydrobenzofurans and benzofurans, classes of compounds that are important to medicinal chemistry and natural product synthesis.
Co-reporter:Jacob S. A. Ishibashi, Alain Dargelos, Clovis Darrigan, Anna Chrostowska, and Shih-Yuan Liu
Organometallics July 24, 2017 Volume 36(Issue 14) pp:2494-2494
Publication Date(Web):May 24, 2017
DOI:10.1021/acs.organomet.7b00296
The first synthesis of a tetracene BN isostere is reported. Comparison with its direct, all-carbon analogue reveals that the BN tetracene isostere exhibits a lower-lying HOMO and a slightly larger optical HOMO–LUMO gap. While all-carbon tetracenes are prone to photodecomposition, the BN tetracene scaffold is less light sensitive, owing in part to its much higher photoluminescence quantum yield. In the context of this larger BN tetracene family, we introduce simple guiding principles for predicting frontier orbital energies as a function of the position of the BN unit within the tetracene scaffold.
Co-reporter:Zachary X. Giustra, Jacob S.A. Ishibashi, Shih-Yuan Liu
Coordination Chemistry Reviews 2016 Volume 314() pp:134-181
Publication Date(Web):1 May 2016
DOI:10.1016/j.ccr.2015.11.006
•Homogeneous catalytic arene and heteroarene hydrogenation is broadly reviewed.•Various hydrogenation mechanistic studies are discussed.•Most recent research efforts have focused on heteroarene substrates.•The current state-of-the-art features primarily Ir-, Rh-, and Ru-based catalysts.•Single catalysts for reversible hydrogenation-dehydrogenation have been reported.In the past few decades, homogeneous catalysis of arene and heteroarene reduction has grown into a mature research field. In particular, hydrogenation of heteroaromatic systems facilitates rapid access to many classes of fine chemicals and pharmaceutically relevant compounds. In this review, we discuss the advancements made in the field of homogeneous metal-catalyzed arene and heteroarene hydrogenation from its early beginnings to the present day. We also review homogeneous catalysts for the reverse dehydrogenation of cyclic saturated species back to their aromatic counterparts, as well as single-catalyst systems capable of performing reversible hydrogenation-dehydrogenation reactions.
Co-reporter:Hyelee Lee, Marcus Fischer, Brian K. Shoichet, and Shih-Yuan Liu
Journal of the American Chemical Society 2016 Volume 138(Issue 37) pp:12021-12024
Publication Date(Web):September 7, 2016
DOI:10.1021/jacs.6b06566
Protein crystallography and calorimetry were used to characterize the binding of 1,2-azaborines to model cavities in T4 lysozyme in direct comparison to their carbonaceous counterparts. In the apolar L99A cavity, affinity for Ab dropped only slightly versus benzene. In the cavity designed to accommodate a single hydrogen bond (L99A/M102Q), Gln102═O···H—N hydrogen bonding for Ab and BEtAb was observed in the crystallographic complexes. The strength of the hydrogen bonding was estimated as 0.94 and 0.64 kcal/mol for Ab and BEtAb, respectively. This work unambiguously demonstrates that 1,2-azaborines can be readily accommodated in classic aryl recognition pockets and establishes one of 1,2-azaborine’s distinguishing features from its carbonaceous isostere benzene: its ability to serve as an NH hydrogen bond donor in a biological setting.
Co-reporter:Senmiao Xu, Yuanzhe Zhang, Bo Li, and Shih-Yuan Liu
Journal of the American Chemical Society 2016 Volume 138(Issue 44) pp:14566-14569
Publication Date(Web):October 24, 2016
DOI:10.1021/jacs.6b09759
A concise synthesis of monobenzofused 1,4-azaborine phosphine ligands (Senphos) is described. These Senphos ligands uniquely support Pd-catalyzed trans-selective hydroboration of terminal and internal 1,3-enynes to furnish corresponding dienylboronates in high efficiency and diastereoselectivity. X-ray structural analysis of the Senphos–Pd(0) complex reveals a κ2-P-η2-BC coordination mode, and this isolated complex has been shown to serve as a competent catalyst for the trans-hydroboration reaction. This work demonstrates that the expanded chemical space provided by the BN/CC isosterism approach translates into the functional space in the context of stereoselective catalytic transformations.
Co-reporter:Colin J. Murphy
The Journal of Physical Chemistry C 2016 Volume 120(Issue 11) pp:6020-6030
Publication Date(Web):January 26, 2016
DOI:10.1021/acs.jpcc.5b11970
We report a combined experimental and theoretical study of the adsorption and assembly of a nitrogen–boron-containing heterocycle, 1,2-dihydro-1,2-azaborine, on Au(111) and Cu(111). Despite the inherent molecular dipole moment, the self-assembly behavior is found to be highly surface dependent, with isolated molecules prevalent on Cu(111) and discrete (“magic”) clusters on Au(111). The ability to form clusters of a particular size can be understood in terms of a balance between attractive intermolecular interactions, including directional B–H···H–N dihydrogen bonding, and repulsive forces from Coulombic interactions between the charged molecules dictated by differences in the charge transfer and Pauli repulsion between the adsorbate and the surface. This work highlights the importance of metal–molecule charge transfer in the adsorption and assembly of dipolar molecules on surfaces and demonstrates that their surface-bound properties cannot be predicted a priori from gas-phase dipole moments alone.
Co-reporter:Amit Kumar;Jacob S. A. Ishibashi;Dr. Thomas N. Hooper;Tanya C. Mikulas; David A. Dixon; Shih-Yuan Liu; Andrew S. Weller
Chemistry - A European Journal 2016 Volume 22( Issue 1) pp:310-322
Publication Date(Web):
DOI:10.1002/chem.201502986
Abstract
The coordination chemistry of the 1,2-BN-cyclohexanes 2,2-R2-1,2-B,N-C4H10 (R2=HH, MeH, Me2) with Ir and Rh metal fragments has been studied. This led to the solution (NMR spectroscopy) and solid-state (X-ray diffraction) characterization of [Ir(PCy3)2(H)2(η2η2-H2BNR2C4H8)][BArF4] (NR2=NH2, NMeH) and [Rh(iPr2PCH2CH2CH2PiPr2)(η2η2-H2BNR2C4H8)][BArF4] (NR2=NH2, NMeH, NMe2). For NR2=NH2 subsequent metal-promoted, dehydrocoupling shows the eventual formation of the cyclic tricyclic borazine [BNC4H8]3, via amino-borane and, tentatively characterized using DFT/GIAO chemical shift calculations, cycloborazane intermediates. For NR2=NMeH the final product is the cyclic amino-borane HBNMeC4H8. The mechanism of dehydrogenation of 2,2-H,Me-1,2-B,N-C4H10 using the {Rh(iPr2PCH2CH2CH2PiPr2)}+ catalyst has been probed. Catalytic experiments indicate the rapid formation of a dimeric species, [Rh2(iPr2PCH2CH2CH2PiPr2)2H5][BArF4]. Using the initial rate method starting from this dimer, a first-order relationship to [amine-borane], but half-order to [Rh] is established, which is suggested to be due to a rapid dimer–monomer equilibrium operating.
Co-reporter:Zachary X. Giustra, Lien-Yang Chou, Chia-Kuang Tsung, and Shih-Yuan Liu
Organometallics 2016 Volume 35(Issue 15) pp:2425-2428
Publication Date(Web):July 27, 2016
DOI:10.1021/acs.organomet.6b00412
Complete −CH2CH2– dehydrogenation of 1,2-dimethyl-1,2-BN-cyclohexene (1) was achieved using a Pd/C catalyst in a gas-phase microreactor. Arrhenius analysis yielded an activation energy (Ea) of 10.3 ± 0.3 kcal mol–1 and a pre-exponential factor (A) of 2.2 ± 0.2 (log A), respectively. These terms reflect a lesser kinetic favorability in comparison to those determined for all-carbon dimethylcyclohexene (Ea = 8.6 ± 0.3 kcal mol–1, log A = 3.6 ± 0.1). Despite being isostructural and isoelectronic with a C═C bond, the B–N bond of 1 thus appears to confer a different measure of activity with respect to Pd-catalyzed −CH2CH2– dehydrogenation.
Co-reporter:Andrew W. Baggett; Monica Vasiliu; Bo Li; David A. Dixon
Journal of the American Chemical Society 2015 Volume 137(Issue 16) pp:5536-5541
Publication Date(Web):April 14, 2015
DOI:10.1021/jacs.5b01916
The first general late-stage functionalization of monocyclic 1,2-azaborines at the C(6) position is described. Ir-catalyzed C–H borylation occurs regioselectively at the C(6) position of B-substituted 1,2-azaborines and is compatible with a range of substitution patterns at boron (e.g., hydride, alkoxide, alkyl, and aryl substituents). Subsequent Suzuki cross coupling with aryl- and heteroaryl bromides furnishes 1,2-azaborine-based biaryl compounds including 6-[pyrid-2-yl]-1,2-azaborines that represent novel κ2-N,N-bidentate ligands. The 6-[pyrid-2-yl]-B-Me-1,2-azaborine ligand has been demonstrated to form an emissive coordination complex with dimesitylboron that exhibits bathochromically shifted absorption and emission maxima and a higher photoluminescence quantum yield compared to its carbonaceous analogue.
Co-reporter:Alec N. Brown; Bo Li
Journal of the American Chemical Society 2015 Volume 137(Issue 28) pp:8932-8935
Publication Date(Web):July 6, 2015
DOI:10.1021/jacs.5b05879
The compatibility of the Negishi cross-coupling reaction with the versatile B–Cl functionality has been demonstrated in the context of late-stage functionalization of 1,2-azaborines. Alkyl-, aryl-, and alkenylzinc reagents have been utilized for the functionalization of the triply orthogonal precursor 3-bromo-1-(tert-butyldimethylsilyl)-2-chloro-1,2-dihydro-1,2-azaborine (2) to furnish new 2,3-substituted monocyclic 1,2-azaborines. This methodology has enabled the synthesis of previously elusive BN-naphthalene and BN-indenyl structures from a common intermediate.
Co-reporter:Andrew W. Baggett;Fang Guo;Dr. Bo Li;Dr. Shih-Yuan Liu;Dr. Frieder Jäkle
Angewandte Chemie 2015 Volume 127( Issue 38) pp:11343-11347
Publication Date(Web):
DOI:10.1002/ange.201504822
Abstract
The regioregular synthesis of the first azaborine oligomers and a corresponding conjugated polymer was accomplished by Suzuki–Miyaura coupling methods. An almost perfectly coplanar syn arrangement of the heterocycles was deduced from an X-ray crystal structure of the dimer, which also suggested that NH⋅⋅⋅π interactions play an important role. Computational studies further supported these experimental observations and indicated that the electronic structure of the longer azaborine oligomers and polymer resembles that of poly(cyclohexadiene) more than poly(p-phenylene). A comparison of the absorption and emission properties of the polymer with those of the oligomers revealed dramatic bathochromic shifts upon chain elongation, thus suggesting highly effective extension of conjugation.
Co-reporter:Dr. Richard J. Burford;Dr. Bo Li;Dr. Monica Vasiliu;Dr. David A. Dixon;Dr. Shih-Yuan Liu
Angewandte Chemie 2015 Volume 127( Issue 27) pp:7934-7938
Publication Date(Web):
DOI:10.1002/ange.201503483
Abstract
Diels–Alder reactions employing 1,2-azaborine heterocycles as 1,3-dienes are reported. Carbocyclic compounds with high stereochemical and functional complexity are produced, as exemplified by the straightforward two-step synthesis of an amino allyl boronic ester bearing four contiguous stereocenters as a single diastereomer. Whereas electron-deficient dienophiles undergo irreversible Diels–Alder reactions, a reversible Diels–Alder reaction with the less electron-deficient methyl acrylate is observed. Both the N and the B substituent of the 1,2-azaborine exert significant influence on the [4+2] cycloaddition reactivity as well as the aromatic character of the heterocycle. The experimentally determined thermodynamic parameters of the reversible Diels–Alder reaction between 1,2-azaborines and methyl acrylate correlate with aromaticity trends and place 1,2-azaborines approximately between furan and thiophene on the aromaticity scale.
Co-reporter:Andrew W. Baggett;Fang Guo;Dr. Bo Li;Dr. Shih-Yuan Liu;Dr. Frieder Jäkle
Angewandte Chemie 2015 Volume 127( Issue 38) pp:
Publication Date(Web):
DOI:10.1002/ange.201507513
Co-reporter:Andrew W. Baggett;Fang Guo;Dr. Bo Li;Dr. Shih-Yuan Liu;Dr. Frieder Jäkle
Angewandte Chemie International Edition 2015 Volume 54( Issue 38) pp:11191-11195
Publication Date(Web):
DOI:10.1002/anie.201504822
Abstract
The regioregular synthesis of the first azaborine oligomers and a corresponding conjugated polymer was accomplished by Suzuki–Miyaura coupling methods. An almost perfectly coplanar syn arrangement of the heterocycles was deduced from an X-ray crystal structure of the dimer, which also suggested that NH⋅⋅⋅π interactions play an important role. Computational studies further supported these experimental observations and indicated that the electronic structure of the longer azaborine oligomers and polymer resembles that of poly(cyclohexadiene) more than poly(p-phenylene). A comparison of the absorption and emission properties of the polymer with those of the oligomers revealed dramatic bathochromic shifts upon chain elongation, thus suggesting highly effective extension of conjugation.
Co-reporter:Andrew W. Baggett;Fang Guo;Dr. Bo Li;Dr. Shih-Yuan Liu;Dr. Frieder Jäkle
Angewandte Chemie International Edition 2015 Volume 54( Issue 38) pp:
Publication Date(Web):
DOI:10.1002/anie.201507513
Co-reporter:Dr. Richard J. Burford;Dr. Bo Li;Dr. Monica Vasiliu;Dr. David A. Dixon;Dr. Shih-Yuan Liu
Angewandte Chemie International Edition 2015 Volume 54( Issue 27) pp:7823-7827
Publication Date(Web):
DOI:10.1002/anie.201503483
Abstract
Diels–Alder reactions employing 1,2-azaborine heterocycles as 1,3-dienes are reported. Carbocyclic compounds with high stereochemical and functional complexity are produced, as exemplified by the straightforward two-step synthesis of an amino allyl boronic ester bearing four contiguous stereocenters as a single diastereomer. Whereas electron-deficient dienophiles undergo irreversible Diels–Alder reactions, a reversible Diels–Alder reaction with the less electron-deficient methyl acrylate is observed. Both the N and the B substituent of the 1,2-azaborine exert significant influence on the [4+2] cycloaddition reactivity as well as the aromatic character of the heterocycle. The experimentally determined thermodynamic parameters of the reversible Diels–Alder reaction between 1,2-azaborines and methyl acrylate correlate with aromaticity trends and place 1,2-azaborines approximately between furan and thiophene on the aromaticity scale.
Co-reporter:Colin J. Murphy
The Journal of Physical Chemistry C 2015 119(26) pp: 14624-14631
Publication Date(Web):April 6, 2015
DOI:10.1021/jp5126427
The chemistry of organoboron compounds has long been dominated by their high reactivity in synthetic organic chemistry. Recently, the incorporation of boron as a structural element in compounds has led to an increased diversity of organic compounds. A promising method of boron incorporation is BN/CC isosterism, where the replacement of a CC unit of the ubiquitous arene, benzene, with the isolectronic BN unit results in azaborine compounds whose properties are intermediate between benzene and borazine. These conjugated boron–nitrogen-containing heteroatom compounds show potential for use as charge transport materials in organic electronic devices in which the molecule–contact interface is a crucial factor of device performance. Therefore, to gain a fundamental understanding of the interaction of azaborines with two common metals, we examined 1,2-dihydro-1,2-azaborine and benzene desorption from Au(111) and Cu(111) by temperature-programmed desorption (TPD). Scanning tunneling microscopy imaging and theoretical calculations aided in the interpretation of the TPD results. Comparison between TPD spectra of 1,2-dihydro-1,2-azaborine and benzene allowed us to benchmark our experiments with literature values for benzene and to accurately quantify the magnitude of both molecule–molecule and molecule–surface interaction strengths. TPD spectra of 1,2-dihydro-1,2-azaborine show three well-defined adsorption states exist on each surface, assigned to mono-, bi-, and multilayers. The multilayer desorption energy of azaborine was found to be approximately 46 kJ/mol, about 4 kJ/mol larger than benzene and the increase is related to both dihydrogen bonding and dipole–dipole interactions. The bilayer formed by 1,2-dihydro-1,2-azaborine is less dense than that formed by benzene, with 0.7 molecules in the bilayer per each molecule in the monolayer on each surface. Importantly, in terms of application, azaborine did not decompose on either Cu or Au surfaces. Our data also reveal that a delicate balance of molecule–surface and molecule–molecule interactions dictate adsorption energetics in the submonolayer regime.
Co-reporter:Gang Chen; Lev N. Zakharov; Mark E. Bowden; Abhijeet J. Karkamkar; Sean M. Whittemore; Edward B. GarnerIII; Tanya C. Mikulas; David A. Dixon; Tom Autrey
Journal of the American Chemical Society 2014 Volume 137(Issue 1) pp:134-137
Publication Date(Web):December 15, 2014
DOI:10.1021/ja511766p
A critical component for the successful development of fuel cell applications is hydrogen storage. For back-up power applications, where long storage periods under extreme temperatures are expected, the thermal stability of the storage material is particularly important. Here, we describe the development of an unusually kinetically stable chemical hydrogen storage material with a H2 storage capacity of 4.7 wt%. The compound, which is the first reported parental BN isostere of cyclohexane featuring two BN units, is thermally stable up to 150 °C both in solution and as a neat material. Yet, it can be activated to rapidly desorb H2 at room temperature in the presence of a catalyst without releasing other detectable volatile contaminants. We also disclose the isolation and characterization of two cage compounds with S4 symmetry from the H2 desorption reactions.
Co-reporter:Anna Chrostowska ; Senmiao Xu ; Audrey Mazière ; Katherine Boknevitz ; Bo Li ; Eric R. Abbey ; Alain Dargelos ; Alain Graciaa
Journal of the American Chemical Society 2014 Volume 136(Issue 33) pp:11813-11820
Publication Date(Web):August 4, 2014
DOI:10.1021/ja5063899
We present a comprehensive electronic structure analysis of two BN isosteres of indole using a combined UV-photoelectron spectroscopy (UV-PES)/computational chemistry approach. Gas-phase He I photoelectron spectra of external BN indole I and fused BN indole II have been recorded, assessed by density functional theory calculations, and compared with natural indole. The first ionization energies of these indoles are natural indole (7.9 eV), external BN indole I (7.9 eV), and fused BN indole II (8.05 eV). The computationally determined molecular dipole moments are in the order: natural indole (2.177 D) > fused BN indole II (1.512 D) > external BN indole I (0.543 D). The λmax in the UV–vis absorption spectra are in the order: fused BN indole II (292 nm) > external BN indole I (282 nm) > natural indole (270 nm). The observed relative electrophilic aromatic substitution reactivity of the investigated indoles with dimethyliminium chloride as the electrophile is as follows: fused BN indole II > natural indole > external BN indole I, and this trend correlates with the π-orbital coefficient at the 3-position. Nucleus-independent chemical shifts calculations show that the introduction of boron into an aromatic 6π-electron system leads to a reduction in aromaticity, presumably due to a stronger bond localization. Trends and conclusions from BN isosteres of simple monocyclic aromatic systems such as benzene and toluene are not necessarily translated to the bicyclic indole core. Thus, electronic structure consequences resulting from BN/CC isosterism will need to be evaluated individually from system to system.
Co-reporter:Jacob S. A. Ishibashi ; Jonathan L. Marshall ; Audrey Mazière ; Gabriel J. Lovinger ; Bo Li ; Lev N. Zakharov ; Alain Dargelos ; Alain Graciaa ; Anna Chrostowska
Journal of the American Chemical Society 2014 Volume 136(Issue 43) pp:15414-15421
Publication Date(Web):October 2, 2014
DOI:10.1021/ja508813v
The synthesis of two parental BN anthracenes, 1 and 2, was developed, and their electronic structure and reactivity behavior were characterized in direct comparison with all-carbon anthracene. Gas-phase UV-photoelecton spectroscopy studies revealed the following HOMO energy trend: anthracene, −7.4 eV; BN anthracene 1, −7.7 eV; bis-BN anthracene 2, −8.0 eV. The λmax of the lower energy band in the UV–vis absorption spectrum is as follows: anthracene, 356 nm; BN anthracene 1, 359 nm; bis-BN anthracene 2, 357 nm. Thus, although the HOMO is stabilized with increasing BN incorporation, the HOMO–LUMO band gap remains unchanged across the anthracene series. The emission λmax values for the three investigated anthracene compounds are at 403 nm. The pKa values of the N-H proton for BN anthracene 1 and bis-BN anthracene 2 were determined to be approximately 26. BN anthracenes 1 and 2 do not undergo heat- or light-induced cycloaddition reactions or Friedel–Crafts acylations. Electrophilic bromination of BN anthracene 1 with Br2, however, occurs regioselectively at the 9-position. The reactivity behavior and regioselectivity of bromination of BN anthracenes are consistent with the electronic structure of these compounds; i.e., (1) the lower HOMO energy levels for BN anthracenes stabilize the molecules against cycloaddition and Friedel–Crafts reactions, and (2) the HOMO orbital coefficients are consistent with the observed bromination regioselectivity. Overall, this work demonstrates that BN/CC isosterism can be used as a molecular design strategy to stabilize the HOMO of acene-type structures while the optical band gap is maintained.
Co-reporter:Alec N. Brown, Lev N. Zakharov, Tanya Mikulas, David A. Dixon, and Shih-Yuan Liu
Organic Letters 2014 Volume 16(Issue 12) pp:3340-3343
Publication Date(Web):June 11, 2014
DOI:10.1021/ol501362w
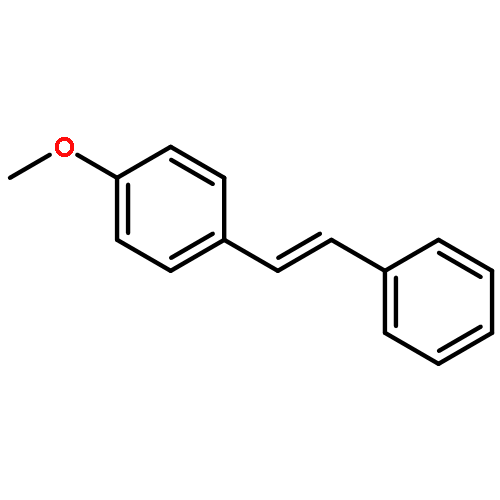
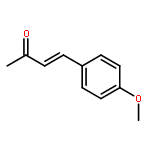
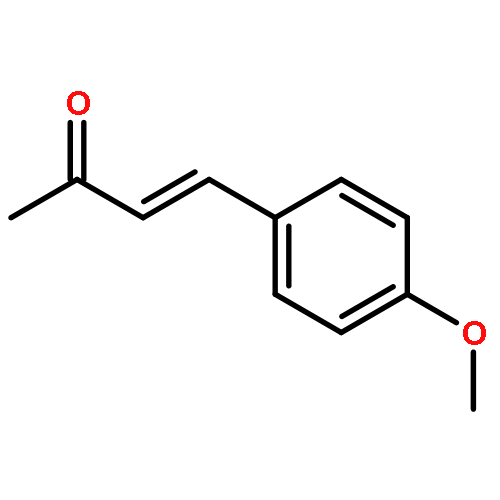
The first example of catalytic B–H activation of azaborines leading to a new family of stilbene derivatives through dehydrogenative borylation is reported. Ten 1,2-azaborine-based BN isosteres of stilbenes have been synthesized using this method, including a BN isostere of a biologically active stilbene. It is demonstrated that BN/CC isosterism in the context of stilbenes can lead to significant changes in the observed photophysical properties such as higher quantum yield and a larger Stokes shift. Direct comparative analysis of BN stilbene 3g and its carbonaceous counterpart 6g is consistent with a stronger charge-transfer character of the excited state exhibited by 3g in which the 1,2-azaborine heterocycle serves as a better electron donor than the corresponding arene.
Co-reporter:Dr. Senmiao Xu;Dr. Fredrik Haeffner;Dr. Bo Li;Dr. Lev N. Zakharov;Dr. Shih-Yuan Liu
Angewandte Chemie International Edition 2014 Volume 53( Issue 26) pp:6795-6799
Publication Date(Web):
DOI:10.1002/anie.201403903
Abstract
We report the first general synthesis of boron-substituted monobenzofused 1,4-azaborines using ring-closing metathesis of an enamine-containing diene as a key synthetic strategy. As part of our investigations, we discovered that the B-C3 moiety in a 1,4-azaborine can serve uniquely as a η2-L-type ligand. This functionality is exemplified by two κ2-N-η2-BC Pt complexes of a boron-pyridyl-substituted monobenzofused-1,4-azaborine. Single-crystal X-ray diffraction analysis of the Pt complexes shows a strong structural contribution from the iminium resonance form of the monobenzofused-1,4-azaborine ligand. We also demonstrate that a palladium(0) complex supported by a 1,4-azaborine-based phosphine ligand can catalyze hydroboration of 1-buten-3-yne with unique selectivity. In view of the importance of arene–metal π-interactions in catalytic applications, this work should open new opportunities for ligand design involving the 1,4-azaborine motif as an arene substitute.
Co-reporter:Dr. Senmiao Xu;Dr. Fredrik Haeffner;Dr. Bo Li;Dr. Lev N. Zakharov;Dr. Shih-Yuan Liu
Angewandte Chemie 2014 Volume 126( Issue 26) pp:6913-6917
Publication Date(Web):
DOI:10.1002/ange.201403903
Abstract
We report the first general synthesis of boron-substituted monobenzofused 1,4-azaborines using ring-closing metathesis of an enamine-containing diene as a key synthetic strategy. As part of our investigations, we discovered that the B-C3 moiety in a 1,4-azaborine can serve uniquely as a η2-L-type ligand. This functionality is exemplified by two κ2-N-η2-BC Pt complexes of a boron-pyridyl-substituted monobenzofused-1,4-azaborine. Single-crystal X-ray diffraction analysis of the Pt complexes shows a strong structural contribution from the iminium resonance form of the monobenzofused-1,4-azaborine ligand. We also demonstrate that a palladium(0) complex supported by a 1,4-azaborine-based phosphine ligand can catalyze hydroboration of 1-buten-3-yne with unique selectivity. In view of the importance of arene–metal π-interactions in catalytic applications, this work should open new opportunities for ligand design involving the 1,4-azaborine motif as an arene substitute.
.jpg)
![2,6-Dimethyl-4'-(trifluoromethyl)-[1,1'-biphenyl]-4-ol](http://img.cochemist.com/ccimg/872300/872258-58-7.png)
![2,6-Dimethyl-4'-(trifluoromethyl)-[1,1'-biphenyl]-4-ol](http://img.cochemist.com/ccimg/872300/872258-58-7_b.png)


![Silane, (1,1-dimethylethyl)[(3E)-3-hexen-5-ynyloxy]dimethyl-](http://img.cochemist.com/ccimg/408400/408305-79-3.png)
![Silane, (1,1-dimethylethyl)[(3E)-3-hexen-5-ynyloxy]dimethyl-](http://img.cochemist.com/ccimg/408400/408305-79-3_b.png)
![Phosphine, [2-(1-naphthalenyl)phenyl]diphenyl-](/data/chemimg/3171500/402822-73-5.png)
![Phosphine, [2-(1-naphthalenyl)phenyl]diphenyl-](/data/chemimg/3171500/402822-73-5_b.png)








![Magnesium, bromo[4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-1-butynyl]-](http://img.cochemist.com/ccimg/178500/178424-11-8.png)
![Magnesium, bromo[4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-1-butynyl]-](http://img.cochemist.com/ccimg/178500/178424-11-8_b.png)






![1,3,2-Benzodioxaborole, 2-[(1E)-3-methyl-1,3-butadienyl]-](http://img.cochemist.com/ccimg/115100/115010-71-4.png)
![1,3,2-Benzodioxaborole, 2-[(1E)-3-methyl-1,3-butadienyl]-](http://img.cochemist.com/ccimg/115100/115010-71-4_b.png)
![Lithium, [2-(diphenylphosphino)phenyl]-](http://img.cochemist.com/ccimg/95100/95048-82-1.png)
![Lithium, [2-(diphenylphosphino)phenyl]-](http://img.cochemist.com/ccimg/95100/95048-82-1_b.png)






























![bis[(2,3,5,6-η)-bicyclo[2.2.1]hepta-2,5-diene]di-μ-chlorodirhodium](http://img.cochemist.com/ccimg/12300/12257-42-0.png)
![bis[(2,3,5,6-η)-bicyclo[2.2.1]hepta-2,5-diene]di-μ-chlorodirhodium](http://img.cochemist.com/ccimg/12300/12257-42-0_b.png)