Co-reporter:Bibek Dhakal, Luis Bohé, and David Crich
The Journal of Organic Chemistry September 15, 2017 Volume 82(Issue 18) pp:9263-9263
Publication Date(Web):August 31, 2017
DOI:10.1021/acs.joc.7b01850
Although the triflate ion is not generally perceived as a nucleophile, many examples of its behavior as such exist in the literature. This Synopsis presents an overview of such reactions, in which triflate may be either a stoichiometric or catalytic nucleophile, leading to the suggestion that nucleophilic catalysis by triflate may be more common than generally accepted, albeit hidden by the typical reactivity of organic triflates which complicates their observation as intermediates.
Co-reporter:Appi Reddy Mandhapati, Guanyu Yang, Takayuki Kato, Dimitri Shcherbakov, Sven N. Hobbie, Andrea Vasella, Erik C. Böttger, and David Crich
Journal of the American Chemical Society October 18, 2017 Volume 139(Issue 41) pp:14611-14611
Publication Date(Web):September 11, 2017
DOI:10.1021/jacs.7b07754
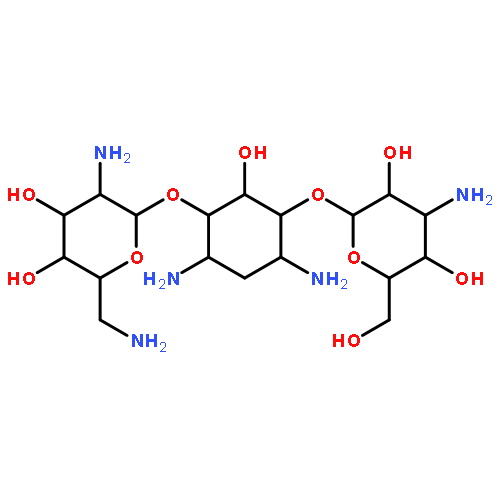
The preparation of a series of four analogues of the aminoglycoside antibiotics neomycin and paromomycin is described in which ring I, involved in critical binding interactions with the ribosomal target, is replaced by an apramycin-like dioxabicyclo[4.4.0]octane system. The effect of this modification is to lock the hydroxymethyl side chain of the neomycin or paromomycin ring I, as part of the dioxabicyclooctane ring, into either the gauche–gauche or the gauche–trans conformation (respectively, axial or equatorial to the bicyclic system). The antiribosomal activity of these compounds is investigated with cell-free translation assays using both bacterial ribosomes and recombinant hybrid ribosomes carrying eukaryotic decoding A site cassettes. Compounds substituted with an equatorial hydroxyl or amino group in the newly formed ring are considerably more active than their axial diastereomers, lending strong support to crystallographically derived models of aminoglycoside–ribosome interactions. One such bicyclic compound carrying an equatorial hydroxyl group has activity equal to that of the parent yet displays better ribosomal selectivity, predictive of an enhanced therapeutic index. A paromomycin analog lacking the hydroxymethyl ring I side chain is considerably less active than the parent. Antibacterial activity against model Gram negative and Gram positive bacteria is reported for selected compounds, as is activity against ESKAPE pathogens and recombinant bacteria carrying specific resistance determinants. Analogues with a bicyclic ring I carrying equatorial amino or hydroxyl groups mimicking the bound side chains of neomycin and paromomycin, respectively, show excellent activity and, by virtue of their novel structure, retain this activity in strains that are insensitive to the parent compounds.
Co-reporter:Peng Wen and David Crich
Organic Letters May 5, 2017 Volume 19(Issue 9) pp:
Publication Date(Web):April 24, 2017
DOI:10.1021/acs.orglett.7b00932
Photocatalytic formation of glycosidic bonds employing stable and readily accessible O-glycosyl derivatives of 2,2,6,6-tetramethylpiperidin-1-ol is presented that employs an iridium-based photocatalyst and blue LEDs. The reaction proceeds at room temperature and in the absence of additives other than 4 Å molecular sieves. Stereoselectivities are modest but nevertheless dependent on the anomeric configuration of the donor, suggesting a substantial degree of concerted character.
Co-reporter:Sandeep Dhanju, Brendan W. Blazejewski, and David Crich
The Journal of Organic Chemistry May 19, 2017 Volume 82(Issue 10) pp:5345-5345
Publication Date(Web):April 28, 2017
DOI:10.1021/acs.joc.7b00717
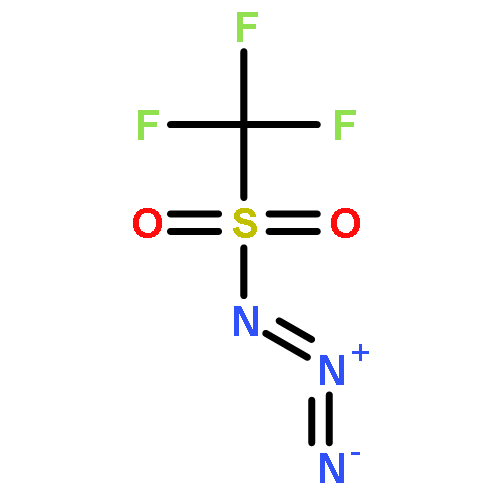
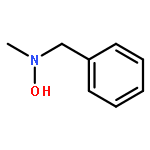
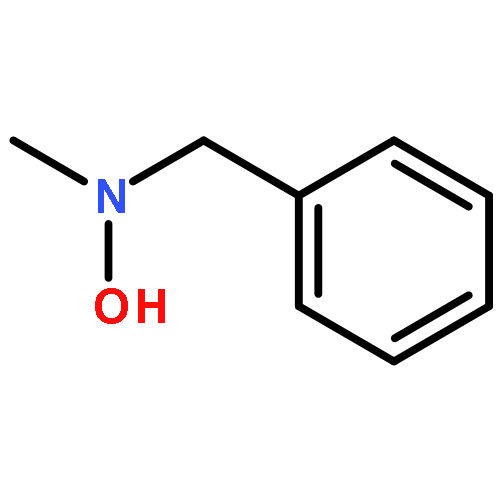
The influence of the electron-withdrawing azide group on the reduction of O-(1-acyloxy-ω-azido)hydroxylamines by triethylsilane in the presence of boron trifluoride etherate is studied and found to increase with increasing proximity to the reaction site, suggesting that the reaction proceeds by way of aminoxocarbenium ion intermediates. The ability to carry azides through the reaction sequence affords O-(ω-azidoalkyl-N,N-dialkylhydroxylamines thereby making such functionality available for use in click chemistry. A series of 4-substituted N-alkoxypiperidines were prepared and studied by variable temperature NMR spectroscopy leading to the conclusion that the rate-determining step in the stereomutation of such piperidines is the piperidine ring flip and not nitrogen inversion or rotation about the N–O bond. The process of N–O bond rotation only becomes rate determining when in the presence of pervasive steric hindrance as is the case with the N-alkoxy-2,2,6,6-tetramethylpiperidines.
Co-reporter:Oskar Popik, Bibek Dhakal, and David Crich
The Journal of Organic Chemistry June 16, 2017 Volume 82(Issue 12) pp:6142-6142
Publication Date(Web):May 22, 2017
DOI:10.1021/acs.joc.7b00746
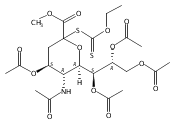
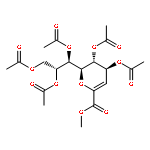
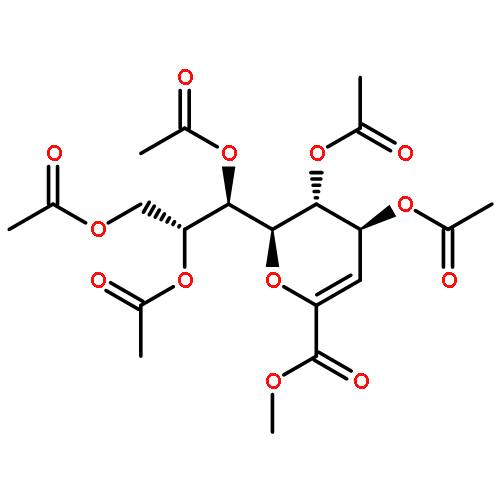
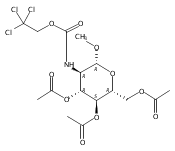
The synthesis of a legionaminic acid donor from N-acetylneuraminic acid in 15 steps and 17% overall yield is described. Activation of the adamantanyl thioglycoside in the donor with N-iodosuccinimide and trifluoromethanesulfonic acid in dichloromethane and acetonitrile at −78 °C in the presence of primary, secondary and tertiary alcohols affords the corresponding glycosides in excellent yield and good to excellent equatorial selectivity. In particular, coupling to the 4-OH of a suitably protected neuraminic acid derivative affords a disaccharide that closely resembles the glycosidic linkage in the polylegionaminic acid from the lipopolysaccharide of the Legionella pneumophila virulence factor. A straightforward deprotection sequence enables conversion of the protected glycosides to the free N,N-diacetyllegionaminic acid glycosides.
Co-reporter:Philip O. Adero, Dean R. Jarois, David Crich
Carbohydrate Research 2017 Volume 449(Volume 449) pp:
Publication Date(Web):8 September 2017
DOI:10.1016/j.carres.2017.06.011
•Hydrogenolytic cleavage in presence of thioglycosides.•Selective hydrogenolysis.•Deprotection of thioglycosides.With the aid of a series of model thioether or thioglycoside containing polyols protected with combinations of benzyl ethers and 2-naphthylmethyl ethers it is demonstrated that the latter are readily cleaved selectively under hydrogenolytic conditions in the presence of the frequently catalyst-poisoning sulfides. These results suggest the possibility of employing 2-naphthylmethyl ethers in place of benzyl ethers in synthetic schemes when hydrogenolytic deprotection is anticipated in the presence of thioether type functionality.Download high-res image (91KB)Download full-size image
Co-reporter:Takayuki Kato, Andrea Vasella, David Crich
Carbohydrate Research 2017 Volume 448(Volume 448) pp:
Publication Date(Web):7 August 2017
DOI:10.1016/j.carres.2017.05.015
•Stereospecific deuterium labelling.•Assignment of pro-R and pro-S hydrogens in side chain.•Side chain conformation.The stereospecifically labeled 6-monodeuterio methyl 2,6-diamino-2,6-dideoxy-α- and β- d-glucopyranosides were synthesized with a view to determining their side chain conformations. NMR studies in D2O at pH 5 and pH 11 reveal both anomers to adopt very predominantly the gt conformation consistent with the gauche conformation of 2-aminoethanol and its acetate salt. In contrast, as also revealed with the help of stereospecifically-labelled monodeuterio isotopomers, the methyl 2-amino-2-deoxy-α- and β- d-glucopyranosides are an approximately 1:1 mixture of gg and gt conformers as is found in glucopyranose itself.Download high-res image (114KB)Download full-size image
Co-reporter:Szymon Buda
Journal of the American Chemical Society 2016 Volume 138(Issue 3) pp:1084-1092
Publication Date(Web):January 5, 2016
DOI:10.1021/jacs.5b13015
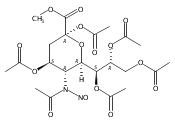
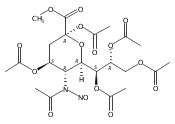
A study of the mechanism of the oxidative deamination of the N-nitroso-N-acetyl sialyl glycosides leading with overall retention of configuration to the corresponding 2-keto-3-deoxy-d-glycero-d-galacto-nonulopyranosidonic acid (KDN) glycosides is described, making use of a series of differentially O-protected N-nitroso-N-acetyl sialyl glycosides and of isotopic labeling studies. No evidence is found for stereodirecting participation by ester groups at the 4- and 7-positions. Comparisons are drawn with oxidative deamination reactions of 4-amino-4-deoxy and 2-amino-2-deoxy hexopyranosides and a common mechanism is formulated involving the intermediacy of 1-oxabicyclo[3.1.0]hexyl oxonium ions following participation by the pyranoside ring oxygen. A minor reaction pathway has been uncovered by labeling studies in the β-thiosialosides that results in the exchange of the 4-O-acetyl group by the glacial acetic acid that serves as external nucleophile in the general oxidative deamination process. A mechanism is proposed for this exchange involving participation by the thioglycoside at the level of an intermediate diazoalkane.
Co-reporter:Sandeep Dhanju and David Crich
Organic Letters 2016 Volume 18(Issue 8) pp:1820-1823
Publication Date(Web):April 7, 2016
DOI:10.1021/acs.orglett.6b00556
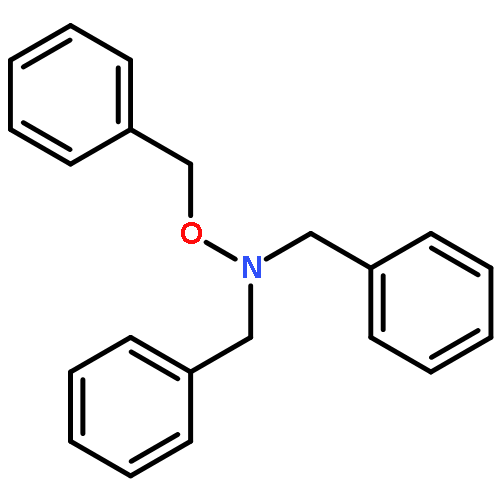
Diverse N,N,O-trisubstituted hydroxylamines, an under-represented group in compound collections, are readily prepared by partial reduction of N-acyloxy secondary amines with diisobutylaluminum hydride followed by acetylation and reduction of the so-formed O-acyl-N,N-disubstituted hydroxylamines with triethylsilane and boron trifluoride etherate. Use of carbon nucleophiles in the last step, including allyltributylstannane, silyl enol ethers, and 2-methylfuran, gives N,N,O-trisubstituted hydroxylamines with branching α- to the O-substituent. N,N-Disubstiuted hydroxylamines are conveniently prepared by reaction of secondary amines with dibenzoyl peroxide followed by diisobutylaluminum hydride reduction.
Co-reporter:Suresh Dharuman, Yichen Wang, David Crich
Carbohydrate Research 2016 Volume 419() pp:29-32
Publication Date(Web):January 2016
DOI:10.1016/j.carres.2015.10.015
•Selective oxidation oxidation of a secondary alcohol in the presence of a hemiacetal is reported.•Stereoselective reduction ketone reduction is reported.•Antibacterial assays of the title compound are reported.•Efficient synthesis of the title compound is reported.A convenient synthesis is described of 5-azido-5-deoxy-2,3-O-isopropylidene-l-rhamnofuranose from l-rhamnose in seven steps and 17% overall yield. A key feature of the synthesis is the selective oxidation of the secondary alcohol in 2,3-O-isopropylidene-l-rhamnofuranose in the presence of the hemiacetal to give the corresponding ketone in good yield using the Parikh–Doering reagent. 5-Azido-5-deoxy-2,3-O-isopropylidene-l-rhamnofuranose is then converted by a literature protocol to 1,5-dideoxy-1,5-imino-l-rhamnitol, which was found to have no significant antimicrobial activity against Pseudomonas aeruginosa, methicillin-resistant Staphylococcus aureus, and Escherichia coli.
Co-reporter:Min Huang, Takayuki Furukawa, Pascal Retailleau, David Crich, Luis Bohé
Carbohydrate Research 2016 Volume 427() pp:21-28
Publication Date(Web):2 June 2016
DOI:10.1016/j.carres.2016.03.028
•Design of cation clocks.•Intramolecular sulfenyl group transfer in glucosylation using sulfoxide donors.•Exploratory use of hydroxylated groups as internal nucleophiles for cation clocks.The use of the 2-O-(2-trimethylsilylmethallyl) group as intramolecular nucleophile and cation clock reaction in the glucopyranose series depends on the nature of the glycosyl donor. As previously reported, with trichloroacetimidates the anticipated intramolecular Sakurai reaction proceeds efficiently and is an effective clock, whereas with sulfoxides complications arise. The source of these complications is now shown to be an intramolecular sulfenyl transfer reaction between the tethered allylsilane and the activated sulfoxide. These results illustrate how a different unimolecular clock reaction may be required for a given cation when it is generated from different donors in order to avoid side reactions. The synthesis and cyclization of a 2-O-(3-hydroxypropyl) glucopyranosyl sulfoxide leading on activation to the formation of a trans-fused acetal is also described. The formation of this crystallographically-established trans-fused acetal is discussed in terms of the high effective concentration of the intramolecular nucleophile which leads to a high degree of a SN2 character in the displacement of the α-glucosyl triflate or at the level of the corresponding α-CIP. The possible use of such intramolecular alcohols as clock reactions and their limitations is discussed.
Co-reporter:Harsha Amarasekara, David Crich
Carbohydrate Research 2016 Volume 435() pp:113-120
Publication Date(Web):29 November 2016
DOI:10.1016/j.carres.2016.09.019
•Intramolecular glycosylation leading to spirodiolide formation.•Stereoselectivity of spirodiolide formation depends on anomeric configuration.•Cyclization yield depends on N5-protecting group.•Trapping of a sialyl nitrilium ion.The synthesis and cyclization reactions, leading to spirocyclic medium ring-sized diolides, of o-(hydroxymethyl)xylylene monoesters of sialyl thioglycosides is described. Cyclization yields and stereoselectivities are found to vary as a function of the anomeric stereochemistry of the thioglycoside and of the N5 protecting group, and these effects are discussed in terms of the reaction mechanism. Cyclization in the presence of acetonitrile results in the isolation and characterization of a Ritter-type N-sialyl acetamide, which affords strong evidence for the participation of acetonitrile in the form of sialyl nitrilium ions.
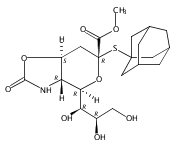
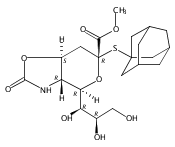
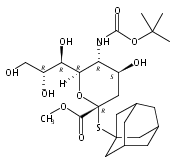
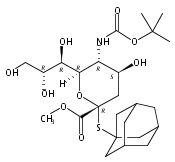
Co-reporter:Bibek Dhakal, Szymon Buda, and David Crich
The Journal of Organic Chemistry 2016 Volume 81(Issue 22) pp:10617-10630
Publication Date(Web):November 2, 2016
DOI:10.1021/acs.joc.6b02221
With a view to the eventual synthesis of glycosyl donors for the stereocontrolled synthesis of pseudaminic acid glycosides, the stereocontrolled synthesis of a d-glycero-d-gulo sialic acid adamantanylthioglycoside carrying an axial azide at the 5-position is described. The synthesis employs levulinic acid as nucleophile in the oxidative deamination of an N-acetylneuraminic acid thioglycoside leading to the formation of a 3-deoxy-d-glycero-d-galacto-2-nonulosonic acid (KDN) derivative selectively protected as 5-O-levulinate. Replacement of the levulinate by triflate enables introduction of the axial azide and hence formation of the glycosyl donor. A shorter synthesis uses trifluoromethanesulfonate as nucleophile in the oxidative deamination step when the 5-O-triflyl KDN derivative is obtained directly. Glycosylation reactions conducted with the 5-azido-d-glycero-d-gulo-configured sialyl adamantanylthioglycoside at −78 °C are selective for the formation of the equatorial glycosides, suggesting that the synthesis of equatorial pseudaminic acid glycosides will be possible as suitable donors become available. A comparable N-acetylneuraminic acid adamantanylthioglycoside carrying an equatorial azide at the 5-position was also found to be selective for equatorial glycoside formation under the same conditions, suggesting that reinvestigation of other azide-protected NeuAc donors is merited. Glycosylation stereoselectivity in the d-glycero-d-gulo series is discussed in terms of the side-chain conformation of the donor.
Co-reporter:Dr. Suresh Dharuman ;Dr. David Crich
Chemistry - A European Journal 2016 Volume 22( Issue 13) pp:4535-4542
Publication Date(Web):
DOI:10.1002/chem.201505019
Abstract
The synthesis of a series of conformationally locked mannopyranosyl thioglycosides in which the C6−O6 bond adopts either the gauche,gauche, gauche,trans, or trans,gauche conformation is described, and their influence on glycosylation stereoselectivity investigated. Two 4,6-O-benzylidene-protected mannosyl thioglycosides carrying axial or equatorial methyl groups at the 6-position were also synthesized and the selectivity of their glycosylation reactions studied to enable a distinction to be made between steric and stereoelectronic effects. The presence of an axial methoxy group at C6 in the bicyclic donor results in a decreased preference for formation of the β-mannoside, whereas an axial methyl group has little effect on selectivity. The result is rationalized in terms of through-space stabilization of a transient intermediate oxocarbenium ion by the axial methoxy group resulting in a higher degree of SN1-like character in the glycosylation reaction. Comparisons are made with literature examples and exceptions are discussed in terms of pervading steric effects layered on top of the basic stereoelectronic effect.
Co-reporter:Philip O. Adero; Takayuki Furukawa; Min Huang; Debaraj Mukherjee; Pascal Retailleau; Luis Bohé
Journal of the American Chemical Society 2015 Volume 137(Issue 32) pp:10336-10345
Publication Date(Web):July 24, 2015
DOI:10.1021/jacs.5b06126
The development of a cation clock method based on the intramolecular Sakurai reaction for probing the concentration dependence of the nucleophile in glycosylation reactions is described. The method is developed for the sulfoxide and trichloroacetimidate glycosylation protocols. The method reveals that O-glycosylation reactions have stronger concentration dependencies than C-glycosylation reactions consistent with a more associative, SN2-like character. For the 4,6-O-benzylidene-directed mannosylation reaction a significant difference in concentration dependence is found for the formation of the β- and α-anomers, suggesting a difference in mechanism and a rationale for the optimization of selectivity regardless of the type of donor employed. In the mannose series the cyclization reaction employed as clock results in the formation of cis and trans-fused oxabicyclo[4,4,0]decanes as products with the latter being strongly indicative of the involvement of a conformationally mobile transient glycosyl oxocarbenium ion. With identical protecting group arrays cyclization in the glucopyranose series is more rapid than in the mannopyranose manifold. The potential application of related clock reactions in other carbenium ion-based branches of organic synthesis is considered.
Co-reporter:Takahiko Matsushita; Weiwei Chen; Reda Juskeviciene; Youjin Teo; Dimitri Shcherbakov; Andrea Vasella; Erik C. Böttger
Journal of the American Chemical Society 2015 Volume 137(Issue 24) pp:7706-7717
Publication Date(Web):May 29, 2015
DOI:10.1021/jacs.5b02248
A series of 20 4′-O-glycosides of the aminoglycoside antibiotic paromomycin were synthesized and evaluated for their ability to inhibit protein synthesis by bacterial, mitochondrial and cytosolic ribosomes. Target selectivity, i.e., inhibition of the bacterial ribosome over eukaryotic mitochondrial and cytosolic ribosomes, which is predictive of antibacterial activity with reduced ototoxicity and systemic toxicity, was greater for the equatorial than for the axial pyranosides, and greater for the d-pentopyranosides than for the l-pentopyranosides and d-hexopyranosides. In particular, 4′-O-β-d-xylopyranosyl paromomycin shows antibacterioribosomal activity comparable to that of paromomycin, but is significantly more selective showing considerably reduced affinity for the cytosolic ribosome and for the A1555G mutant mitochondrial ribosome associated with hypersusceptibility to drug-induced ototoxicity. Compound antibacterioribosomal activity correlates with antibacterial activity, and the ribosomally more active compounds show activity against Escherichia coli, Klebsiella pneumonia, Enterobacter cloacae, Acinetobacter baumannii, and methicillin-resistant Staphylococcus aureus (MRSA). The paromomycin glycosides retain activity against clinical strains of MRSA that are resistant to paromomycin, which is demonstrated to be a consequence of 4′-O-glycosylation blocking the action of 4′-aminoglycoside nucleotidyl transferases by the use of recombinant E. coli carrying the specific resistance determinant.
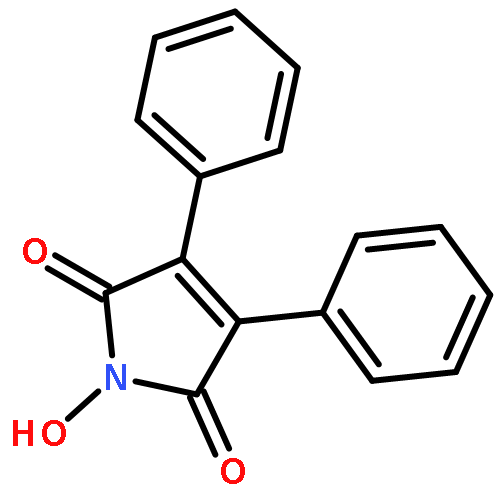
Co-reporter:Girish C. Sati and David Crich
Organic Letters 2015 Volume 17(Issue 16) pp:4122-4124
Publication Date(Web):August 12, 2015
DOI:10.1021/acs.orglett.5b02079
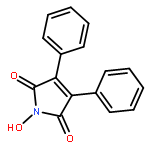
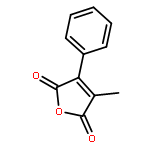
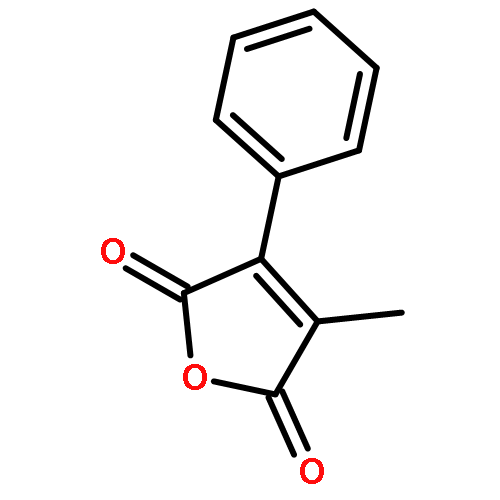
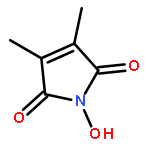
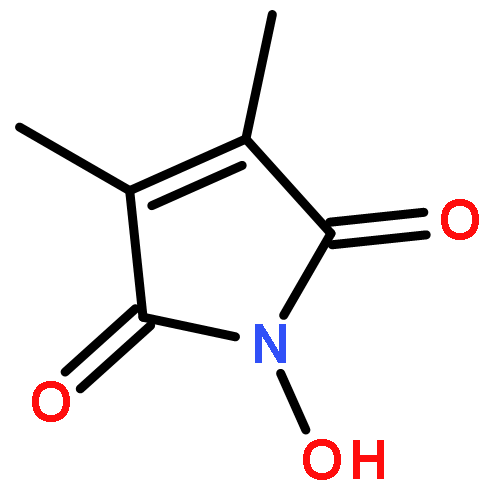
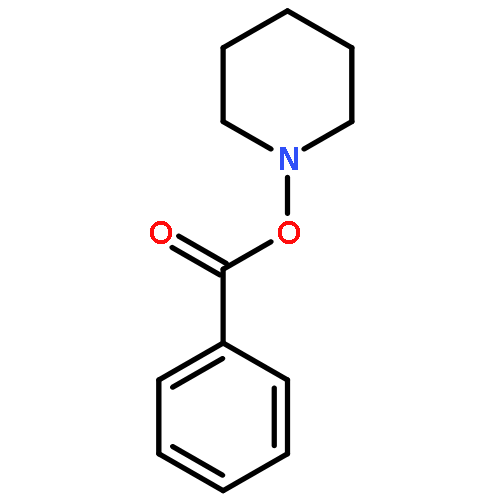
Reaction of variously substituted N-trifluoromethanesulfonyloxy maleimides with primary amines in the presence of potassium carbonate in DMF at room temperature results in the formation of 3-N-alkyl pyrimidin-2,4-diones in good yield.
Co-reporter:Amandine Noel, Bernard Delpech and David Crich
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 35) pp:9324-9324
Publication Date(Web):20 Aug 2015
DOI:10.1039/C5OB90143J
Correction for ‘Comparison of the reactivity of β-thiolactones and β-lactones toward ring-opening by thiols and amines’ by Amandine Noel et al., Org. Biomol. Chem., 2012, 10, 6480–6483.
Co-reporter:Luis Bohé, David Crich
Carbohydrate Research 2015 Volume 403() pp:48-59
Publication Date(Web):11 February 2015
DOI:10.1016/j.carres.2014.06.020
•Mechanistic and spectroscopic evidence for glycosyl oxocarbenium ions.•Computational work on glycosyl oxocarbenium ions.•Kinetic studies on glycosylation reactions.•Counterion and additive effects.•Non-traditional methods.An overview of recent advances in glycosylation with particular emphasis on mechanism is presented. The mounting evidence for both the existence of glycosyl oxocarbenium ions as fleeting intermediates in some reactions, and the crucial role of the associated counterion in others is discussed. The extremes of the SN1 and SN2 manifolds for the glycosylation reaction are bridged by a continuum of mechanisms in which it appears likely that most examples are located.
Co-reporter:Appi Reddy Mandhapati, Takayuki Kato, Takahiko Matsushita, Bashar Ksebati, Andrea Vasella, Erik C. Böttger, and David Crich
The Journal of Organic Chemistry 2015 Volume 80(Issue 3) pp:1754-1763
Publication Date(Web):January 5, 2015
DOI:10.1021/jo502677a
The synthesis of a series of α-trifluoromethylcyclohexanols and analogous trimethylsilyl ethers by addition of the Ruppert–Prakash reagent to substituted cyclohexanones is presented. A method for the assignment of configuration of such compounds, of related α-trifluoromethylcyclohexylamines and of quaternary trifluoromethyl-substituted carbons is described based on the determination of the 3JCH coupling constant between the fluorine-decoupled 13CF3 resonance and the vicinal hydrogens. This method is dubbed fluorine-decoupled carbon spectroscopy and abbreviated FDCS. The method is also applied to the configurational assignment of substances bearing mono-, di-, and perfluoroalkyl rather than trifluoromethyl groups. The configuration of all substances was verified by either 19F−1H heteronuclear Overhauser spectroscopy (HOESY) or X-ray crystallography. The relative merits of FDCS and HOESY are compared and contrasted. 2JCH, 3JCH, and 4JCH coupling constants to 19F decoupled CF3 groups in alkenes and arenes have also been determined and should prove to be useful in the structural assignment of trifluoromethylated alkenes and arenes.
Co-reporter:Takayuki Kato, Guanyu Yang, Youjin Teo, Reda Juskeviciene, Déborah Perez-Fernandez, Harish M. Shinde, Sumanth Salian, Bruno Bernet, Andrea Vasella, Erik C. Böttger, and David Crich
ACS Infectious Diseases 2015 Volume 1(Issue 10) pp:479
Publication Date(Web):July 28, 2015
DOI:10.1021/acsinfecdis.5b00069
Chemistry for the efficient modification of the kanamycin class of 4,6-aminoglycosides at the 4′-position is presented. In all kanamycins but kanamycin B, 4′-O-alkylation is strongly detrimental to antiribosomal and antibacterial activity. Ethylation of kanamycin B at the 4″-position entails little loss of antiribosomal and antibacterial activity, but no increase of ribosomal selectivity. These results are contrasted with those for the 4,5-aminoglycosides, where 4′-O-alkylation of paromomycin causes only a minimal loss of activity but results in a significant increase in selectivity with a concomitant loss of ototoxicity.Keywords: antibacterial activity; decoding A site; mitochondrial rRNA; ototoxicity; ribosomal selectivity
Co-reporter:Peng Wen and David Crich
The Journal of Organic Chemistry 2015 Volume 80(Issue 24) pp:12300-12310
Publication Date(Web):November 13, 2015
DOI:10.1021/acs.joc.5b02203
The preparation of a series of mannopyranosyl donors carrying 2-O-(2-oxoalkyl) ethers and their use in glycosylation reactions are described. The formation of cyclic products with the simple 2-O-phenacyl ether and with the 2-O-(t-butoxycarbonylmethyl) ether establishes the stereoelectronic feasibility of participation in such systems. The high β-selectivities observed with the bis-trifluoromethyl phenacyl ether indicate that participation can be suppressed through the introduction of electron-withdrawing substituents. The high β-selectivities and absence of cyclic products observed with the 2-O-(methoxycarbonylmethyl) ether exclude the effective participation of esters through six-membered cyclic intermediates in this series. The results are discussed in terms of the conformation of cyclic dioxenium ions (E,E-, E,Z-, or Z,Z-) and in the context of “neighboring group” participation by nonvicinal esters in glycosylation. Methods for the deprotection of the 2-O-phenacyl and 2-O-(methoxycarbonylmethyl) ethers are described.
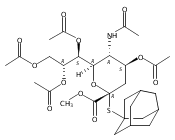
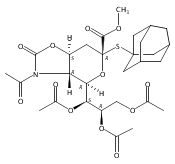
Co-reporter:Appi Reddy Mhapati;Dr. Salla Rajender;Jonathan Shaw ;Dr. David Crich
Angewandte Chemie International Edition 2015 Volume 54( Issue 4) pp:1275-1278
Publication Date(Web):
DOI:10.1002/anie.201409797
Abstract
The preparation of a crystalline, peracetyl adamantanyl thiosialoside donor protected by an isothiocyanate group is described. On activation at −78 °C in the presence of typical carbohydrate acceptors, this donor gives high yields of the corresponding sialosides with exquisite α-selectivity. The high selectivity extends to the 4-O-benzyl-protected 3-OH acceptors, which are typically less reactive and selective than galactose 3,4-diols. Treatment of the α-sialosides with tris(trimethylsilyl)silane or allyltris(trimethylsilyl)silane results in replacement of the C5N5 bond by a CH or a CC bond. The reaction of the isothiocyanate-protected sialosides with thioacids generates amides, while reaction with an amine gives a thiourea, which can be converted into a guanidine. The very high α-selectivities observed with the new donor and the rich chemistry of the isothiocyante function considerably extend the scope for optimization at the sialoside 5-position.
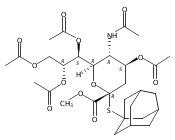
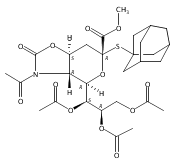
Co-reporter:Appi Reddy Mhapati;Dr. Salla Rajender;Jonathan Shaw ;Dr. David Crich
Angewandte Chemie 2015 Volume 127( Issue 4) pp:1291-1294
Publication Date(Web):
DOI:10.1002/ange.201409797
Abstract
The preparation of a crystalline, peracetyl adamantanyl thiosialoside donor protected by an isothiocyanate group is described. On activation at −78 °C in the presence of typical carbohydrate acceptors, this donor gives high yields of the corresponding sialosides with exquisite α-selectivity. The high selectivity extends to the 4-O-benzyl-protected 3-OH acceptors, which are typically less reactive and selective than galactose 3,4-diols. Treatment of the α-sialosides with tris(trimethylsilyl)silane or allyltris(trimethylsilyl)silane results in replacement of the C5N5 bond by a CH or a CC bond. The reaction of the isothiocyanate-protected sialosides with thioacids generates amides, while reaction with an amine gives a thiourea, which can be converted into a guanidine. The very high α-selectivities observed with the new donor and the rich chemistry of the isothiocyante function considerably extend the scope for optimization at the sialoside 5-position.
Co-reporter:Angélique Ferry ; Gaëlle Malik ; Xavier Guinchard ; Václav Vĕtvička
Journal of the American Chemical Society 2014 Volume 136(Issue 42) pp:14852-14857
Publication Date(Web):September 23, 2014
DOI:10.1021/ja507084t
Di- and trimeric hydroxylamine-based mimetics of β-(1→3)-glucans have been accessed by an asymmetric synthesis route featuring an iterative double ring-closing reductive amination reaction. These oligomeric hydroxylamines are demonstrated to inhibit the staining of human neutrophils and of mouse macrophages by fluorescent anti-CR3 and anti-dectin-1 antibodies, respectively, and to stimulate phagocytosis, all in a linkage-dependent manner suggestive of binding to the lectin domains of complement receptor 3 (CR3) and dectin-1. The ability of these relatively short mimetics to bind to CR3 and dectin-1, as compared to the greater degree of polymerization required in β-(1→3)-glucans, is discussed in terms of the increased hydrophobicity of the α-face on replacement of the glycosidic bond by the hydroxylamine linkage.
Co-reporter:Pavan K. Kancharla ; Takayuki Kato
Journal of the American Chemical Society 2014 Volume 136(Issue 14) pp:5472-5480
Publication Date(Web):March 7, 2014
DOI:10.1021/ja501276r
A method for the investigation of the influence of protecting groups on the anomeric equilibrium in the sialic acid glycosides has been developed on the basis of the equilibration of O-sialyl hydroxylamines by reversible homolytic scission of the glycosidic bond following the dictates of the Fischer–Ingold persistent radical effect. It is found that a trans-fused 4O,5N-oxazolidinone group stabilizes the equatorial glycoside, i.e., reduces the anomeric effect, when compared to the 4O,5N-diacetyl protected systems. This effect is discussed in terms of the powerful electron-withdrawing nature of the oxazolidinone system, which in turn is a function of its strong dipole moment in the mean plane of the pyranose ring system. The new equilibration method displays a small solvent effect and is most pronounced in less polar media consistent with the anomeric effect in general. The unusual (for anomeric radicals) poor kinetic selectivity of anomeric sialyl radicals is discussed in terms of the planar π-type structure of these radicals and of competing 1,3-diaxial interactions in the diastereomeric transition states for trapping on the α- and β-faces of the radical.
Co-reporter:Weiwei Chen, Takahiko Matsushita, Dimitri Shcherbakov, Heithem Boukari, Andrea Vasella, Erik C. Böttger and David Crich
MedChemComm 2014 vol. 5(Issue 8) pp:1179-1187
Publication Date(Web):07 Apr 2014
DOI:10.1039/C4MD00119B
A series of 4′-O-glycopyranosyl paromomycin analogs and a 4′-O-(glucosyloxymethyl) analog were synthesized and evaluated for their ribosomal activity to determine the influence of the glycosyl moiety on drug activity and selectivity. Antibacterial activity against clinical strains of Escherichia coli and Staphylococcus aureus was also investigated. While all compounds were less active than paromomycin itself, differences in activity were seen between the gluco-, manno-, and galactopyranosyl series and between individual anomers. These differences in activity, which are discussed in terms of variations in affinity for the ribosomal decoding A site, may prove useful in the design of subsequent generations of improved aminoglycoside antibiotics with reduced toxicity.
Co-reporter:Amandine Noel, Bernard Delpech, and David Crich
The Journal of Organic Chemistry 2014 Volume 79(Issue 9) pp:4068-4077
Publication Date(Web):April 9, 2014
DOI:10.1021/jo500577c
The synthesis of a series of di-, tri-, and tetraalkyl β-thiolactones and β-lactones is described as well as their thermal decomposition with extrusion of carbon oxysulfide and carbon dioxide in two solvents of opposite polarities. The β-thiolactones are considerably more thermally stable than the β-lactones and require higher temperatures for efficient decomposition in both solvents, whatever the degree of substitution. The results are interpreted in terms of a zwitterionic mechanism for fragmentation with a change in the rate-determining step between the two series.
Co-reporter:Appi Reddy Mhapati;Dimitri Shcherbakov;Stefan Duscha;Andrea Vasella;Erik C. Böttger
ChemMedChem 2014 Volume 9( Issue 9) pp:
Publication Date(Web):
DOI:10.1002/cmdc.201490032
Co-reporter:Appi Reddy Mhapati;Dimitri Shcherbakov;Stefan Duscha;Andrea Vasella;Erik C. Böttger
ChemMedChem 2014 Volume 9( Issue 9) pp:2074-2083
Publication Date(Web):
DOI:10.1002/cmdc.201402146
Abstract
A series of apramycin derivatives was prepared and investigated for antibacterial activity and the ability to inhibit protein synthesis in cell-free translation assays. The effect of various modifications at the 6′- and N7′-positions on antiribosomal activity is discussed in terms of their influence on drug binding to specific residues in the decoding A-site. These studies contribute to the development of a structure–activity relationship for the antibacterial activity of the apramycin class of aminoglycosides and to the future design and development of more active and less toxic antibiotics.
Co-reporter:Myriame Moumé-Pymbock ; Takayuki Furukawa ; Sujit Mondal
Journal of the American Chemical Society 2013 Volume 135(Issue 38) pp:14249-14255
Publication Date(Web):August 28, 2013
DOI:10.1021/ja405588x
The effect of a 4,6-O-alkylidene acetal on the rate of acid-catalyzed hydrolysis of methyl galactopyranosides and of spontaneous hydrolysis of 2,4-dinitrophenyl galactopyranosides has been studied through the synthesis and hydrolysis of analogs in which O6 is replaced by a methoxymethylene unit in which the methoxy group adopts either an equatorial or an axial position according to the configuration. Consistent with earlier studies under both acid-catalyzed and spontaneous hydrolysis conditions, the alkylidene acetal, or its 7-carba analog, retards hydrolysis with respect to comparable systems lacking the cyclic protecting group. The configuration at C6 in the 7-carba analogs does not influence the rate of acid-catalyzed hydrolysis but has a minor influence on the rate of spontaneous hydrolysis of the 2,4-dinitrophenyl galactosides, confirming earlier studies on the role played by the hydroxymethyl group conformation on glycoside reactivity. The benzylidene acetal is found to stabilize the α-anomer of galactopyranose derivatives relative to monocyclic analogs. Reasons for the α-selectivity of 4,6-O-benzylidene-protected galactopyranosyl donors bearing neighboring group-active protecting groups at O2 are discussed.
Co-reporter:Pavan K. Kancharla
Journal of the American Chemical Society 2013 Volume 135(Issue 50) pp:18999-19007
Publication Date(Web):November 21, 2013
DOI:10.1021/ja410683y
Two N-acetyl 4O,5N-oxazolidinone-protected sialyl thioglycosides epimeric at the 7-position have been synthesized and their reactivity and stereoselectivity in glycosylation reactions have been compared. It is demonstrated that the natural 7S-donor is both more reactive and more α-selective than the unnatural 7R-isomer. The difference in reactivity is attributed to the side chain conformation and specifically to the proximity of O7 to the anomeric center. In the natural 7S-isomer, O7 is closer to the anomeric center than in its unnatural 7R-epimer and, therefore, better able to support incipient positive charge at the locus of reaction. The difference in selectivity is also attributed to the side conformation, which in the unnatural 7R-series is placed perpendicularly above the α-face of the donor and so shields it to a greater extent than in the 7S-series. These observations are consistent with earlier conclusions on the influence of the side chain conformation on reactivity and selectivity derived from conformationally locked models in the glucose and galactose series and corroborate the suggestion that those effects are predominantly stereoelectronic rather than torsional. The possible relevance of side chain conformation as a factor in the influence of glycosylation stereoselectivity by remote protecting groups and as a control element in enzymic processes for glycosidic bond formation and hydrolysis are discussed. Methods for assignment of the anomeric configuration in the sialic acid glycosides are critically surveyed.
Co-reporter:Fabien Fécourt, Bernard Delpech, Oleg Melnyk, and David Crich
Organic Letters 2013 Volume 15(Issue 14) pp:3758-3761
Publication Date(Web):July 1, 2013
DOI:10.1021/ol401677a
Se-(9-Fluorenylmethyl) selenoesters are readily prepared, stable precursors to selenocarboxylates, which they liberate on treatment with DBU. Fm selenoesters are compatible with the use of TFA for the removal of Boc groups and with simple peptide bond forming reactions. Amino acid derived selenocarboxylates condense directly with amines to give amides, react smoothly with isocyanates and isothiocyanates to give amides, and couple with electron-deficient azides also to give amides.
Co-reporter:Cécile Ouairy, Thierry Cresteil, Bernard Delpech, David Crich
Carbohydrate Research 2013 Volume 377() pp:35-43
Publication Date(Web):9 August 2013
DOI:10.1016/j.carres.2013.05.011
•New tetrahydroimidazopyridine-type glycosidase inhibitors have been synthesized.•Two of the glycoimidazoles were assayed for inhibitory activity of α- and β-gluco- and mannosidase.•The 3-hydroxy group is critical in the key substrate–enzyme interactions enzymes.Three tetrahydropyridoimidazole-type glycosidase inhibitors have been synthesized with the 3-deoxy ribo- and arabino-, and 3-deoxy-3-fluoro gluco-configurations and two of them screened for activity against α- and β-gluco- and mannosidase enzymes. Only one substance, the 3-deoxy-3-fluoro-derivative of the gluco-configured tetrahydropyridoimidazole was found to have any activity against a single enzyme, sweet almond β-glucosidase, and even then at a level 100-fold lower than that of the corresponding simple gluco-configured tetrahydropyridoimidazole thereby underlining the importance of the 3-hydroxy group in the key substrate–enzyme interactions.
Co-reporter:Angélique Ferry, Gaëlle Malik, Pascal Retailleau, Xavier Guinchard, and David Crich
The Journal of Organic Chemistry 2013 Volume 78(Issue 14) pp:6858-6867
Publication Date(Web):June 21, 2013
DOI:10.1021/jo400864s
An improved strategy for the synthesis of P-chiral gluco- and manno-phosphonite-borane complexes is described on the basis of the addition of diethyl phosphonite-borane to a glucal-derived aldehyde, followed by a cyclization coupled with an ethyl/methyl exchange. This direct P(III) strategy facilitates the obtention of various P-chiral phosphonite-boranes, of which further coupling reactions are described leading to the selective synthesis of two phostone dimers.
Co-reporter:Sébastien Picard, David Crich
Tetrahedron 2013 69(26) pp: 5501-5510
Publication Date(Web):
DOI:10.1016/j.tet.2013.04.094
Co-reporter:Dr. Chrasekhar Navuluri ;Dr. David Crich
Angewandte Chemie International Edition 2013 Volume 52( Issue 43) pp:11339-11342
Publication Date(Web):
DOI:10.1002/anie.201303781
Co-reporter:Dr. Chrasekhar Navuluri ;Dr. David Crich
Angewandte Chemie 2013 Volume 125( Issue 43) pp:11549-11552
Publication Date(Web):
DOI:10.1002/ange.201303781
Co-reporter:Dr. Gaëlle Malik;Angélique Ferry;Dr. Xavier Guinchard;Dr. Thierry Cresteil;Dr. David Crich
Chemistry - A European Journal 2013 Volume 19( Issue 6) pp:2168-2179
Publication Date(Web):
DOI:10.1002/chem.201202374
Abstract
A series of novel polyhydroxylated N-alkoxypiperidines has been synthesized by ring-closing double reductive amination (DRA) of highly functionalized 1,5-dialdehydes with various hydroxylamines. The required saccharide-based dialdehydes were prepared efficiently from sodium cyclopentadienylide in seven steps. A two-step protocol has been developed for the DRA; it led, after deprotection, to isofagomine, 3-deoxyisofagomine, and numerous other N-alkoxy analogues. The barrier to inversion in these polyhydroxylated N-alkoxypiperidine derivatives was found by variable-temperature NMR methods to be approximately 15 kcal mol−1. With the exception of N-hydroxyisofagomine itself, none of the compounds prepared showed significant inhibitory activity against sweet almond β-glucosidase.
Co-reporter:Angélique Ferry ; Xavier Guinchard ; Pascal Retailleau
Journal of the American Chemical Society 2012 Volume 134(Issue 29) pp:12289-12301
Publication Date(Web):June 29, 2012
DOI:10.1021/ja305104b
Stereoselective syntheses of P-chiral ammonium phosphonite-borane complexes in the gluco- and manno-like series have been developed from P(V) phostone derivatives. The coupling reactions of these phostone donors with alcohols have been investigated with particular emphasis on the influence of protecting groups and conditions on stereoselectivity. The phosphonite–borane complexes may be applied directly in the coupling reactions and the products oxidized in situ to give phostone-mimetics of disaccharides. On the basis of these studies, successful protocols were established for the synthesis of β-gluco and α- and β-manno-configured phostones of primary alcohols. Deprotection of the dimeric compounds leads to novel families of α- or β-(1→6)-linked glycomimetics.
Co-reporter:Min Huang ; Pascal Retailleau ; Luis Bohé
Journal of the American Chemical Society 2012 Volume 134(Issue 36) pp:14746-14749
Publication Date(Web):August 24, 2012
DOI:10.1021/ja307266n
The use of a cationic cyclization reaction as a probe of the glycosylation mechanism has been developed and applied to the 4,6-O-benzylidene-protected mannopyranoside system. Cyclization results in the formation of both cis- and trans-fused tricyclic systems, invoking an intermediate glycosyl oxocarbenium ion reacting through a boat conformation. Competition reactions with isopropanol and trimethyl(methallyl)silane are interpreted as indicating that β-O-mannosylation proceeds via an associative SN2-like mechanism, whereas α-O-mannosylation and β-C-mannosylation are dissociative and SN1-like. Relative rate constants for reactions going via a common intermediate can be estimated.
Co-reporter:Gaëlle Malik, Xavier Guinchard, and David Crich
Organic Letters 2012 Volume 14(Issue 2) pp:596-599
Publication Date(Web):December 23, 2011
DOI:10.1021/ol203213f
A de novo synthesis of novel polyhydroxylated N-alkoxypiperidines based on the ring-closing double reductive amination of 1,5-dialdehydes, obtained by oxidative cleavage of cyclopentene derivatives, with O-substituted hydroxylamines is reported. Isofagomine was accessed by cleavage of the N–O bond of an N-alkoxypiperidine.
Co-reporter:Amandine Noel, Bernard Delpech, and David Crich
Organic Letters 2012 Volume 14(Issue 5) pp:1342-1345
Publication Date(Web):February 15, 2012
DOI:10.1021/ol300255q
5-N-Acetyl-5-N,4-O-oxazolidinone protected α- and β-sialyl phosphates react with allyltributylstannane and a variety of trimethylsilyl enol ethers to give α-sialyl C-glycosides in high yield and excellent selectivity. Elimination to give the 2,3-glycal is minimized by the presence of the oxazolidinone ring. The oxazolidinone ring can be subsequently cleaved under mild conditions at room temperature leaving in place the native acetamide group.
Co-reporter:Amandine Noel, Bernard Delpech, and David Crich
Organic Letters 2012 Volume 14(Issue 16) pp:4138-4141
Publication Date(Web):July 30, 2012
DOI:10.1021/ol301779e
N-Acetyl 4-O,5-N-oxazolidinone protected sialyl phosphates of either anomeric configuration are excellent donors for the formation of α-S-sialosides at −78 °C in dichloromethane with primary, secondary, and tertiary thiols including galactose 3-, 4-, and 6-thiols. The reactions, which proceed under typical Lewis acid promoted glycosylation conditions, are highly α-selective and do not suffer from competing elimination of the phosphate.
Co-reporter:Amandine Noel, Bernard Delpech and David Crich
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 32) pp:6480-6483
Publication Date(Web):14 Jun 2012
DOI:10.1039/C2OB25640A
An investigation into the comparative reactivity of simple β-lactones and β-thiolactones toward a thiol and a primary amine is reported. A simple 3-mercaptomethyl-2-oxetanone is found to undergo rearrangement in the presence of aqueous base to give the corresponding thietane-3-carboxylic acid rather than the 3-hydroxymethyl-2-thietanone. Implications for the use of β-thiolactones in bioorganic and medicinal chemistry are discussed.
Co-reporter:Sylvain Aubry, Geneviève Aubert, Thierry Cresteil and David Crich
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 13) pp:2629-2632
Publication Date(Web):18 Jan 2012
DOI:10.1039/C2OB06976H
The synthesis of β-thiolactone and β-lactam analogs of tetrahydrolipstatin is described from a common late-stage β-lactone derivative. These analogs, and a cis-disubstituted β-lactone analog of tetrahydrolipstatin, were screened for activity against porcine pancreatic lipase and for inhibition of cell growth of a panel of four human cancer lines.
Co-reporter:Indrajeet Sharma, Luis Bohé, David Crich
Carbohydrate Research 2012 Volume 357() pp:126-131
Publication Date(Web):1 August 2012
DOI:10.1016/j.carres.2012.05.025
It is reported that the replacement of the 4- and 6-O-benzyl ethers in 2,3,4,6-tetra-O-benzyl-α,β-mannopyranose by a 4,6-O-benzylidene acetal results in an increased population of the β-anomer at equilibrium in CDCl3 solution. The phenomenon is considered to arise from the lower steric bulk of the benzylidene acetal that, through diminished buttressing interactions, reduces steric interactions normally present in the β-anomer.
Co-reporter:Myriame Moumé-Pymbock and David Crich
The Journal of Organic Chemistry 2012 Volume 77(Issue 20) pp:8905-8912
Publication Date(Web):September 25, 2012
DOI:10.1021/jo3011655
Unlike alcohols, the reaction of C-nucleophiles with 2-O-benzyl-4,6-O-benzylidene-protected 3-deoxy-gluco- and mannopyranosyl thioglycosides is highly stereoselective providing the α-C-glycosides in the gluco-series and the β-C-glycosides in the manno-series. Conformational analysis of nucleophilic attack of putative intermediate glycosyl oxocarbenium ions suggests that the observed selectivities for C-glycoside formation can be explained by preferential attack on the opposite face of the oxocarbenium to the C2–H2 bond and that eclipsing interactions with this bond are the main stereodetermining factor. It is argued that the steric interactions in the attack of alcohols (sp3-hybridized O) and of typical carbon-based nucleophiles (sp2 C) on oxocarbenium ions are very different, with the former being less severe, and thus that there is no a priori reason to expect O- and C-glycosylation to exhibit parallel stereoselectivities for attack on a given oxocarbenium ion.
Co-reporter:Dr. Pavan K. Kancharla;Chrasekhar Navuluri ;Dr. David Crich
Angewandte Chemie 2012 Volume 124( Issue 44) pp:11267-11271
Publication Date(Web):
DOI:10.1002/ange.201204400
Co-reporter:Dr. Pavan K. Kancharla;Chrasekhar Navuluri ;Dr. David Crich
Angewandte Chemie International Edition 2012 Volume 51( Issue 44) pp:11105-11109
Publication Date(Web):
DOI:10.1002/anie.201204400
Co-reporter:Sylvain Aubry, Kaname Sasaki, Laure Eloy, Geneviève Aubert, Pascal Retailleau, Thierry Cresteil and David Crich
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 20) pp:7134-7143
Publication Date(Web):18 Jul 2011
DOI:10.1039/C1OB05967J
A series of novel peptide-based β-thiolactones were synthesized and assayed for cytotoxicity against several human cancer cell lines, where they showed greater activity than the corresponding β-lactones and β-lactams. Several of the β-thiolactones prepared showed strong inhibitory activity in vitro against human cathepsins B and L.
Co-reporter:David Crich and Md. Yeajur Rahaman
The Journal of Organic Chemistry 2011 Volume 76(Issue 21) pp:8611-8620
Publication Date(Web):September 28, 2011
DOI:10.1021/jo201780e
Tetra-, hexa-, and octasaccharide subunits of the [→4)-β-d-Manp-(1→4)-β-d-Xylp-(1→]n xylomannan motif proposed as the structure of a novel nonprotein, thermal hysteresis-producing antifreeze substance from the freeze-tolerant Alaskan beetle Upis ceramboides have been accessed by total chemical synthesis. Comparison of their NMR spectral data with data of the isolate lends strong support to the proposed structure. Synthetic tetrasaccharides representing various linkage isomers considered (α- rather than β-manno, and linkage through mannose O3 rather than O4) show more significant chemical shift differences with the isolate and are therefore excluded from further consideration.
Co-reporter:David Crich
The Journal of Organic Chemistry 2011 Volume 76(Issue 22) pp:9193-9209
Publication Date(Web):September 15, 2011
DOI:10.1021/jo2017026
This Perspective outlines work in the Crich group on the diastereoselective synthesis of the so-called difficult classes of glycosidic bond: the 2-deoxy-β-glycopyranosides, the β-mannopyranosides, the α-sialosides, the α-glucopyranosides, and the β-arabinofuranosides with an emphasis on the critical interplay between mechanism and methodology development.
Co-reporter:Indrajeet Sharma and David Crich
The Journal of Organic Chemistry 2011 Volume 76(Issue 16) pp:6518-6524
Publication Date(Web):June 30, 2011
DOI:10.1021/jo200497j
Attachment of a growing peptide chain to a glycylaminomethyl resin via a thioglycinamide bond is compatible with Fmoc-chemistry solid-phase peptide synthesis. Subsequent S-alkylation of the thioamide gives a thioimide that, on treatment with aqueous trifluoroacetic acid, releases the peptide from the resin in the form of a C-terminal thioester.
Co-reporter:David Crich, Ming Li, Prasanna Jayalath
Carbohydrate Research 2009 Volume 344(Issue 1) pp:140-144
Publication Date(Web):5 January 2009
DOI:10.1016/j.carres.2008.10.007
Dimethylthexylsilyl 2-acetamido-3-O-allyl-2-deoxy-6-O-(4-methoxybenzyl)-β-d-glucopyranoside was prepared by reduction of the corresponding 4,6-O-(4-methoxybenzylidene) acetal with sodium cyanoborohydride and trifluoroacetic acid. This alcohol was coupled to 2-O-benzoyl-3,4,6-tri-O-benzyl-α-d-glucopyranosyl trichloroacetimidate to give a β-glucoside that was converted to dimethylthexylsilyl 3,4,6-tri-O-benzyl-β-d-mannopyranosyl-(1→4)-2-acetamido-3-O-allyl-2-deoxy-6-O-(4-methoxybenzyl)-β-d-glucopyranoside by saponification, Dess–Martin oxidation, and sodium borohydride reduction. Sulfonylation then gave dimethylthexylsilyl 2-O-(benzylsulfonyl)-3,4,6-tri-O-benzyl-β-d-mannopyranosyl-(1→4)-2-acetamido-3-O-allyl-2-deoxy-6-O-(4-methoxybenzyl)-β-d-glucopyranoside.
Co-reporter:Amr Sonousi
Organic Letters () pp:
Publication Date(Web):August 4, 2015
DOI:10.1021/acs.orglett.5b01902
Selective protection of secondary amines as triazenes in the presence of multiple primary amines is demonstrated, with subsequent protection of the primary amines as either azides or carbamates in the same pot. Aminoglycoside antibiotic examples reveal broad functional group compatibility. The triazene group is removed with trifluoroacetic acid and, because of the low barrier to rotation, affords sharp 1H NMR spectra at room temperature.
Co-reporter:Amandine Noel, Bernard Delpech and David Crich
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 35) pp:NaN9324-9324
Publication Date(Web):2015/08/20
DOI:10.1039/C5OB90143J
Correction for ‘Comparison of the reactivity of β-thiolactones and β-lactones toward ring-opening by thiols and amines’ by Amandine Noel et al., Org. Biomol. Chem., 2012, 10, 6480–6483.
Co-reporter:Amandine Noel, Bernard Delpech and David Crich
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 32) pp:NaN6483-6483
Publication Date(Web):2012/06/14
DOI:10.1039/C2OB25640A
An investigation into the comparative reactivity of simple β-lactones and β-thiolactones toward a thiol and a primary amine is reported. A simple 3-mercaptomethyl-2-oxetanone is found to undergo rearrangement in the presence of aqueous base to give the corresponding thietane-3-carboxylic acid rather than the 3-hydroxymethyl-2-thietanone. Implications for the use of β-thiolactones in bioorganic and medicinal chemistry are discussed.
Co-reporter:Sylvain Aubry, Geneviève Aubert, Thierry Cresteil and David Crich
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 13) pp:NaN2632-2632
Publication Date(Web):2012/01/18
DOI:10.1039/C2OB06976H
The synthesis of β-thiolactone and β-lactam analogs of tetrahydrolipstatin is described from a common late-stage β-lactone derivative. These analogs, and a cis-disubstituted β-lactone analog of tetrahydrolipstatin, were screened for activity against porcine pancreatic lipase and for inhibition of cell growth of a panel of four human cancer lines.
Co-reporter:Sylvain Aubry, Kaname Sasaki, Laure Eloy, Geneviève Aubert, Pascal Retailleau, Thierry Cresteil and David Crich
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 20) pp:NaN7143-7143
Publication Date(Web):2011/07/18
DOI:10.1039/C1OB05967J
A series of novel peptide-based β-thiolactones were synthesized and assayed for cytotoxicity against several human cancer cell lines, where they showed greater activity than the corresponding β-lactones and β-lactams. Several of the β-thiolactones prepared showed strong inhibitory activity in vitro against human cathepsins B and L.







![1H-Pyrrole-2,5-dione, 3,4-dimethyl-1-[[(trifluoromethyl)sulfonyl]oxy]-](http://img.cochemist.com/ccimg/204100/204009-42-7.png)
![1H-Pyrrole-2,5-dione, 3,4-dimethyl-1-[[(trifluoromethyl)sulfonyl]oxy]-](http://img.cochemist.com/ccimg/204100/204009-42-7_b.png)
![Silane, [2-(iodomethyl)-2-propenyl]trimethyl-](http://img.cochemist.com/ccimg/80200/80121-73-9.png)
![Silane, [2-(iodomethyl)-2-propenyl]trimethyl-](http://img.cochemist.com/ccimg/80200/80121-73-9_b.png)

methanone](http://img.cochemist.com/ccimg/52800/52742-32-2.png)
methanone](http://img.cochemist.com/ccimg/52800/52742-32-2_b.png)


![Benzenemethanamine, N-[4-(1,1-dimethylethyl)cyclohexylidene]-](http://img.cochemist.com/ccimg/27800/27721-51-3.png)
![Benzenemethanamine, N-[4-(1,1-dimethylethyl)cyclohexylidene]-](http://img.cochemist.com/ccimg/27800/27721-51-3_b.png)


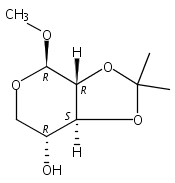
![(3aR,4R,6S,7S,7aR)-4-Methoxy-2,2,6-trimethyltetrahydro-3aH-[1,3]dioxolo[4,5-c]pyran-7-ol](http://img.cochemist.com/ccimg/14200/14133-63-2.png)
![(3aR,4R,6S,7S,7aR)-4-Methoxy-2,2,6-trimethyltetrahydro-3aH-[1,3]dioxolo[4,5-c]pyran-7-ol](http://img.cochemist.com/ccimg/14200/14133-63-2_b.png)



![D-Streptamine,O-2-amino-2-deoxy-a-D-glucopyranosyl-(1®4)-O-[3-amino-3-deoxy-a-D-glucopyranosyl-(1®6)]-2-deoxy-](http://img.cochemist.com/ccimg/2300/2280-32-2.png)
![D-Streptamine,O-2-amino-2-deoxy-a-D-glucopyranosyl-(1®4)-O-[3-amino-3-deoxy-a-D-glucopyranosyl-(1®6)]-2-deoxy-](http://img.cochemist.com/ccimg/2300/2280-32-2_b.png)












![a-D-Mannopyranoside, phenyl4,6-O-[(R)-phenylmethylene]-1-thio-](http://img.cochemist.com/ccimg/159500/159407-19-9.png)
![a-D-Mannopyranoside, phenyl4,6-O-[(R)-phenylmethylene]-1-thio-](http://img.cochemist.com/ccimg/159500/159407-19-9_b.png)





















![(3S,4S,5S,6S)-6-[4-(2-aminoethyl)-2-hydroxy-phenoxy]-3,4,5-trihydroxy-tetrahydropyran-2-carboxylic acid](http://img.cochemist.com/ccimg/36000/35954-65-5.png)
![(3S,4S,5S,6S)-6-[4-(2-aminoethyl)-2-hydroxy-phenoxy]-3,4,5-trihydroxy-tetrahydropyran-2-carboxylic acid](http://img.cochemist.com/ccimg/36000/35954-65-5_b.png)


![[(3R,4S,5S,6S)-3,4,5-triacetoxy-6-[(3R,4S,5S,6R)-3,5-diacetoxy-2-(acetoxymethyl)-6-bromo-tetrahydropyran-4-yl]oxy-tetrahydropyran-2-yl]methyl acetate](http://img.cochemist.com/ccimg/23300/23202-66-6.png)
![[(3R,4S,5S,6S)-3,4,5-triacetoxy-6-[(3R,4S,5S,6R)-3,5-diacetoxy-2-(acetoxymethyl)-6-bromo-tetrahydropyran-4-yl]oxy-tetrahydropyran-2-yl]methyl acetate](http://img.cochemist.com/ccimg/23300/23202-66-6_b.png)
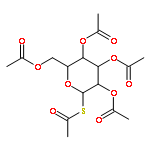
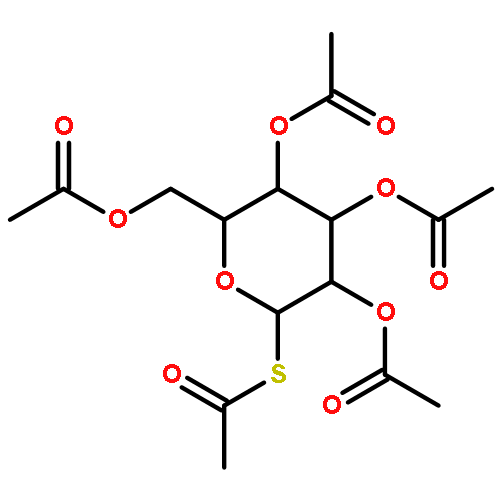
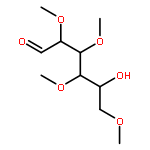
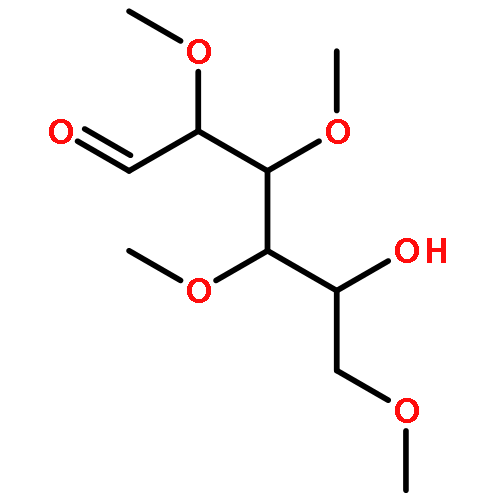





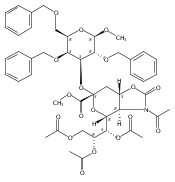


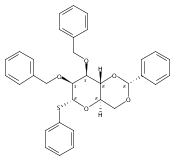
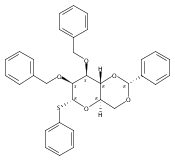
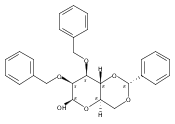
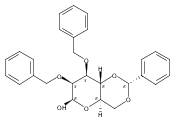
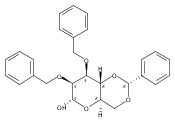
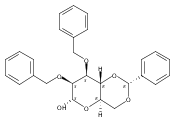
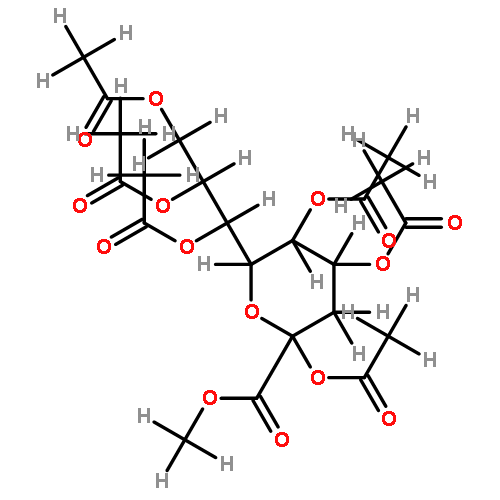
![β-D-Glucopyranoside, phenyl 2,3-bis-O-(phenylmethyl)-4,6-O-[(R)-phenylmethylene]-1-thio-](/data/chemimg/1372400/87470-70-0.png)
![β-D-Glucopyranoside, phenyl 2,3-bis-O-(phenylmethyl)-4,6-O-[(R)-phenylmethylene]-1-thio-](/data/chemimg/1372400/87470-70-0_b.png)