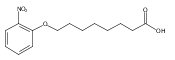
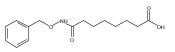
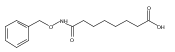
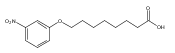
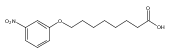
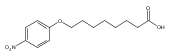
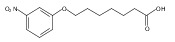
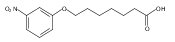
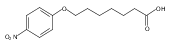
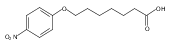
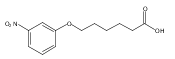
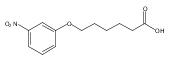
Co-reporter:Jian-Bei Xi, Yan-Fen Fang, Brendan Frett, Meng-Li Zhu, Tong Zhu, Yan-Nan Kong, Feng-Jie Guan, Yun Zhao, Xiong-Wen Zhang, Hong-yu Li, Ming-Liang Ma, Wenhao Hu
European Journal of Medicinal Chemistry 2017 Volume 126(Volume 126) pp:
Publication Date(Web):27 January 2017
DOI:10.1016/j.ejmech.2016.12.026
•A series of novel and potent Nek2 inhibitors based on a new imidazo[1,2-a]pyridine scaffold were designed and synthesized.•MBM-17 and MBM-55 effectively inhibited the proliferation of cancer cells by inducing cell cycle arrest and apoptosis.•MBM-17S and MBM-55S showed good selectivity and in vitro and in vivo antitumor activity without apparent toxicity.We present herein the discovery and development of novel and potent Nek2 inhibitors with distinctive in vitro and in vivo antitumor activity based on an imidazo[1,2-a]pyridine scaffold. Our studies identified a nonlinear SAR for activity against both Nek2 and cancer cells. Bioisostere and structure-based design techniques were employed to identify compounds 42c (MBM-17, IC50 = 3.0 nM) and 42g (MBM-55, IC50 = 1.0 nM), which displayed low nanomolar activity and excellent selectivity for Nek2. Both compounds effectively inhibited the proliferation of cancer cells by inducing cell cycle arrest and apoptosis. Importantly, the salts form of these two compounds (MBM-17S and MBM-55S) significantly suppressed tumor growth in vivo without apparent toxicity based on appearance and changes in body weight. In summary, MBM-17 and MBM-55 displayed the potential for substantial therapeutic application in cancer treatment.Download high-res image (227KB)Download full-size image
Co-reporter:Yanfen Fang, Wanli Zhang, Mengli Zhu, Shiguang Chen, Xuan Liu, Wei Lu, Xiongwen Zhang
Journal of Photochemistry and Photobiology B: Biology 2017 Volume 166() pp:264-271
Publication Date(Web):January 2017
DOI:10.1016/j.jphotobiol.2016.11.021
•GLUT-1 was overexpressed in hepatoma HepG2 cells and gastric cancer NCI-N87 cells.•The cellular uptake of N2 was significant higher in cancer cells than normal cells.•d-glucose or GLUT-1 inhibitor effectively inhibited the cellular uptake of N2.•The cellular uptake of N2 was in a concentration- and time-dependent manner.Cancer cells are usually characterized with an increase in glucose uptake when compared with normal cells, which is known as Warburg effect. Near-infrared (NIR) fluorescent glucose analogues have been previously synthesized and been applied in cancer cell imaging. However, most NIR dyes usually have one or more charge in their structures, which may cause low cell membrane permeability and hamper their application in cell imaging. Here we reported a novel glucose analogue N2, which was designed and synthesized based on a new type of NIR dye, DCPO. As expected, higher level of N2 uptake was observed in hepatic carcinoma cells (HepG2) and gastric cancer cells (NCI-N87) than their equivalent cells from normal tissues of the same origin, respectively. The accumulation of N2 in cancer cells was in consistent with the overexpression of glucose transporter GLUT-1 in these cells. The cellular uptake of N2 was then confirmed to be dependent on GLUT-1, which was evidenced by the decreased uptake of N2 in the presence of d-glucose or GLUT-1 inhibitor phloretin. Moreover, uptake of N2 in cancer cells was found to be in a concentration- and time-dependent manner. In all, our study demonstrated that N2, as a novel DCPO-conjugated bioprobe, could be used to monitor cellular glucose consumption, and therefore might be applied in cancer cell bioimaging and bioassay in cancer studies.
Co-reporter:Xuan Zhang, Kaiyong Tang, Hong Wang, Yaqian Liu, Bin Bao, Yanfen Fang, Xiongwen Zhang, and Wei Lu
Bioconjugate Chemistry 2016 Volume 27(Issue 5) pp:1267
Publication Date(Web):April 12, 2016
DOI:10.1021/acs.bioconjchem.6b00099
Traditional antitumor drugs such as camptothecin and paclitaxel derivatives are widely used in cancer chemotherapy. However, the major defects of those agents include severe toxicity and poor water solubility. With these in mind, a novel multifunctional linker was designed and two Cathepsin B (CTB) sensitive CPT conjugates (9a and 9b) were synthesized. Through click chemistry, additional functional group mPEG2000 can be easily introduced into these conjugates. The introduction of mPEG2000 fragment resulted in the formation of nanoparticles 1a and 1b (average particle sizes were 216.9 and 257.9 nm, respectively) with significantly increased water solubility (more than 19 000-fold). The release of therapeutic drug SN-38 in the presence of CTB was confirmed by HPLC and prodrug 1a showed potent in vitro cytotoxicity against all tested cell lines. Impressively, compared with irinotecan, CTB sensitive prodrug 1a displayed similar in vivo efficacy with remarkable decreased in vivo toxicity.
Co-reporter:Tong Zhu;Xuan Liu;Yu Lu;Xiaoping Chai;Xiao Liu;Yanfen Fang;Wang Liu;Xihan Wu;Xiongwen Zhang
Journal of Experimental & Clinical Cancer Research 2016 Volume 35( Issue 1) pp:
Publication Date(Web):2016/12/01
DOI:10.1186/s13046-016-0464-2
Overexpression of Aurora A and B has been reported in a wide range of tumor types, including gastric cancer. Anti-angiogenesis has been considered as an important therapeutic modality in advanced gastric cancer. Here we identified a novel compound TY-011 with promising antitumor activity by targeting mitotic kinases (Aurora A and B) and angiogenic receptor tyrosine kinase (VEGFR2).HTRF® KinEASE™ assay was used to detect the effect of TY-011 against Aurora A, Aurora B and VEGFR2 activities. Docking simulation study was performed to predict the binding mode of TY-011 with Aurora A and B kinases. CCK-8 assay was used to test cell growth. Cell cycle and cell apoptosis was analyzed by flow cytometry. Gastric cancer cell xenograft mouse models were used for in vivo study. TUNEL kit was used to determine the apoptosis of tumor tissues. Immunohistochemistry analysis and HUVEC tube formation assay were performed to determine the anti-angiogenesis ability. Immunofluorescence and western blot were used to test protein expression.TY-011 was identified as a potential Aurora A and B inhibitor by HTRF® KinEASE™ assay. It effectively inhibited cellular Aurora A and B activities in a concentration-dependent manner. TY-011 occupied the ATP-binding site of both Aurora A and B kinases. TY-011 demonstrated prominent inhibitory effects on proliferation of gastric cancer cells. TY-011 treatment induced an obvious accumulation of cells at G2/M phase and a modest increase of cells with >4 N DNA content, which then underwent apoptosis. Meaningfully, orally administration of TY-011 demonstrated superior efficacy against the tumor growth in gastric cancer cell xenograft, with ~90% inhibition rate and 100% tumor regression at 9 mg/kg dose, and TY-011 did not affect the body weight of mice. Interestingly, we observed that TY-011 also antagonized tumor angiogenesis by targeting VEGFR2 kinase.These results indicate that TY-011 is a well-tolerated, orally active compound that targets mitosis and angiogenesis in tumor growth, and provides strong preclinical support for use as a therapeutic for human gastric cancers.
.jpg)
![L-Ornithinamide, N-[(1,1-dimethylethoxy)carbonyl]-L-valyl-N5-(aminocarbonyl)-N-[4-(hydroxymethyl)-3-[(2-propyn-1-yloxy)methyl]phenyl]-](/data/chemimg/3723600/1771753-61-7.png)
![L-Ornithinamide, N-[(1,1-dimethylethoxy)carbonyl]-L-valyl-N5-(aminocarbonyl)-N-[4-(hydroxymethyl)-3-[(2-propyn-1-yloxy)methyl]phenyl]-](/data/chemimg/3723600/1771753-61-7_b.png)
![L-Ornithinamide, N-[(phenylmethoxy)carbonyl]-L-valyl-N5-(aminocarbonyl)-N-[4-(hydroxymethyl)-3-[(2-propyn-1-yloxy)methyl]phenyl]-](/data/chemimg/3723600/1771753-60-6.png)
![L-Ornithinamide, N-[(phenylmethoxy)carbonyl]-L-valyl-N5-(aminocarbonyl)-N-[4-(hydroxymethyl)-3-[(2-propyn-1-yloxy)methyl]phenyl]-](/data/chemimg/3723600/1771753-60-6_b.png)
![Benzenemethanol, 4-amino-2-[(2-propyn-1-yloxy)methyl]-](/data/chemimg/3723600/1771753-87-7.png)
![Benzenemethanol, 4-amino-2-[(2-propyn-1-yloxy)methyl]-](/data/chemimg/3723600/1771753-87-7_b.png)
![Benzoic acid, 4-amino-2-[(2-propyn-1-yloxy)methyl]-, methyl ester](/data/chemimg/3723600/1771753-86-6.png)
![Benzoic acid, 4-amino-2-[(2-propyn-1-yloxy)methyl]-, methyl ester](/data/chemimg/3723600/1771753-86-6_b.png)
![Benzoic acid, 4-nitro-2-[(2-propyn-1-yloxy)methyl]-, methyl ester](/data/chemimg/3723600/1771753-85-5.png)
![Benzoic acid, 4-nitro-2-[(2-propyn-1-yloxy)methyl]-, methyl ester](/data/chemimg/3723600/1771753-85-5_b.png)




![L-Ornithine, N-[(phenylmethoxy)carbonyl]-L-valyl-N5-(aminocarbonyl)-](http://img.cochemist.com/ccimg/159900/159858-06-7.png)
![L-Ornithine, N-[(phenylmethoxy)carbonyl]-L-valyl-N5-(aminocarbonyl)-](http://img.cochemist.com/ccimg/159900/159858-06-7_b.png)
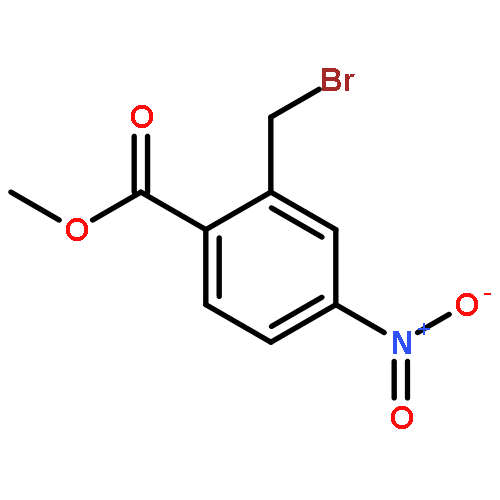
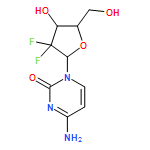
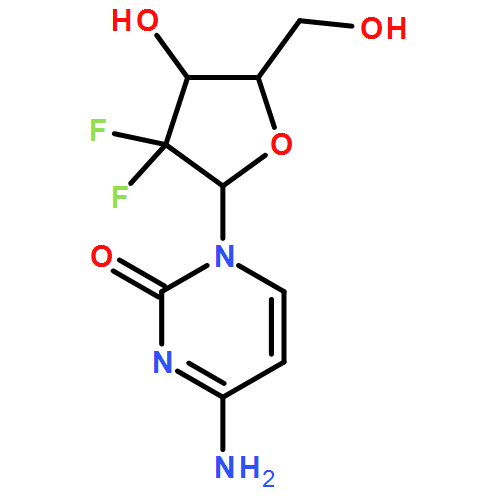




![BENZO[A]HEPTALEN-9(5H)-ONE,7-AMINO-6,7-DIHYDRO-1,2,3,10-TETRAMETHOXY-, (7S)-](http://img.cochemist.com/ccimg/3500/3476-50-4.png)
![BENZO[A]HEPTALEN-9(5H)-ONE,7-AMINO-6,7-DIHYDRO-1,2,3,10-TETRAMETHOXY-, (7S)-](http://img.cochemist.com/ccimg/3500/3476-50-4_b.png)