Co-reporter:Robert Otto; Robert Penzis; Friedemann Gaube; Oliver Adolph; Karl J. Föhr; Paul Warncke; Dina Robaa; Dorothea Appenroth; Christian Fleck; Christoph Enzensperger; Jochen Lehmann;Thomas Winckler
Journal of Medicinal Chemistry 2015 Volume 58(Issue 16) pp:6710-6715
Publication Date(Web):August 17, 2015
DOI:10.1021/acs.jmedchem.5b00958
Neurodegenerative diseases represent a challenge for biomedical research due to their high prevalence and lack of mechanism-based treatments. Because of the complex pathology of neurodegenerative disorders, multifunctional drugs have been increasingly recognized as potential treatments. We identified homobivalent γ-carbolinium salts as potent inihitors of both cholinesterases, N-methyl-d-aspartate receptors, and monoamine oxidases. Homobivalent γ-carbolines displayed similar structure–activity relationships on all tested targets and may present promising designed multiple ligands for the treatment of neurodegenerative disorders.
Co-reporter:Yao Chen, Jianfei Sun, Zhangjian Huang, Hong Liao, Sixun Peng, Jochen Lehmann, Yihua Zhang
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 9) pp:2462-2470
Publication Date(Web):1 May 2013
DOI:10.1016/j.bmc.2013.03.005
To search for multifunctional anti-Alzheimer’s disease (AD) agents with good safety, the previously synthesized tacrine–flurbiprofen hybrids 1a and 1b were modified into tacrine–flurbiprofen–nitrate trihybrids 3a–h. These compounds displayed comparable or higher cholinesterase inhibitory activity relative to the bivalent hybrids. Compound 3a was the most potent, which released moderate NO, exerted blood vessel relaxative activity, and showed significant Aβ inhibitory effects whereas tacrine and flurbiprofen did not exhibit any Aβ inhibitory activity at the same dose. In addition, 3a was active in improving memory impairment in vivo. More importantly, the hepatotoxicity study showed that 3a was much safer than tacrine, suggesting it might be a promising anti-AD agent for further investigation.Eight tacrine–flurbiprofen–nitrate trihybrids were synthesized and biologically evaluated and most of them exhibited good performance. Particularly, compound 3a showed higher in vitro activity (Ki for AChE = 16.1 nM; Ki for BuChE = 1.7 nM) and lower hepatotoxicity than tacrine.
Co-reporter:Yao Chen;Jianfei Sun;Sixun Peng;Hong Liao;Yihua Zhang
Archiv der Pharmazie 2013 Volume 346( Issue 12) pp:865-871
Publication Date(Web):
DOI:10.1002/ardp.201300074
Abstract
Five tacrine–flurbiprofen hybrid compounds (3a–e) were synthesized as multi-target-directed compounds for the treatment of Alzheimer's disease. Compared to tacrine, two compounds (3d and 3e) showed better acetylcholinesterase (AChE) inhibitory activity and others (3b–e) better or the same butyrylcholinesterase (BuChE) inhibitory activity. Notably, 3d showed a mixed-type inhibitory action for AChE, indicating a “dual-binding site action” of both toward the catalytic active site (CAS) and the peripheral anionic site (PAS), whereas for BuChE, a competitive inhibitory action was observed. Furthermore, a cell-based assay on amyloid-β inhibition demonstrated that the selected target compound 3d effectively inhibits the formation of amyloid-β in vitro.
Co-reporter:Yao Chen, Jianfei Sun, Zhangjian Huang, Hong Liao, Sixun Peng, Jochen Lehmann, Yihua Zhang
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 11) pp:3162-3165
Publication Date(Web):1 June 2013
DOI:10.1016/j.bmcl.2013.04.008
To search for potent anti-Alzheimer’s disease (AD) agents with multifunctional effects, 12 NO-donating tacrine–flurbiprofen hybrid compounds (2a–l) were synthesized and biologically evaluated. It was found that all the new target compounds showed selective butyrylcholinesterase (BuChE) inhibitory activity in vitro comparable or higher than tacrine and the tacrine–flurbiprofen hybrid compounds 1a–c, and released moderate amount of NO in vitro. The kinetic study suggests that one of the most active and highest BuChE selective compounds 2d may not only compete with the substrate for the same catalytic active site (CAS) but also interact with a second binding site. Furthermore, 2d and 2l exhibited significant vascular relaxation effect, which is beneficial for the treatment of AD. All the results suggest that 2d and 2l might be promising lead compounds for further research.
Co-reporter:Yao Chen ; Jianfei Sun ; Lei Fang ; Mei Liu ; Sixun Peng ; Hong Liao ; Jochen Lehmann ;Yihua Zhang
Journal of Medicinal Chemistry 2012 Volume 55(Issue 9) pp:4309-4321
Publication Date(Web):April 18, 2012
DOI:10.1021/jm300106z
In search of multifunctional cholinesterase inhibitors as potential anti-Alzheimer drug candidates, tacrine–ferulic acid–NO donor trihybrids were synthesized and tested for their cholinesterase inhibitory activities, release of nitric oxide, vasodilator properties, cognition improving potency, and hepatotoxicity. All of the novel target compounds show higher in vitro cholinesterase inhibitory activity than tacrine. Three selected compounds (3a, 3f, and 3k) produce moderate vasorelaxation in vitro, which correlates with the release of nitric oxide. Compared to its non-nitrate dihybrid analogue (3u), the trihybrid 3f exhibits better performance in improving the scopolamine-induced cognition impairment (mice) and, furthermore, less hepatotoxicity than tacrine.
Co-reporter:Yao Chen, Lei Fang, Sixun Peng, Hong Liao, Jochen Lehmann, Yihua Zhang
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 9) pp:3181-3187
Publication Date(Web):1 May 2012
DOI:10.1016/j.bmcl.2012.03.046
Acetylcholinesterase (AChE) is considered to be one of the most important targets for the treatment of Alzheimer’s disease (AD). Previously our group has reported a series of tacrine-based hybrids as potent AChE inhibitors (AChEI). To discover more novel scaffolds, molecular docking and dynamics stimulation were applied to acquire the binding models of AChE with the most prominent compounds from our work. A structure-based pharmacophore model plus shape constraints was generated from the binding models and it was then employed to virtually screen commercial databases, giving a focused hit list of candidates. Subsequently, we scored the hit compounds by their molecular binding energies, which were calculated by MM/PBSA method. Fifteen compounds were selected and purchased for testing their anti-AChE effects, while seven of them showed inhibitory effects with IC50 values ranging from 1.5 to 9.8 μM. The drug-like properties of these compounds, including Log D, A log P, molecular volume and Lipinski rule of five, were also calculated. Compounds 12 and 16 (IC50 = 2.5 and 1.5 μM, respectively) exhibited potent activity and acceptable drug-like properties, thus might serve as leads for further modification. The data suggest that the presented model might be a valid approach for identification and development of new AChEIs.Structure-based pharmacophore model generated from two potent acetylcholinesterase inhibitors previously reported by our laboratory.
Co-reporter:Dina Robaa ; Christoph Enzensperger ; Shams ElDin AbulAzm ; Mohamed M. Hefnawy ; Hussein I. El-Subbagh ; Tanveer A. Wani
Journal of Medicinal Chemistry 2011 Volume 54(Issue 20) pp:7422-7426
Publication Date(Web):September 5, 2011
DOI:10.1021/jm200676f
Racemic and enantiopure 8-substituted derivatives of the lead dopamine receptor antagonist LE 300 (1) were prepared, and their affinities for the dopamine receptors (D1–D5) were tested. The separate enantiomers showed significantly different affinities; the (8S)-methyl and (8R)-hyroxymethyl derivatives where the substituents point below the reference plane of the indolo[3,2-f][3]benzazecine scaffold were markedly more active than their enantiomeric counterparts. The racemic 8-carboxy derivative was shown to be selective for the D5-receptor, even against D1.
Co-reporter:Dina Robaa ; Christoph Enzensperger ; Shams El Din Abul Azm ; El Sayeda El Khawass ; Ola El Sayed
Journal of Medicinal Chemistry 2010 Volume 53(Issue 6) pp:2646-2650
Publication Date(Web):February 24, 2010
DOI:10.1021/jm901291r
On the basis of the D1/5-selective dopamine antagonist LE 300 (1), an indolo[3,2-f]benzazecine derivative, we changed the annulation pattern of the heterocycles. The target compounds represent novel heterocyclic ring systems. The most constrained indolo[4,3a,3-ef]benzazecine 2 was inactive, but the indolo[4,3a,3-fg]benzazacycloundecene 3 showed antagonistic properties (functional Ca2+ assay) with nanomolar affinities (radioligand binding) for all dopamine receptor subtypes, whereas the indolo[2,3-f]benzazecine 4 displayed a selectivity profile similar to 3 but with decreased affinities.
Co-reporter:Maria Schulze, Oliver Siol, Michael Decker, Jochen Lehmann
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 9) pp:2946-2949
Publication Date(Web):1 May 2010
DOI:10.1016/j.bmcl.2010.03.011
Three different types of homobivalent compounds, 5,8,9,13b-tetrahydro-6H-isoqino[1,2-a]isoquinolines bearing tertiary N-atoms, their quaternary ammonium salts and their dibenzazecine analogues, connected by alkylene spacers of various lengths were synthesized. Compared to the therapeutically used inhibitor galanthamine, some of the bivalent compounds showed much higher inhibitory activities at both cholinesterases in the Ellman test. Surprisingly, not only the quaternary salts, but also the uncharged tertiary compounds exhibited IC50 values at butyrylcholinesterase in the nanomolar range. Selectivity toward BChE of up to 76-fold was observed.
Co-reporter:Kathrin Lange, Andreas Koenig, Carolin Roegler, Andreas Seeling, Jochen Lehmann
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 11) pp:3141-3144
Publication Date(Web):1 June 2009
DOI:10.1016/j.bmcl.2008.04.057
The vasodilators glyceryl trinitrate (GTN) and pentaerythrityl tetranitrate (PETN) are supposed to be degraded in vivo to the lower nitrates PETriN, PEDN, PEMN, 1,2-GDN, 1,3-GDN, 1-GMN, and 2-GMN. We synthesized these bioactive metabolites as reference compounds for pharmacokinetic studies. The use of HPLC-methods for monitoring the stepwise reduction of PETN to lower nitrates and the syntheses of the glyceryl dinitrates proved advantageous. Furthermore, we measured the vasorelaxant properties of all metabolites by performing organ bath experiments with porcine pulmonary arteries. In general, the vasodilator potency increases with the number of nitrate moieties in the compound.The bioactive metabolites of GTN and PETN were synthesized and their vasorelaxant potencies measured by performing organ bath experiments with porcine pulmonary arteries.
Co-reporter:Lei Fang ; Dorothea Appenroth ; Michael Decker ●; Michael Kiehntopf ∞; Amelie Lupp ; Sixun Peng ; Christian Fleck ; Yihua Zhang
Journal of Medicinal Chemistry 2008 Volume 51(Issue 24) pp:7666-7669
Publication Date(Web):November 21, 2008
DOI:10.1021/jm801131a
A series of tacrine−NO donor hybrid compounds are synthesized and evaluated for cholinesterase inhibitory activity, cognition improving activity, and hepatotoxicity. The pharmacological results indicate that hybrid compounds 1, 2, and 3a potently inhibit cholinesterase in vitro and significantly improve the scopolamine-induced cognition impairment, whereas an analogue (3h) of 2 without the NO donor moiety does not. Compared to tacrine, 1 and 2 show much less hepatotoxicity. Molecular modeling studies suggest that 2 may interact with the catalytic and the peripheral anionic site of acetylcholinesterase.
Co-reporter:Lei Fang ; Dorothea Appenroth ; Michael Decker ; Michael Kiehntopf ; Carolin Roegler ; Thomas Deufel ; Christian Fleck ; Sixun Peng ; Yihua Zhang
Journal of Medicinal Chemistry 2008 Volume 51(Issue 4) pp:713-716
Publication Date(Web):January 31, 2008
DOI:10.1021/jm701491k
In search of safer anti-Alzheimer drugs, 14 NO-donor-tacrine hybrids (1−14) were synthesized and evaluated for their ability to inhibit cholinesterases and for vasorelaxation effects. Compounds 1−13 showed good cholinesterases inhibitory activities in vitro, while 14, particularly, was highly selective, preferring butyrylcholinesterase rather than acetylcholinesterase. Four selected compounds (1, 9, 11, and 14) moderately relaxed the porcine pulmonary arteries in organ bath. In the hepatotoxicity study, significant hepatotoxicity was caused by tacrine but not by 9.
Co-reporter:Joerg Konter, Ute Möllmann, Jochen Lehmann
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 17) pp:8294-8300
Publication Date(Web):1 September 2008
DOI:10.1016/j.bmc.2008.05.008
Novel hybrid compounds combining the antifungal drug ketoconazole with a diazen-1-ium-1,2-diolate or an organic nitrate moiety and the corresponding NO-donors without ketoconazole were synthesized and their activities against a broad variety of fungal strains were tested. Hybridization modifies the spectrum of antimicrobial activities and generally, the ketoconazole–NO-donor hybrids are more potent than ketoconazole. The NO-donors alone show insufficient effectiveness.Antimicrobial activities of ketoconazole–NO-donor hybrids (MIC-values) against eight different fungal strains range from 0.6 to 1000 μM.
Co-reporter:Christoph Enzensperger, Tilo Görnemann, Heinz H. Pertz, Jochen Lehmann
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 13) pp:3809-3813
Publication Date(Web):1 July 2008
DOI:10.1016/j.bmcl.2008.04.081
Dibenzo- and benzindolo-azecines represent a novel class of high-affinity dopamine receptor antagonists. To further characterize these drugs as potential neuroleptics, we selected a set of azecine derivatives and ring expanded homologues and we measured their antagonist activity at the 5-HT2A receptor in the porcine coronary artery. SARs found for the 5-HT2A receptor resemble those for the D1 but not the D2 receptor. The protein–ligand interactions were discussed with respect to the different binding pockets.A cross-target SAR was conducted with 13 azecine-styled compounds on D1, D2 and 5-HT2A receptors. Surprisingly, molecular modifications affect the affinity for the D1 receptor in the same manner as the 5-HT2A receptor. The protein–ligand interactions were discussed with respect to the different binding pockets.
Co-reporter:Ali El-Emam;Joerg Konter;Gamal El-Din A. A. Abuo-Rahma
European Journal of Organic Chemistry 2007 Volume 2007(Issue 4) pp:616-624
Publication Date(Web):27 NOV 2006
DOI:10.1002/ejoc.200600662
Nitrogen-bound diazen-1-ium-1,2-diolates (diazeniumdiolates, “NONOates”, “solid NO”) are generally prepared from secondary amines and nitric oxide and are compounds of first choice for the direct release of nitric oxide (NO). First, we report on the relationships between the structures of the amines and the formation rates of the corresponding NONOates, second on the structures of the NONOates and the rate of NO release and finally between the rates of NONOate formation and NO release from these species. A series of differently sized and substituted cyclic and aliphatic amines were used to quantify the reactivity of amines towards NO by monitoring the decrease in NO pressure with the NOtizer, an apparatus developed for this study. The release of NO was measured amperometrically with an NO-sensitive electrode and the half-lives of novel diazeniumdiolates were determined by UV spectroscopy. It was found that steric hindrance and heteroatomic substituents in the amines' side chains reduce the formation rates of the sodium salts of the NONOates and also slow down NO release from them. Exceptions were found with piperidine-2-carboxylic acid derivatives which react to give NONOates more slowly than piperidine but release NO much faster despite steric hindrance. A secondary amine carrying an additional primary amine reacted quickly with NO, but the corresponding NONOate showed slower releaser of NO owing to the formation of an intramolecular NONOate salt as well as the sodium salt derivative. Azetidine reacts faster with NO than all of the other amines, but decomposition of the corresponding NONOate proved to be unexpectedly slow.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2007)
Co-reporter:Andreas Koenig, Carolin Roegler, Kathrin Lange, Andreas Daiber, Erika Glusa, Jochen Lehmann
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 21) pp:5881-5885
Publication Date(Web):1 November 2007
DOI:10.1016/j.bmcl.2007.08.046
The vasoactive properties of 14 organic mononitrates were investigated in vitro using PGF2α-precontracted porcine pulmonary arteries. A surprisingly wide range of vasorelaxant potencies was observed (pD2: 3.36–7.50). Activities showed to be highly sensitive to the molecular structure and the substituents at the molecular carrier of the nitrate group. A correlation between lipophilicity and vasorelaxant potency could not be recognized. 2-Nitrooxyethylammoniumnitrate (1) was found to be slightly superior to the high potency trinitrate GTN.The vasoactive properties of 14 organic mononitrates were investigated in vitro using PGF2α-precontracted porcine pulmonary arteries. Activities showed to be highly sensitive to the structure and substituents. Compound 1 was found to be slightly superior to the high potency trinitrate GTN.
Co-reporter:Lei Fang, Yihua Zhang, Jochen Lehmann, You Wang, Hui Ji, Dayong Ding
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 4) pp:1062-1066
Publication Date(Web):15 February 2007
DOI:10.1016/j.bmcl.2006.11.018
A series of furoxan-based nitric oxide-releasing glucocorticoid derivatives was synthesized. The pharmacological assays indicated that three compounds, including I4, I5, and I6, had anti-inflammatory activity. Furthermore compared with the leading compound hydrocortisone the safety of I6 was greatly improved. Due to releasing NO in vivo the side effects of glucocorticoids, including hypertension and osteoporosis, were effectively avoided.A series of furoxan-based nitric oxide-releasing glucocorticoid derivatives was synthesized and evaluated for their anti-inflammatory activity and safety.
Co-reporter:Yvonne Schott, Michael Decker, Hans Rommelspacher, Jochen Lehmann
Bioorganic & Medicinal Chemistry Letters 2006 Volume 16(Issue 22) pp:5840-5843
Publication Date(Web):15 November 2006
DOI:10.1016/j.bmcl.2006.08.067
In the course of studies directed toward the discovery of novel acetyl- and butyrylcholinesterase (AChE and BChE) inhibitors for the treatment of Alzheimer‘s disease, we focused on β-carbolines (BCs). 6-Oxygenated β-carboline and β-carbolinium derivatives based on the serotonin template were synthesized and tested in vitro for their ability to inhibit AChE and BChE, respectively. Particularly the carbolinium salts, which can be formed by intracerebral methylation out of the tertiary-BC prodrugs, show inhibitory activity levels reaching those of galantamine, physostigmine, and rivastigmine.A series of β-carbolines and β-carbolinium salts were synthesized and their inhibitory activities towards AChE and BChE measured. Micromolar inhibitors with some selectivity toward AChE were identified.
Co-reporter:Gamal El Din A.A. Abuo-Rahma, Axel Horstmann, Mohamed F. Radwan, Ali El-Emam, Erika Glusa, Jochen Lehmann
European Journal of Medicinal Chemistry 2005 Volume 40(Issue 3) pp:281-287
Publication Date(Web):March 2005
DOI:10.1016/j.ejmech.2004.11.003
Diazeniumdiolates (NONOates), among them a ciprofloxacin-diazeniumdiolate hybrid compound, were synthesized and the pH-, temperature- and structure-dependent liberation of nitric oxide (NO) was monitored by laser magnetic resonance spectroscopy (LMRS). The compounds induced a transient and reversible relaxation (EC50 8.3–150 nM) of pulmonary arteries independently from intact endothelium by stimulation of guanylyl cyclase (sGC). Increase in vascular cGMP was observed and blocking sGC with ODQ, an inhibitor of the NO-sensitive guanylyl cyclase, induced a rightward shift of the concentration–response curves. Repeated exposure did not show homologous desensitization. ADP-induced platelet aggregation (IC50 = 0.15–3 μM, IC50 for SNP: 2 μM) and collagen-induced aggregation were potently inhibited. Preincubation with ODQ also diminished these inhibitory effects.
Co-reporter:Michael Decker, Klaus-Jürgen Schleifer, Martin Nieger, Jochen Lehmann
European Journal of Medicinal Chemistry 2004 Volume 39(Issue 6) pp:481-489
Publication Date(Web):June 2004
DOI:10.1016/j.ejmech.2004.02.001
-LE300, a benz[d]indolo[2,3-g]azecine with nanomolar affinities to the hD1 receptor family, has been further pharmacologically characterized and its modelled structure was compared to its X-ray structure in order to explain NMR data, that was not in accordance to the X-ray structure. Moderate affinity at the hD3 receptor was determined, nanomolar affinities were found at the 5-HT2A and 5-HT2C receptors, micromolar affinity at the 5-HT1A receptor using binding assays. Functional studies indicate moderate antagonist activity at the 5-HT2A site . No activity was found at dopamine, serotonin and norepinephrine transporters. These results suggested the use of LE300 for cocaine addiction treatment. High activities were found using in vivo testing: LE300 suppressed spontanous locomotor activity with an ID50 of 1.24 mg/kg and attenuated locomotor activity induced by 20 mg/kg cocaine with an AD50 of 1.50 mg/kg. It failed to substitute for the discriminative stimulus effects produced by cocaine.
Co-reporter:Christoph Weßler, Alexander Homann, Uwe Fricke, Jochen Lehmann
European Journal of Medicinal Chemistry 2003 Volume 38(Issue 6) pp:581-586
Publication Date(Web):June 2003
DOI:10.1016/S0223-5234(03)00079-5
A series of substituted benzylnitrates (1) and the formally but not chemically similar cyclohexylmethylnitrate (CHMN) have been synthesised. Vasodilating activities were measured on endothelium-intact and NG-nitro-l-arginine (l-NNA)-blocked porcine right coronary arteries, precontracted with prostaglandin F2α (PGF2α). Glyceroltrinitrate (GTN) was used as reference. In intact coronary arteries the vasodilating activities of all benzylnitrates are lower compared with GTN, but higher compared with CHMN. However, blocking the function of the endothelium by l-NNA, the activity of all benzylnitrates increased, whereas that of CHMN and GTN remained nearly unaffected. Under these conditions, the mononitrates 4-nitro-benzylnitrate (1c) and 4-nitrooxymethyl-benzonitrile (1h) even showed higher vasodilator activities than the trinitrate GTN and in general, vasorelaxation by the benzylnitrates as defined by the concentrations for half maximal effects (EC50 values) was found to be 2–3 orders of magnitude higher than that induced by CHMN. The study demonstrates that the in vitro activities of organic nitrates do not correlate with the number of nitrate groups within the molecule nor to the lipophilicity of the molecules. Instead, vasodilator activity is highly sensitive to the structure and the type of the substituents in the molecular carrier of the nitrate group.
Co-reporter:Michael Decker
Archiv der Pharmazie 2003 Volume 336(Issue 10) pp:
Publication Date(Web):23 OCT 2003
DOI:10.1002/ardp.200300777
A number of 5-phenyl-1, 2, 3, 4, 5, 6-hexahydro-azepino-[4, 5-b]indoles 3 were synthesized with different substituents at the azepine-N position (methyl-, allyl-, 2-phenyl-ethyl-, cyclopropylmethyl- and unsubstituted). Furthermore, the indole-N-methylated compound was generated and by using norephedrines and norpseudoephedrines as a chiral pool, 4-methyl-5-phenyl-1, 2, 3, 4, 5, 6-hexahydro-azepino-[4, 5-b]indoles were prepared which contained racemisation at the reacting C-atom. These compounds, as well as the ring-open amino-alcohols, were screened for their affinity to the hD1-, hD5-, hD2L-, and hD4-receptors (ç please check sentence). They had micromolar affinities for the receptors and showed the highest affinity to the D1-subtype family. The cyclic compounds possessed the highest affinity, with the cyclopropylmethyl-(3c) and methyl-substituents (3e) being the most active of the tested compounds. Based on an intracellular cAMP-assay, the unsubstituted compound (at the azepine-N position) turned out to be an agonist for the D1-and D5-subtype family, whereas the substituted compounds showed (partial) agonistic, or even inverse agonistic activity.
Co-reporter:Ashraf H. Abadi;Stefan Lankow;Barbara Hoefgen;Michael Decker;Matthias U. Kassack
Archiv der Pharmazie 2002 Volume 335(Issue 8) pp:
Publication Date(Web):21 OCT 2002
DOI:10.1002/1521-4184(200211)335:8<367::AID-ARDP367>3.0.CO;2-C
A series of 7, 7′—alkylene-bridged dimers(7a—e) of the benz [d]indolo[3, 2-f]azecine derivative LE300 was synthesized. Affinity and functional activity at dopamine D1 and D2 receptors were estimated by radioligand binding and a functional Ca2+ assay. All the new bivalent ligands showed significant binding affinities to both D1 and D2 receptorswith an optimal distance between the two monomeric recognition unitsof 6 methylene moieties. The D1/D2-selectivity pattern was dependent on the spacer length. No (7a, b) or only moderate (7c—e) functional activity wasdetected for all bivalent compounds by measuring the inhibition of agonist-induced increase in intracellular Ca2+.
Co-reporter:Hussein El-Subbagh;Thomas Wittig;Michael Decker;Sigurd Elz;Martin Nieger
Archiv der Pharmazie 2002 Volume 335(Issue 9) pp:
Publication Date(Web):22 NOV 2002
DOI:10.1002/1521-4184(200212)335:9<443::AID-ARDP443>3.0.CO;2-U
LE 300 represents a structurally novel type of antagonist acting preferentially at the dopamine D1/D5 receptors and the serotonin 5-HT2A receptor. The compound consists of a 10-membered central azecine ring fused to indole on one and to benzene on the other side.To estimate the importance of the indole moiety in this highly active benz-indolo-azepine, the indole has to be removed and the “de-indolised” analog reinvestigated pharmacologically. Accordingly, we synthesized 3-benzazecines and in addition some homologuous 3-benzazonines. Methoxylated β-phenylethylamines were treated with ethyl ω-bromo-butanoates and -pentanoates, respectively, to give the corresponding lactams which were cyclized (POCl3) and reduced (NaBH4), yielding the cis-annelated (X-ray) benzindolizines and -quinolizines. The 10- and 9-membered rings were obtained by cleavage of the central C—N bond, which was performed in the following two ways: Quarternisaion with methyl iodide and cleavage with sodium in liquid ammonia gave the NCH3 derivatives, reaction with benzyloxycarbonyl chloride/NaBH4 followed by catalytic debenzylation yielded a corresponding NH compound. Functional experiments on rat artery segments precontracted with ketanserin and radioligand binding experiments using human cloned dopamine receptor subtypes were conducted with all of the benzazecine and benzazonine derivatives. In contrast to the benz-indolo-compound LE 300 they did not show any significant affinity towards the D1, D2, D4, andD5 receptors and only moderate antagonistic activity at the 5-HT2A receptor. It can be concluded from our study that an indole moiety or at least another second aromatic system at the central azecine ring is part of the pharmacophore and thus essential for high biological activity.

![Thieno[2,3-f][3]benzazecine, 4,5,6,7,8,13-hexahydro-6-methyl-](http://img.cochemist.com/ccimg/874700/874656-31-2.png)
![Thieno[2,3-f][3]benzazecine, 4,5,6,7,8,13-hexahydro-6-methyl-](http://img.cochemist.com/ccimg/874700/874656-31-2_b.png)
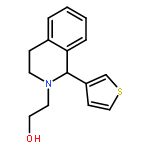
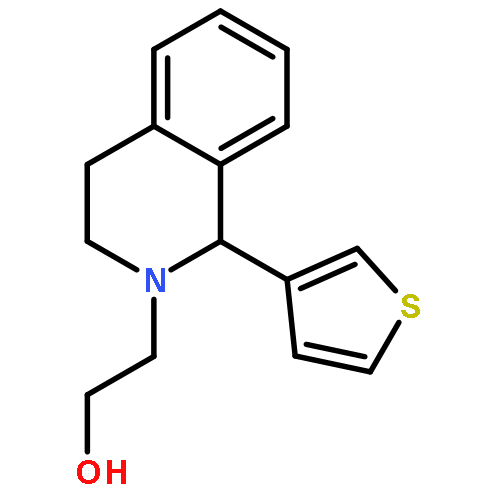
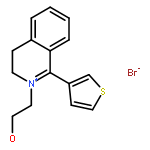
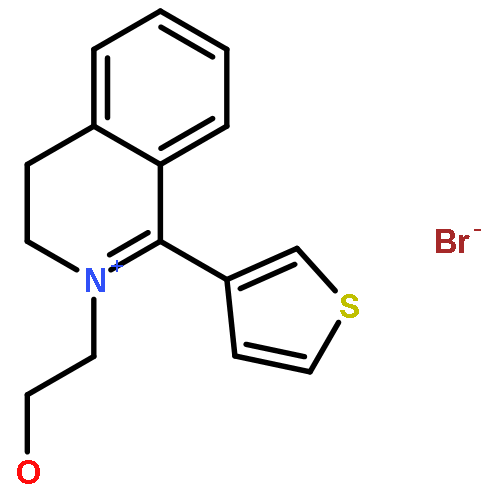
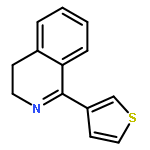
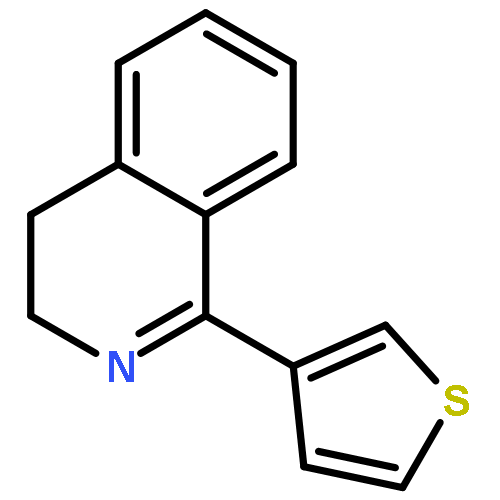
![6H-Benzo[a][1]benzothieno[3,2-h]quinolizine, 5,8,9,14b-tetrahydro-](http://img.cochemist.com/ccimg/107100/107042-09-1.png)
![6H-Benzo[a][1]benzothieno[3,2-h]quinolizine, 5,8,9,14b-tetrahydro-](http://img.cochemist.com/ccimg/107100/107042-09-1_b.png)
![5H-Benzo[a]thieno[3,2-h]quinolizine, 4,7,8,12b-tetrahydro-](http://img.cochemist.com/ccimg/107100/107042-01-3.png)
![5H-Benzo[a]thieno[3,2-h]quinolizine, 4,7,8,12b-tetrahydro-](http://img.cochemist.com/ccimg/107100/107042-01-3_b.png)
![Isoquinolinium, 3,4-dihydro-2-[2-(3-thienyl)ethyl]-, bromide](http://img.cochemist.com/ccimg/107100/107041-99-6.png)
![Isoquinolinium, 3,4-dihydro-2-[2-(3-thienyl)ethyl]-, bromide](http://img.cochemist.com/ccimg/107100/107041-99-6_b.png)
![5H-Pyrido[4,3-b]indole, 5-methyl-](http://img.cochemist.com/ccimg/61500/61406-19-7.png)
![5H-Pyrido[4,3-b]indole, 5-methyl-](http://img.cochemist.com/ccimg/61500/61406-19-7_b.png)
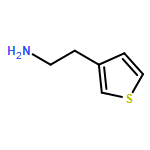
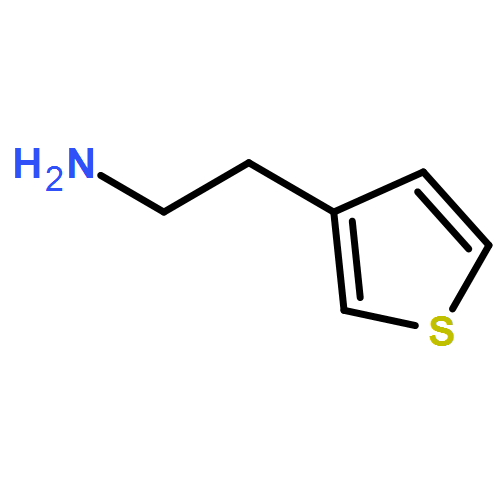
![2-(Benzo[b]thiophen-3-yl)ethanamine](http://img.cochemist.com/ccimg/14600/14585-66-1.png)
![2-(Benzo[b]thiophen-3-yl)ethanamine](http://img.cochemist.com/ccimg/14600/14585-66-1_b.png)



![9-methyl-9H-Pyrido[3,4-b]indole](http://img.cochemist.com/ccimg/2600/2521-07-5.png)
![9-methyl-9H-Pyrido[3,4-b]indole](http://img.cochemist.com/ccimg/2600/2521-07-5_b.png)
![5H-Pyrido[4,3-b]indole](http://img.cochemist.com/ccimg/300/244-69-9.png)
![5H-Pyrido[4,3-b]indole](http://img.cochemist.com/ccimg/300/244-69-9_b.png)