Co-reporter:Yong Wang, Zhong-Liang Xu, Jing Ai, Xia Peng, Jian-Ping Lin, Yin-Chun Ji, Mei-Yu Geng and Ya-Qiu Long
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 9) pp:1545-1562
Publication Date(Web):31 Oct 2012
DOI:10.1039/C2OB26710A
The 1,6-naphthyridine motif is a multivalent scaffold in medicinal chemistry presenting various bioactivities when properly substituted. By incorporating a cyclic urea pharmacophore into the 1,6-naphthyridine framework through conformationally constraining the 7,8-positions, the resulting 1H-imidazo[4,5-h][1,6]naphthyridin-2(3H)-one was identified as a new class of c-Met kinase inhibitor. A comprehensive SAR study indicated that an N-1 alkyl substituent bearing a terminal free amino group, a hydrophobic substituted benzyl group at the N-3 position and the tricyclic core were essential for retaining effective Met inhibition of the 1H-imidazo[4,5-h][1,6]naphthyridin-2(3H)-one chemotype. Further introduction of a 4′-carboxamide phenoxy group at the C-5 position significantly improved the potency. The best c-Met kinase inhibitory activity was exemplified by 2t with an IC50 = 2.6 μM, which also displayed effective inhibition against TPR-Met phosphorylation and the proliferation of the BaF3-TPR-Met cells at low micromolar concentrations.
Co-reporter:Zhongjie Liang, Jing Ai, Xiao Ding, Xia Peng, Dengyou Zhang, Ruihan Zhang, Ying Wang, Fang Liu, Mingyue Zheng, Hualiang Jiang, Hong Liu, Meiyu Geng, and Cheng Luo
ACS Medicinal Chemistry Letters 2013 Volume 4(Issue 4) pp:408
Publication Date(Web):February 25, 2013
DOI:10.1021/ml4000047
The aberrant function of c-Met kinase signaling pathway is ubiquitously involved in a broad spectrum of human cancers; thus, a strong rationale exists for targeting the kinase pathway in cancer therapy. Via integration of computational and experimental studies, anthraquinone derivatives were identified for the first time as potent c-Met kinase inhibitors in this research. The aberrant activation of the c-Met kinase pathway results from (TPR)-Met, MET gene mutation, or amplification and a hepatocyte growth factor (HGF)/scatter factor-dependent autocrine or paracrine mechanism. However, anthraquinone derivatives exclusively suppressed c-Met phosphorylation stimulated by HGF in A549 cells, indicating that the compounds possess the ability to block the extracellular HGF-dependent pathway. A surface plasmon resonance assay revealed that the most potent compound, 2a, shows a high binding affinity for HGF with an equilibrium dissociation constant of 1.95 μM. The dual roles of compound 2a demonstrate the potency of anthraquinone derivatives and provide a new design solution for the c-Met kinase signaling pathway.Keywords: Anthraquinone derivatives; binding affinity with HGF; c-Met kinase inhibitors
Co-reporter:Dengyou Zhang, Xiaowei Zhang, Jing Ai, Yun Zhai, Zhongjie Liang, Ying Wang, Yi Chen, Chunpu Li, Fei Zhao, Hualiang Jiang, Meiyu Geng, Cheng Luo, Hong Liu
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 21) pp:6804-6820
Publication Date(Web):1 November 2013
DOI:10.1016/j.bmc.2013.07.032
A series of 2-amino-N-benzylpyridine-3-carboxnamides, 2-amino-N-benzylpyridine-3-sulfonamides and 2-amino-3-benzylthiopyridines against c-Met were designed by means of bioisosteric replacement and docking analysis. Optimization of the 2-amino-3-benzylthiopyridine scaffold led to the identification of compound (R)-10b displaying c-Met inhibition with an IC50 up to 7.7 nM. In the cytotoxic evaluation, compound (R)-10b effectively inhibited the proliferation of c-Met addictive human cancer cell lines (IC50 from 0.19 to 0.71 μM) and c-Met activation-mediated cell metastasis. At a dose of 100 mg/Kg, (R)-10b evidently inhibited tumor growth (45%) in NIH-3T3/TPR-Met xenograft model. Of note, (R)-10b could overcome c-Met-activation mediated gefitinib-resistance, which indicated its potential use for drug combination. Taken together, 2-amino-3-benzylthiopyridine scaffold was first disclosed and exhibited promising pharmacological profiles against c-Met, which left room for further exploration.A series of 2-amino-N-benzylpyridine-3-carboxnamides, 2-amino-N-benzylpyridine-3-sulfonamides and 2-amino-3-benzylthiopyridines against c-Met were designed by means of bioisosteric replacement and docking analysis. Optimization of the 2-amino-3-benzylthiopyridine scaffold identified compound (R)-10b displaying c-Met inhibition with an IC50 up to 7.7 nM. In the cytotoxic evaluation, compound (R)-10b effectively inhibited the proliferation of c-Met addictive human cancer cell lines (IC50 from 0.19 to 0.71 μM) and c-Met activation-mediated cell metastasis. Of note, (R)-10b could overcome c-Met-activation mediated gefitinib-resistance. (R)-10b could exhibit moderate inhibition of tumor growth (45%) in NIH-3T3/TPR-Met xenograft model probably due to its high clearance and low bioavailability.
Co-reporter:Dengyou Zhang, Jing Ai, Zhongjie Liang, Wei Zhu, Xia Peng, Xianjie Chen, YinChun Ji, Hualiang Jiang, Cheng Luo, Meiyu Geng, Hong Liu
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 8) pp:2408-2413
Publication Date(Web):15 April 2013
DOI:10.1016/j.bmcl.2013.02.037
A series of novel 5-(benzyloxy)pyridin-2(1H)-ones were designed, synthesized and biologically evaluated for c-Met inhibition. Various amides and benzoimidazoles at C-3 position were investigated. A potent compound 12b with a c-Met IC50 of 12 nM was identified. This compound exhibited potent inhibition of EBC-1 cell associated with c-Met constitutive activation and showed high selectivity for c-Met than other tested 11 kinases. The binding model 12b with c-Met was disclosed by docking analysis.A series of novel 5-(benzyloxy)pyridin-2(1H)-ones were designed, synthesized and biologically evaluated for c-Met inhibition. The carbonyl oxygen of the pyridin-2(1H)-one was demonstrated to be an important factor for c-Met binding affinity. Various amides and benzoimidazoles at C-3 position of the pyridin-2(1H)-one core were investigated. A potent compound 12b with a c-Met IC50 of 12 nM was identified. This compound exhibited potent inhibition of EBC-1 cell associated with c-Met constitutive activation and showed high selectivity for c-Met than other tested 11 kinases.
Co-reporter:Zhongjie Liang, Xiao Ding, Jing Ai, Xiangqian Kong, Limin Chen, Liang Chen, Cheng Luo, Meiyu Geng, Hong Liu, Kaixian Chen and Hualiang Jiang
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 2) pp:421-430
Publication Date(Web):30 Sep 2011
DOI:10.1039/C1OB06186K
The receptortyrosine kinase c-Met is an attractive target for therapeutic treatment of cancers nowadays. The discovery of small molecule inhibitors is of special interest in the blockade of the c-Met kinase pathway. Here, we initiated our study from compound 1a, a novel inhibitor against c-Met kinase. A substructure similarity search against the SPECS database and chemical synthesis methods were performed to obtain a series of pyrazolidine-3,5-dione derivatives. Through the enzyme-based assay against c-Met kinase, 4 compounds (1c, 1e, 1m and 1o) showed potential inhibitory activity, with IC50 values mostly less than 10 μM. Based on the structure–activity relationship (SAR) and binding mode analysis, a focused combinatorial library was designed by the LD1.0 program. Taking into account ADMET properties and synthesis accessibility, seven candidate compounds (5a–g) were successfully synthesized. The activity of the most potent compounds 5b (IC50 = 0.46 μM) was 20 fold higher than that of the lead 1a. Taken together, our findings identified the pyrazolidine-3,5-dione derivatives as potent inhibitors against c-Met kinase and demonstrated the efficiency of the strategy in the development of small molecules against c-Met kinase.
Co-reporter:Dengyou Zhang, Jing Ai, Zhongjie Liang, Chunpu Li, Xia Peng, YinChun Ji, Hualiang Jiang, Meiyu Geng, Cheng Luo, Hong Liu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 17) pp:5169-5180
Publication Date(Web):1 September 2012
DOI:10.1016/j.bmc.2012.07.007
A series of 2-aminopyridine-3-carboxamide derivatives against c-Met were designed and synthesized by employing bioisosteric replacement of heterocyclic moieties with the amide bond. The structure–activity relationship (SAR) at various positions of the scaffold was explored. In this study, a promising compound (S)-24o with a c-Met IC50 of 0.022 μM was identified. The compound exhibited dose-dependent inhibition of the phosphorylation of c-Met as well as downstream signaling in EBC-1 cells. Furthermore, the interactive binding model of (S)-24o with c-Met was elucidated by virtue of a molecular modeling study.A series of 2-aminopyridine-3-carboxamide derivatives against c-Met were designed and synthesized by employing bioisosteric replacement of heterocyclic moieties with the amide bond. The SAR exploration led to the identification of a potent compound (S)-24o with a c-Met IC50 of 0.022 μM. This compound exhibited dose-dependent inhibition of the phosphorylation of c-Met as well as c-Met downstream signaling in EBC-1 cells. The interactive binding model of (S)-24o was elucidated using AutoDock4.2.
Co-reporter:Kui Wu, Jing Ai, Qiufeng Liu, TianTian Chen, Ailing Zhao, Xia Peng, Yuanxiang Wang, Yinchun Ji, Qizheng Yao, Yechun Xu, Meiyu Geng, Ao Zhang
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 20) pp:6368-6372
Publication Date(Web):15 October 2012
DOI:10.1016/j.bmcl.2012.08.075
Two series of new analogues were designed by replacing the quinoline scaffold of our earlier lead 2 (zgwatinib) with quinoxaline and pyrido[2,3-d]pyrimidine frameworks. Moderate c-Met inhibitory activity was observed in the quinoxaline series. Among the pyrido[2,3-d]pyrimidine series, compounds 13a–c possessing an O-linkage were inactive, whilst the N-linked analogues 15a–c retained c-Met inhibitory potency. Highest activity was observed in the 3-nitrobenzyl analog 15b that showed an IC50 value of 6.5 nM. Further structural modifications based on this compound were undergoing.
Co-reporter:Keqiang Fan, Xia Zhang, Hongchun Liu, Hui Han, Yuanming Luo, Qian Wang, Meiyu Geng and Keqian Yang
The Journal of Antibiotics 2012 65(9) pp:449-452
Publication Date(Web):June 20, 2012
DOI:10.1038/ja.2012.48
The jadomycins are a unique family of angucycline-derived antibiotics with interesting cytotoxic activities. In this work, six new jadomycin derivatives were produced in vivo by providing non-natural amino acids in fermentation media. They were further purified and identified by MS and NMR analyses. The cytotoxicities of these derivatives were evaluated against tumor cell lines MCF-7 and HCT116, as well as the normal human microvascular epithelial cells. The derivatives with alkyl side chains showed similar levels of cytotoxicity as jadomycin B and other known derivatives with nonpolar side chains, with IC50 ranging from 1.3 to 10 μM; but the activities are not selective as these compounds also showed similar levels of cytotoxicity toward the normal human microvascular epithelial cells in the same concentration range. For the first time, derivatives with amino side chains (jadomycin Orn and K) were prepared and evaluated. Significantly, jadomycin Orn showed differential activity against normal and tumor cell lines. This result points to a new direction to modify jadomycin structure. The insights on the structure–activity relationship of jadomycins will guide further efforts to generate new and improved jadomycin derivatives against tumor cells.
Co-reporter:Fang Chen;Ying Wang;Dr. Jing Ai;Zhengsheng Zhan;Yongcong Lv;Zhongjie Liang;Dr. Cheng Luo;Dr. Desheng Mei; Meiyu Geng; Wenhu Duan
ChemMedChem 2012 Volume 7( Issue 7) pp:1276-1285
Publication Date(Web):
DOI:10.1002/cmdc.201200145
Abstract
The HGF/c-Met signaling pathway mediates a variety of important biological activities, but dysregulation of the pathway is also closely associated with poor prognosis in a wide range of human cancers. c-Met is considered to be among the most promising therapeutic targets for anticancer drug discovery. Herein we report the discovery of a series of O-linked triazolotriazines that show sub-nanomolar inhibition of c-Met activity. Among these new compounds, 6 a exhibits high c-Met inhibitory potency in both enzymatic and cellular assays with great selectivity.
Co-reporter:Dr. Danqi Chen;Ying Wang;Yuchi Ma;Dr. Bing Xiong;Dr. Jing Ai; Yi Chen;Dr. Meiyu Geng;Dr. Jingkang Shen
ChemMedChem 2012 Volume 7( Issue 6) pp:
Publication Date(Web):
DOI:10.1002/cmdc.201290028
Co-reporter:Dr. Danqi Chen;Ying Wang;Yuchi Ma;Dr. Bing Xiong;Dr. Jing Ai; Yi Chen;Dr. Meiyu Geng;Dr. Jingkang Shen
ChemMedChem 2012 Volume 7( Issue 6) pp:1057-1070
Publication Date(Web):
DOI:10.1002/cmdc.201200120
Abstract
To identify novel c-Met inhibitors, sequences and crystal structures of the human kinome were analyzed to find interesting hinge binders that have been underexplored within the tyrosine kinase subfamily. Through this study, the imidazolopyridine ring was selected as a novel c-Met hinge-binding inhibitor scaffold. A series of derivatives was prepared, and the structure–activity relationships were studied. Among these, one compound in particular showed excellent activities in enzymatic and cellular assays, good in vitro metabolic stability, and favorable pharmacokinetic parameters. When administered orally, the compound inhibited tumor growth in an NIH-3T3/TPR-Met xenograft model and did not show adverse effects on body weight. The present work not only conceptually demonstrates a new route for designing novel kinase inhibitors by using known structural information of ligand–hinge interactions but also provides a series of imidazolopyridine derivatives as potent c-Met inhibitors.
Co-reporter:Yuanxiang Wang ; Jing Ai ; Ying Wang ; Yi Chen ; Lu Wang ; Gang Liu ; Meiyu Geng ;Ao Zhang
Journal of Medicinal Chemistry 2011 Volume 54(Issue 7) pp:2127-2142
Publication Date(Web):March 15, 2011
DOI:10.1021/jm101340q
By use of an improved synthetic strategy, a series of 3,5-disubstituted and 3,5,7-trisubstituted quinolines were readily prepared. 3,5,7-Trisubstituted quinolines 21a−c, 21l, and 27a−c were identified as the most potent c-Met inhibitors with IC50 of less than 1.0 nM. Compound 21b showed the most promising overall PK profile and has high potency and extraordinary selectivity to c-Met against c-Met family member Ron and 12 other tyrosine kinases. It produced constitutive inhibition of c-Met phosphorylation in c-Met dependent cell lines. At doses of 100 mg/kg, compound 21b showed statistically significant tumor growth inhibition (68−69%) in both NIH-3T3-TPR-Met and U-87 MG human gliobastoma xenograft models. These results clearly indicated that compound 21b is a potent and highly selective c-Met inhibitor. Its favorable in vitro and in vivo profiles warrant further investigation.
Co-reporter:Lin Du ; Hong-Chun Liu ; Wei Fu ; De-Hai Li ; Qiu-Ming Pan ; Tian-Jiao Zhu ; Mei-Yu Geng ;Qian-Qun Gu
Journal of Medicinal Chemistry 2011 Volume 54(Issue 16) pp:5796-5810
Publication Date(Web):July 15, 2011
DOI:10.1021/jm200511x
Fifteen citrinin derivatives (1–4, 6–16), including two unprecedented citrinin trimers tricitrinols A (3) and B (4), were isolated from Penicillium citrinum HGY1–5. The six-membered ring A system is essential for the cytotoxicity of active dimers (1, 2, and 5) and trimers (3 and 4). Tricitrinol B (4) showed extensive cytotoxicity in 17 tumor cells with comparable low-micromolar IC50 values (1–10 μM) and potential antimultidrug resistance capabilities. Tricitrinol B (4) induced cell apoptosis in HL60 and HCT116 cells via mainly extrinsic pathways and G2/M arrest. Further antitumor mechanism study and computational docking analysis indicated that tricitrinol B (4) works as an intercalating topoisomerase IIα (topo IIα) poison, which inhibits the enzyme activity of topo IIα by interfering predominantly with the topo IIα-mediated poststrand-passage cleavage/religation equilibrium over with the prestrand-passage one and induced DNA damage. Tricitrinol B (4) represents a novel class of topo IIα-inhibitory skeletons for developing new chemotherapeutic agents.
Co-reporter:Yuanxiang Wang, Jing Ai, Gang Liu, Meiyu Geng and Ao Zhang
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 17) pp:5930-5933
Publication Date(Web):01 Jul 2011
DOI:10.1039/C1OB05830D
An effective one-pot synthesis of quinolines bearing diverse C3-piperazinyl functions was developed by using a modified Friedländer's protocol. The method not only enables the synthesis of our early reported c-Met inhibitor on a large scale, but also provides a way to generate novel multi-substituted quinolines for further structure–activity relationship (SAR) study.
Co-reporter:Ailing Zhao, Xin Gao, Yuanxiang Wang, Jing Ai, Ying Wang, Yi Chen, Meiyu Geng, Ao Zhang
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 13) pp:3906-3918
Publication Date(Web):1 July 2011
DOI:10.1016/j.bmc.2011.05.038
A series of thieno[2,3-d]pyrimidines and furo[2,3-d]pyrimidines were synthesized and evaluated for the c-Met inhibition. Thieno[2,3-d]pyrimidine 6b stood out as the most potent showing an IC50 of 35.7 nM. This compound displayed high inhibitory effect on cell proliferation in BaF3-TPR-Met cells and showed high selectivity for c-Met family against other 14 tested kinases. However, compound 6b was found ineffective in the c-Met-dependent U-87MG human gliobastoma xenograft model that may be relevant to its poor PK profile.A series of thieno[2,3-d]pyrimidines and furo[2,3-d]pyrimidines were synthesized and evaluated for the c-Met inhibition. Thieno[2,3-d]pyrimidine 6b stood out as the most potent showing an IC50 of 35.7 nM. This compound displayed high inhibitory effect on cell proliferation in BaF3-TPR-Met cells and showed high selectivity for c-Met family against other 14 tested kinases. However, compound 6b was found ineffective in the c-Met-dependent U-87MG human gliobastoma xenograft model that may be relevant to its poor PK profile.
Co-reporter:Shaopeng Chen;Xiaowei Zhang;Junlei Wang;Shengbiao Wan;Meiyu Geng;Tao Jiang
Chemical Biology & Drug Design 2011 Volume 78( Issue 6) pp:1006-1013
Publication Date(Web):
DOI:10.1111/j.1747-0285.2011.01209.x
A new series of potential epidermal growth factor receptor inhibitors possessing bisquinazoline and saccharide moieties were designed and synthesized. The biological results demonstrated that the synthetic derivatives significantly inhibited epidermal growth factor receptor enzymatic activity in vitro. Of them, compound 14b showed the highest inhibitory rate toward epidermal growth factor receptor protein tyrosine kinase (81.36%) at a concentration of 1 μm. Further molecular simulation predicted that 14b offered its saccharide moieties hydrogen bonding to ATP-binding pocket.
Co-reporter:Zhongjie Liang, Dengyou Zhang, Jing Ai, Limin Chen, Hengshuai Wang, Xiangqian Kong, Mingyue Zheng, Hong Liu, Cheng Luo, Meiyu Geng, Hualiang Jiang, Kaixian Chen
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 12) pp:3749-3754
Publication Date(Web):15 June 2011
DOI:10.1016/j.bmcl.2011.04.064
A series of N′-(2-oxoindolin-3-ylidene)hydrazide derivatives were identified as moderately potent inhibitors against c-Met kinase by pharmacophore-based virtual screening and chemical synthesis methods. The structure–activity relationship (SAR) at various positions of the scaffold was investigated and its binding mode with c-Met kinase was analyzed by molecular modeling studies. In this study, two potent compounds D2 and D25, with IC50 value at 1.3 μM and 2.2 μM against c-Met kinase respectively, were identified. Finally, based on the clues extracted from this study, future development for the optimization of this scaffold was discussed.Compound D2 was discovered by pharmacophore-based virtual screening against SPECS database. Based on the SAR and binding modes analysis, modifications on the N′-(2-oxoindolin-3-ylidene)hydrazide scaffold were proposed.
Co-reporter:Yu Jiao, Hongchun Liu, Meiyu Geng, Wenhu Duan
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 7) pp:2071-2074
Publication Date(Web):1 April 2011
DOI:10.1016/j.bmcl.2011.02.005
A series of 10-arylcamptothecin derivatives was designed and synthesized. The key step of the synthesis was achieved by employing Suzuki cross-coupling chemistry. All of the new derivatives were tested for cytotoxicity against three human tumor cell lines, BEL-7402, A549, and HL-60; most of the derivatives exhibited potent cytotoxicity. The stability study showed that compound 30 was more stable than its lead compound 10-hydroxycamptothecin under the physiological condition. Mechanistic study demonstrated that compound 30 and its hydrochloride 31 had a pharmacological profile similar with camptothecin.
Co-reporter:Min Gui;Da-Kuo Shi;Min Huang;Yu Zhao;Qi-Ming Sun
Investigational New Drugs 2011 Volume 29( Issue 5) pp:800-810
Publication Date(Web):2011 October
DOI:10.1007/s10637-010-9425-3
Glycosylated natural products are reliable platforms for the development of anticancer drugs, simply due to the important features added by sugar appendages to the shape and the stereoelectronic properties of natural scaffolds. Herein, we indentified D11, a novel diphyllin glycoside with acetylated D-quinovose sugar moiety, as a potent topoisomerase IIα (Topo IIα) inhibitior. This peculiar sugar moiety endows D11 an optimal conformation with a high binding affinity for Topo IIα via hydrogen bonding to the entrance of ATPase pocket, thereby helping achieve more potent Topo II inhibition activity compared to the aglycon diphyllin. Further biochemical insights manifested that D11 significantly inhibited Topo IIα ATPase-catalyzed ATP hydrolysis in an ATP-dependent, but a DNA-independent manner. All these underlie the consequent superiority of D11 in the in vitro proliferation inhibition, apoptosis induction and the in vivo remarkable antitumor potency in xenograft mouse model. Moreover, D11 treatment also displayed no obvious body weight loss and other apparent toxicities, indicative of the restricted side effects by the virtue of sugar attachment. Taken together, a defined glycosylated manipulation of diphyllin may be a promising alternative approach in the development of novel Topo II inhibitors.
Co-reporter:Qin Nie;Xiao-guang Du;Mei-yu Geng
Acta Pharmacologica Sinica 2011 32(5) pp:545-551
Publication Date(Web):2011-04-18
DOI:10.1038/aps.2011.14
Amyloid β (Aβ) peptides have long been viewed as a potential target for Alzheimer's disease (AD). Aggregation of Aβ peptides in the brain tissue is believed to be an exclusively pathological process. Therefore, blocking the initial stages of Aβ peptide aggregation with small molecules could hold considerable promise as the starting point for the development of new therapies for AD. Recent rapid progresses in our understanding of toxic amyloid assembly provide a fresh impetus for this interesting approach. Here, we discuss the problems, challenges and new concepts in targeting Aβ peptides.
Co-reporter:Caihua Zhu;Qin Chen;Zuoquan Xie;Jing Ai;Linjiang Tong
Journal of Molecular Medicine 2011 Volume 89( Issue 3) pp:279-289
Publication Date(Web):2011 March
DOI:10.1007/s00109-010-0701-7
Histone deacetylases (HDACs) play fundamental roles in the epigenetic regulation of gene expression and contribute to the growth, differentiation, and apoptosis of cancer cells. Although HDACs are recognized to be closely related to cancer development and altered expression of certain HDACs is observed in tumor samples, the arcane characters of HDACs in tumorigenesis have not been fully illustrated. Herein, we report that HDAC7 is a crucial player in cancer cell proliferation. Knockdown of HDAC7 resulted in significant G1/S arrest in different cancer cell lines. Subsequent investigations indicated that HDAC7 silencing blocked cell cycle progression through suppressing c-Myc expression and increasing p21 and p27 protein levels. The ectopic expression of c-Myc in turn antagonized the cell cycle arrest and repressed the elevation of p21 and p27 in HDAC7 silencing setting. Of note, HDAC7 deficiency was further identified to induce cellular senescence program, which was also reversed by c-Myc re-expression. Further chromatin immunoprecipitation assays indicated that HDAC7 directly binds with c-Myc gene and HDAC7 silencing decreased c-Myc mRNA level via reducing histone H3/H4 acetylation and repressing the association of RNA polymerase II (RNAP II) with c-Myc gene. Taken together, our findings highlight for the first time an unrecognized link between HDAC7 and c-Myc and offer a novel mechanistic insight into the contribution of HDAC7 to tumor progression.
Co-reporter:Qin Nie;Xiao-guang Du;Mei-yu Geng
Acta Pharmacologica Sinica 2011 32(5) pp:545-551
Publication Date(Web):2011-04-18
DOI:10.1038/aps.2011.14
Amyloid β (Aβ) peptides have long been viewed as a potential target for Alzheimer's disease (AD). Aggregation of Aβ peptides in the brain tissue is believed to be an exclusively pathological process. Therefore, blocking the initial stages of Aβ peptide aggregation with small molecules could hold considerable promise as the starting point for the development of new therapies for AD. Recent rapid progresses in our understanding of toxic amyloid assembly provide a fresh impetus for this interesting approach. Here, we discuss the problems, challenges and new concepts in targeting Aβ peptides.
Co-reporter:Gaopeng Song, Hongchun Liu, Wei Zhang, Meiyu Geng, Yingxia Li
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 14) pp:5183-5193
Publication Date(Web):15 July 2010
DOI:10.1016/j.bmc.2010.05.064
A series of anthracene l-rhamnopyranosides were designed and synthesized in a practical way and their cytotoxic activity was examined in vitro. Most compounds exhibited both potent cytotoxicity against several tumor cell lines and high DNA binding capacity. The preliminary results showed that subtle modifications of rhamnosyl moiety in anthracene rhamnosides with acetyl group had a selective toxicity for different tumor cells and the displacement of C-10 carbonyl group in emodin by acetylmethylene group was helpful to improve the inhibitory activity. Lipophilicity of the anthracene glycosides was not a crucial factor for cytotoxicity and most molecules with good cytotoxicity could inhibit the catalytic activity of Top2α.A series of anthracene l-rhamnopyranosides were designed and synthesized in a practical way. Most compounds exhibited both potent cytotoxicity against several tumor cell lines and high DNA binding capacity.
Co-reporter:Yunli Peng;Jing Li;Meiyu Geng
Molecular and Cellular Biochemistry 2010 Volume 340( Issue 1-2) pp:143-152
Publication Date(Web):2010 July
DOI:10.1007/s11010-010-0411-z
Endothelium plays a vital role in the logistics of the immune system, as well as the maintenance of the homeostasis. The major objective of this study is to unravel the relationship between expression changes of carbohydrate structures and the dysfunction of human umbilical vein endothelial cells (HUVEC) stimulated with tumor-conditioned medium (TCM), which is involved in tumor cell extravasation. Using flow cytometry (FCM) assay, the expression profiles of a selected group of 9 carbohydrate structures have been determined in HUVEC under control conditions and TCM-treated conditions, six of which increased significantly in expression after induction. Particularly, the expression level of β-1,6-GlcNAc branching glycan was extremely higher after the stimulation. In parallel, the conformation change of HUVEC monolayer has been detected with inverted phase contrast microscopy and confocal microscopy. Under TCM stimulation, the actin cytoskeleton underwent rearrangement and formed abundant stress fiber within cells; therefore cell contraction was induced, which resulted in paracellular gap formation and barrier dysfunction. We furthered our study to investigate the mechanism underlying the conformation change of HUVEC. The results demonstrated that TCM induced the increase in β-1,6-GlcNAc branching expression of PECAM-1, accompanied by the tyrosine phosphorylation of PECAM-1. The downstream effector RhoA was activated in consequence of the activation of PECAM-1. In conclusion, our results strongly suggested that the carbohydrate composition of endothelial cell surface is very important for the cells to exert their physiological effects correlated with cancer extravasation.
Co-reporter:Sheng-Ping Yang ; Xiao-Wei Zhang ; Jing Ai ; Li-She Gan ; Jin-Biao Xu ; Ying Wang ; Zu-Shang Su ; Lu Wang ; Jian Ding ; Mei-Yu Geng ;Jian-Min Yue
Journal of Medicinal Chemistry () pp:
Publication Date(Web):August 30, 2012
DOI:10.1021/jm3007454
Eucalyptin A (1), together with two known compounds 2 and 3 exhibiting potent inhibition on HGF/c-Met axis, was discovered from the fruits of Eucalyptus globulus. 1 possessed an unprecedented carbon framework of phloroglucinol-coupled sesquiterpenoid, and its structure was elucidated by spectroscopic method and ECD calculation. A brief structure–activity relationship discussion indicated that the coupling of a phloroglucinol and a sesquiterpenoid is essential for the activity.
Co-reporter:Yuanxiang Wang, Jing Ai, Gang Liu, Meiyu Geng and Ao Zhang
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 17) pp:NaN5933-5933
Publication Date(Web):2011/07/01
DOI:10.1039/C1OB05830D
An effective one-pot synthesis of quinolines bearing diverse C3-piperazinyl functions was developed by using a modified Friedländer's protocol. The method not only enables the synthesis of our early reported c-Met inhibitor on a large scale, but also provides a way to generate novel multi-substituted quinolines for further structure–activity relationship (SAR) study.
Co-reporter:Zhongjie Liang, Xiao Ding, Jing Ai, Xiangqian Kong, Limin Chen, Liang Chen, Cheng Luo, Meiyu Geng, Hong Liu, Kaixian Chen and Hualiang Jiang
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 2) pp:NaN430-430
Publication Date(Web):2011/09/30
DOI:10.1039/C1OB06186K
The receptortyrosine kinase c-Met is an attractive target for therapeutic treatment of cancers nowadays. The discovery of small molecule inhibitors is of special interest in the blockade of the c-Met kinase pathway. Here, we initiated our study from compound 1a, a novel inhibitor against c-Met kinase. A substructure similarity search against the SPECS database and chemical synthesis methods were performed to obtain a series of pyrazolidine-3,5-dione derivatives. Through the enzyme-based assay against c-Met kinase, 4 compounds (1c, 1e, 1m and 1o) showed potential inhibitory activity, with IC50 values mostly less than 10 μM. Based on the structure–activity relationship (SAR) and binding mode analysis, a focused combinatorial library was designed by the LD1.0 program. Taking into account ADMET properties and synthesis accessibility, seven candidate compounds (5a–g) were successfully synthesized. The activity of the most potent compounds 5b (IC50 = 0.46 μM) was 20 fold higher than that of the lead 1a. Taken together, our findings identified the pyrazolidine-3,5-dione derivatives as potent inhibitors against c-Met kinase and demonstrated the efficiency of the strategy in the development of small molecules against c-Met kinase.
Co-reporter:Yong Wang, Zhong-Liang Xu, Jing Ai, Xia Peng, Jian-Ping Lin, Yin-Chun Ji, Mei-Yu Geng and Ya-Qiu Long
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 9) pp:NaN1562-1562
Publication Date(Web):2012/10/31
DOI:10.1039/C2OB26710A
The 1,6-naphthyridine motif is a multivalent scaffold in medicinal chemistry presenting various bioactivities when properly substituted. By incorporating a cyclic urea pharmacophore into the 1,6-naphthyridine framework through conformationally constraining the 7,8-positions, the resulting 1H-imidazo[4,5-h][1,6]naphthyridin-2(3H)-one was identified as a new class of c-Met kinase inhibitor. A comprehensive SAR study indicated that an N-1 alkyl substituent bearing a terminal free amino group, a hydrophobic substituted benzyl group at the N-3 position and the tricyclic core were essential for retaining effective Met inhibition of the 1H-imidazo[4,5-h][1,6]naphthyridin-2(3H)-one chemotype. Further introduction of a 4′-carboxamide phenoxy group at the C-5 position significantly improved the potency. The best c-Met kinase inhibitory activity was exemplified by 2t with an IC50 = 2.6 μM, which also displayed effective inhibition against TPR-Met phosphorylation and the proliferation of the BaF3-TPR-Met cells at low micromolar concentrations.

![3H-Indolium, 2-[5-[1-[6-[(2,5-dioxo-1-pyrrolidinyl)oxy]-6-oxohexyl]-1,3-dihydro-3,3-dimethyl-5-sulfo-2H-indol-2-ylidene]-1,3-pentadien-1-yl]-1-ethyl-3,3-](/data/chemimg/105700/146368-14-1.png)
![3H-Indolium, 2-[5-[1-[6-[(2,5-dioxo-1-pyrrolidinyl)oxy]-6-oxohexyl]-1,3-dihydro-3,3-dimethyl-5-sulfo-2H-indol-2-ylidene]-1,3-pentadien-1-yl]-1-ethyl-3,3-](/data/chemimg/105700/146368-14-1_b.png)
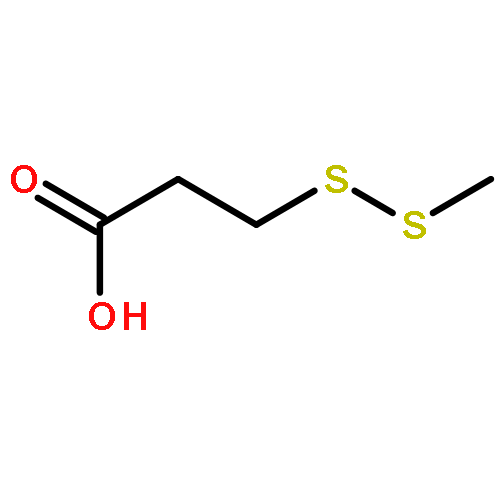
![2,5-Pyrrolidinedione, 1-[3-(methyldithio)-1-oxopropoxy]-](http://img.cochemist.com/ccimg/403600/403518-14-9.png)
![2,5-Pyrrolidinedione, 1-[3-(methyldithio)-1-oxopropoxy]-](http://img.cochemist.com/ccimg/403600/403518-14-9_b.png)












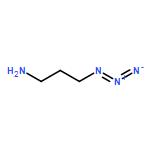
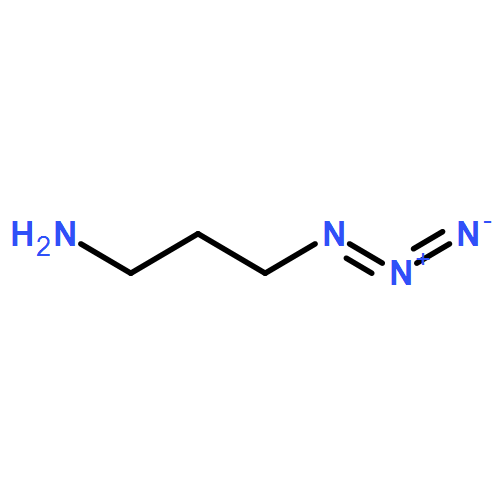
![Carbamic acid, [2-[(2,5-dioxo-1-pyrrolidinyl)oxy]-2-oxoethoxy]-,1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/80400/80366-85-4.png)
![Carbamic acid, [2-[(2,5-dioxo-1-pyrrolidinyl)oxy]-2-oxoethoxy]-,1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/80400/80366-85-4_b.png)