Co-reporter:Seth B. Herzon and Christopher D. Vanderwal
Chemical Reviews September 27, 2017 Volume 117(Issue 18) pp:11649-11649
Publication Date(Web):September 27, 2017
DOI:10.1021/acs.chemrev.7b00520
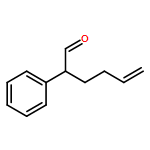
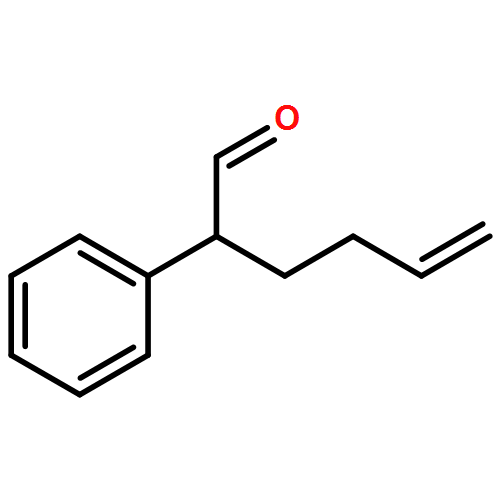
Co-reporter:Mary Elisabeth Daub, Philipp C. Roosen, and Christopher D. Vanderwal
The Journal of Organic Chemistry May 5, 2017 Volume 82(Issue 9) pp:4533-4533
Publication Date(Web):April 12, 2017
DOI:10.1021/acs.joc.7b00448
Since their discovery in the 1970s, the striking architectures and the unusual isonitrile functional groups of the isocyanoterpenes have attracted the interest of many organic chemists. The more recent revelation of their potent in vitro antiplasmodial activity sparked new endeavors to synthesize members of this family of secondary metabolites. In this Synopsis, we discuss three distinct strategies that each address multiple structurally different members of the isocyanoterpenes, ending with some of our group’s recent contributions in this area.
Co-reporter:Andrew M. White, Kathy Dao, Darius Vrubliauskas, Zef A. Könst, Gregory K. Pierens, Attila Mándi, Katherine T. Andrews, Tina S. Skinner-Adams, Mary E. Clarke, Patrick T. Narbutas, Desmond C.-M. Sim, Karen L. Cheney, Tibor Kurtán, Mary J. Garson, and Christopher D. Vanderwal
The Journal of Organic Chemistry December 15, 2017 Volume 82(Issue 24) pp:13313-13313
Publication Date(Web):November 10, 2017
DOI:10.1021/acs.joc.7b02421
Three new isocyanoditerpenes (5–7) have been characterized from Australian specimens of the nudibranch Phyllidiella pustulosa. The planar structure and (3R,6S,7R) absolute configuration of pustulosaisonitrile-1 were deduced by spectroscopic analyses at 900 MHz informed by molecular modeling, DFT calculations, and computational NMR chemical shift predictions and by comparison of experimental electronic circular dichroism (ECD) data with TDDFT-ECD calculations for the truncated model compound 8. A catalyst-controlled enantio- and diastereoselective total synthesis of the two most likely diastereomeric candidates for the structure of 5 solidified its (3R,6S,7R,10S,11R,14R) absolute configuration. Three individual enantioselective methods provided stereochemical control at key positions, permitting an unambiguous final structural assignment. Isocyanide 5 and synthetic diastereomers 5a and 5c showed activity against Plasmodium falciparum malaria parasites (IC50 ∼1 μM).
Co-reporter:Mary Elisabeth Daub, Jacques Prudhomme, Choukri Ben Mamoun, Karine G. Le Roch, and Christopher D. Vanderwal
ACS Medicinal Chemistry Letters 2017 Volume 8(Issue 3) pp:
Publication Date(Web):February 16, 2017
DOI:10.1021/acsmedchemlett.7b00013
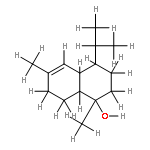
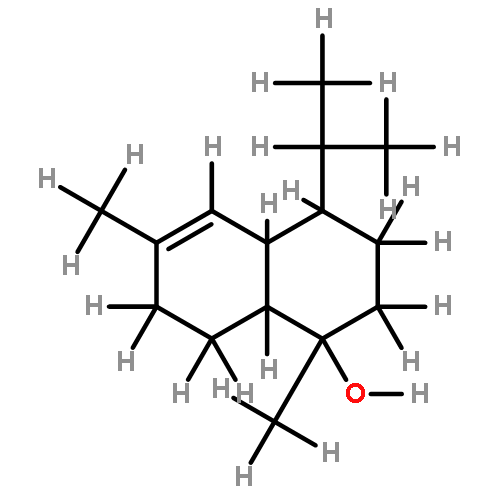
Several kalihinol natural products, members of the broader isocyanoterpene family of antimalarial agents, are potent inhibitors of Plasmodium falciparum, the agent of the most severe form of human malaria. Our previous total synthesis of kalihinol B provided a blueprint to generate many analogues within this family, some as complex as the natural product and some much simplified and easier to access. Each analogue was tested for blood-stage antimalarial activity using both drug-sensitive and -resistant P. falciparum strains. Many considerably simpler analogues of the kalihinols retained potent activity, as did a compound with a different decalin scaffold made in only three steps from sclareolide. Finally, one representative compound showed reasonable stability toward microsomal metabolism, suggesting that the isonitrile functional group that is critical for activity is not an inherent liability in these compounds.Keywords: Antimalarial; isonitrile; natural product synthesis; structure−activity relationship; terpenoid;
Co-reporter:Allen Y. Hong, Christopher D. Vanderwal
Tetrahedron 2017 Volume 73, Issue 29(Issue 29) pp:
Publication Date(Web):20 July 2017
DOI:10.1016/j.tet.2016.11.004
Driven by a new biogenetic hypothesis, the first total synthesis of alsmaphorazine B and several related indole alkaloids has been achieved. Numerous early approaches proved unsuccessful owing to unproductive side reactivity; nevertheless, they provided important clues that guided the evolution of our strategy. Critical to our success was a major improvement in our Zincke aldehyde cycloaddition strategy, which permitted the efficient gram-scale synthesis of akuammicine. The sequential chemoselective oxidations of akuammicine leading up to the key oxidative rearrangement also yielded several biogenetically related indole alkaloids en route to alsmaphorazine B.Download high-res image (173KB)Download full-size image
Co-reporter:Alexander R. White, Brendan M. Duggan, Shiou-Chuan Tsai, and Christopher D. Vanderwal
Organic Letters 2016 Volume 18(Issue 5) pp:1124-1127
Publication Date(Web):February 18, 2016
DOI:10.1021/acs.orglett.6b00230
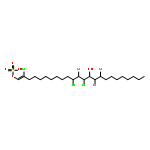
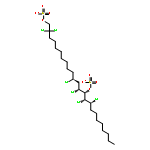
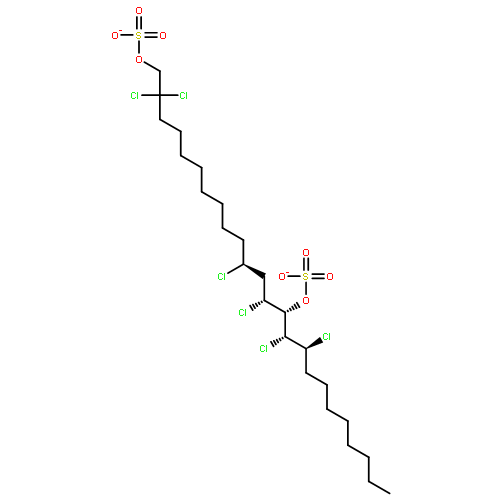
Many halogenases interchangeably incorporate chlorine and bromine into organic molecules. On the basis of an unsubstantiated report that the alga Ochromonas danica, a prodigious producer of chlorosulfolipids, was able to produce bromosulfolipids, we have investigated the promiscuity of its halogenases toward bromine incorporation. We have found that bromosulfolipids are produced with the exact positional and stereochemical selectivity as in the chlorosulfolipid danicalipin A when this alga is grown under modified conditions containing excess bromide ion.
Co-reporter:Philipp C. Roosen ; Christopher D. Verwal
Angewandte Chemie 2016 Volume 128( Issue 25) pp:7296-7299
Publication Date(Web):
DOI:10.1002/ange.201603581
Abstract
7,20-Diisocyanoadociane (DICA) is a potent antimalarial isocyanoterpene endowed with a fascinating tetracyclic structure composed of fused chair cyclohexanes. We report a highly stereocontrolled synthesis of a late-stage intermediate, the “Corey dione”, from which DICA has been made previously. This formal synthesis features a rapid buildup of much of the complexity of the target through a sequence of enone tandem vicinal difunctionalization, Friedel–Crafts cyclodehydration, and sequential stereocontrolled reductions. Most importantly, this success establishes the broader feasibility of our previously developed general synthesis approach to the isocyanoterpene family and provides a blueprint for a very direct synthesis of DICA and related natural products.
Co-reporter:Evan J. Horn, Joel S. Silverston, and Christopher D. Vanderwal
The Journal of Organic Chemistry 2016 Volume 81(Issue 5) pp:1819-1838
Publication Date(Web):February 10, 2016
DOI:10.1021/acs.joc.5b02550
Significant efforts were made to complete a synthesis of the complex norcembranoid ineleganolide via a seemingly attractive strategy involving late-stage creation of the central seven-membered ring. While the two key enantioenriched building blocks were made via high-yielding sequences and their convergent union was efficient, the critical C4–C5 bond of this sterically congested natural product could never be forged. Several interesting examples of unexpected acid–base behavior and unanticipated proximity-induced reactivity accounted for most of the problems in the execution of the synthesis plan.
Co-reporter:Philipp C. Roosen ; Christopher D. Verwal
Angewandte Chemie International Edition 2016 Volume 55( Issue 25) pp:7180-7183
Publication Date(Web):
DOI:10.1002/anie.201603581
Abstract
7,20-Diisocyanoadociane (DICA) is a potent antimalarial isocyanoterpene endowed with a fascinating tetracyclic structure composed of fused chair cyclohexanes. We report a highly stereocontrolled synthesis of a late-stage intermediate, the “Corey dione”, from which DICA has been made previously. This formal synthesis features a rapid buildup of much of the complexity of the target through a sequence of enone tandem vicinal difunctionalization, Friedel–Crafts cyclodehydration, and sequential stereocontrolled reductions. Most importantly, this success establishes the broader feasibility of our previously developed general synthesis approach to the isocyanoterpene family and provides a blueprint for a very direct synthesis of DICA and related natural products.
Co-reporter:Ryan K. Quinn; Zef A. Könst; Sharon E. Michalak; Yvonne Schmidt; Anne R. Szklarski; Alex R. Flores; Sangkil Nam; David A. Horne; Christopher D. Vanderwal;Erik J. Alexanian
Journal of the American Chemical Society 2015 Volume 138(Issue 2) pp:696-702
Publication Date(Web):December 22, 2015
DOI:10.1021/jacs.5b12308
Methods for the practical, intermolecular functionalization of aliphatic C–H bonds remain a paramount goal of organic synthesis. Free radical alkane chlorination is an important industrial process for the production of small molecule chloroalkanes from simple hydrocarbons, yet applications to fine chemical synthesis are rare. Herein, we report a site-selective chlorination of aliphatic C–H bonds using readily available N-chloroamides and apply this transformation to a synthesis of chlorolissoclimide, a potently cytotoxic labdane diterpenoid. These reactions deliver alkyl chlorides in useful chemical yields with substrate as the limiting reagent. Notably, this approach tolerates substrate unsaturation that normally poses major challenges in chemoselective, aliphatic C–H functionalization. The sterically and electronically dictated site selectivities of the C–H chlorination are among the most selective alkane functionalizations known, providing a unique tool for chemical synthesis. The short synthesis of chlorolissoclimide features a high yielding, gram-scale radical C–H chlorination of sclareolide and a three-step/two-pot process for the introduction of the β-hydroxysuccinimide that is salient to all the lissoclimides and haterumaimides. Preliminary assays indicate that chlorolissoclimide and analogues are moderately active against aggressive melanoma and prostate cancer cell lines.
Co-reporter:Mary Elisabeth Daub; Jacques Prudhomme; Karine Le Roch
Journal of the American Chemical Society 2015 Volume 137(Issue 15) pp:4912-4915
Publication Date(Web):March 27, 2015
DOI:10.1021/jacs.5b01152
Of the 50+ kalihinane diterpenoids reported to date, only five had been tested for antimalarial activity, in spite of the fact that kalihinol A is the most potent among the members of the larger family of antimalarial isocyanoterpenes. We have validated a strategy designed to access many of the kalihinanes with a 12-step enantioselective synthesis of kalihinol B, the tetrahydrofuran isomer of kalihinol A (a tetrahydropyran). Kalihinol B shows similarly high potency against chloroquine-resistant Plasmodium falciparum.
Co-reporter:Hung V. Pham; Alexander S. Karns; Christopher D. Vanderwal;K. N. Houk
Journal of the American Chemical Society 2015 Volume 137(Issue 21) pp:6956-6964
Publication Date(Web):May 11, 2015
DOI:10.1021/jacs.5b03718
The fascinating intramolecular arene/allene cycloaddition discovered by Himbert affords dearomatized, polycyclic intermediates with sufficient strain energy to drive rearrangement processes of the newly formed ring system. We disclose a detailed examination of a thermally induced stepwise dyotropic skeletal rearrangement of these cycloadducts, a reaction also first described by Himbert. We offer computational evidence for a two-stage mechanism for this formal dyotropic rearrangement and provide rationalizations for the significant substitution-dependent rate differences observed in experiments. These investigations led to the development of a Lewis-acid-catalyzed rearrangement of precursors that were unreactive under simple thermal instigation. The isolation of the product of an “interrupted” rearrangement under Lewis acidic conditions provides further support for the proposed stepwise mechanism. Computational results also matched experiments in terms of regiochemical preferences in unsymmetrical rearrangement precursors and explained why lactam O-, S-, and C-heterologues do not easily undergo this rearrangement.
Co-reporter:Allen Y. Hong
Journal of the American Chemical Society 2015 Volume 137(Issue 23) pp:7306-7309
Publication Date(Web):June 2, 2015
DOI:10.1021/jacs.5b04686
An N-oxide fragmentation/hydroxylamine oxidation/intramolecular 1,3-dipolar cycloaddition cascade efficiently converted an oxidized congener of akuammicine into the complex, hexacyclic architecture of the alsmaphorazine alkaloids. This dramatic structural change shows the chemical feasibility of our novel proposal for alsmaphorazine biogenesis. Critical to these endeavors was a marked improvement in our previously reported Zincke aldehyde cycloaddition approach to indole alkaloids, which permitted the gram-scale synthesis of akuammicine. The chemoselective oxidations of akuammicine leading up to the key rearrangement also generated several biogenetically related alkaloids of the alstolucine and alpneumine families.
Co-reporter:Jonathan K. Lam, Scott B. Joseph, Christopher D. Vanderwal
Tetrahedron Letters 2015 Volume 56(Issue 23) pp:3165-3168
Publication Date(Web):3 June 2015
DOI:10.1016/j.tetlet.2014.12.089
We have developed a Zincke-aldehyde-based approach to the complex alkaloid gelsemine. It features a key thermal pericyclic cascade that converts a Zincke aldehyde bearing a pendant alkene into a hydroisoindolone product that shares many structural features with the target molecule. Our efforts to convert these intermediates to gelsemine are also discussed.
Co-reporter:Philipp C. Roosen and Christopher D. Vanderwal
Organic Letters 2014 Volume 16(Issue 17) pp:4368-4371
Publication Date(Web):August 8, 2014
DOI:10.1021/ol502205m
An anionic oxy-Cope/transannular conjugate addition approach to the potent antimalarial 7,20-diisocyanoadociane is presented. The unexpected formation of undesired diastereomers in the key reaction led to the structural reassignment of previous products of this type of cascade and a reevaluation of the reversibility of the transannular ring closure. During efforts to coax the reaction toward the desired product, a transannular ene reaction provided tricyclic compounds relevant to the kempane diterpenoids.
Co-reporter:Samuel S. Tartakoff and Christopher D. Vanderwal
Organic Letters 2014 Volume 16(Issue 5) pp:1458-1461
Publication Date(Web):February 26, 2014
DOI:10.1021/ol500265v
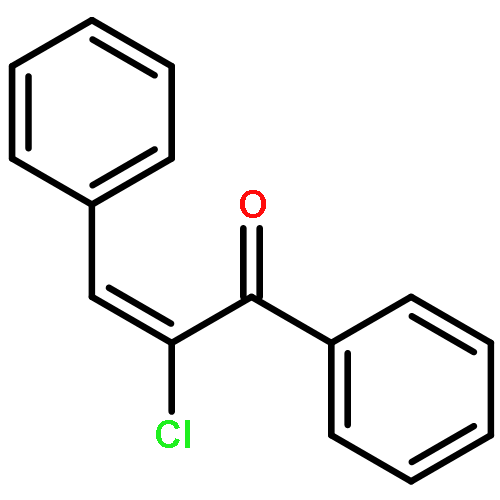
A synthesis of the ABC tricyclic ring system of the clionastatins, an unusual pair of highly chlorinated androstane steroids, has been accomplished. This work provides strong support for the original structural proposal. An unexpected substrate-dependent reversal in alkene chlorination diastereoselectivity was critical to success. This approach should be amenable to an eventual enantioselective synthesis of the natural products themselves.
Co-reporter:Carl V. Vogel;Halina Pietraszkiewicz;Omar M. Sabry; William H. Gerwick;Dr. Frederick A. Valeriote; Christopher D. Verwal
Angewandte Chemie 2014 Volume 126( Issue 45) pp:12401-12405
Publication Date(Web):
DOI:10.1002/ange.201407726
Abstract
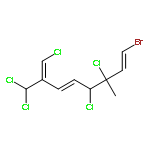
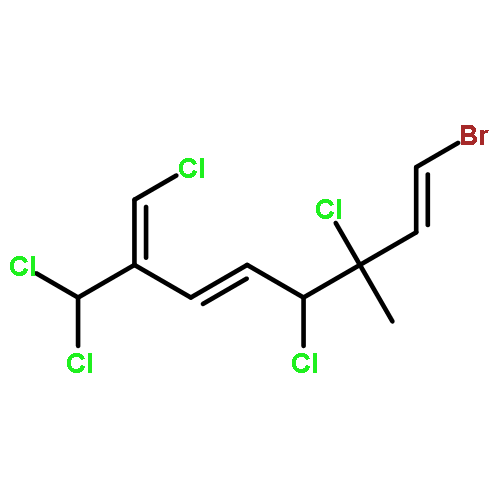
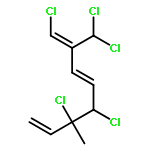
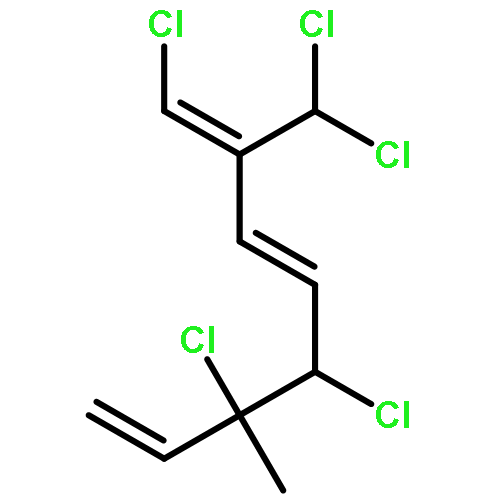
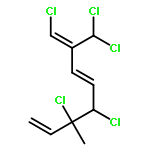
The family of polyhalogenated monoterpenes from Plocamium counts over a hundred known members. Using glyceraldehyde acetonide as a chiral-pool precursor, an enantioselective and divergent strategy was developed that provides a blueprint for the synthesis of many of the small yet complex acyclic members of this family. The broad applicability of this approach is demonstrated with the short, eight-step synthesis of four natural products and three analogues. These syntheses are the first of any members of the acyclic polyhalogenated Plocamium monoterpenes and permitted the evaluation of their selectivity against a range of tumor cell lines.
Co-reporter:Won-jin Chung, Joseph S. Carlson, and Christopher D. Vanderwal
The Journal of Organic Chemistry 2014 Volume 79(Issue 5) pp:2226-2241
Publication Date(Web):February 4, 2014
DOI:10.1021/jo5000829
A second-generation synthesis of three structurally related chlorosulfolipids has been developed. Key advances include highly stereocontrolled additions to α,β-dichloroaldehydes, kinetic resolutions of complex chlorinated vinyl epoxide intermediates, and Z-selective alkene cross metatheses of cis-vinyl epoxides. This strategy facilitated the synthesis of enantioenriched danicalipin A, mytilipin A, and malhamensilipin A in nine, eight, and 11 steps, respectively.
Co-reporter:Carl V. Vogel;Halina Pietraszkiewicz;Omar M. Sabry; William H. Gerwick;Dr. Frederick A. Valeriote; Christopher D. Verwal
Angewandte Chemie International Edition 2014 Volume 53( Issue 45) pp:12205-12209
Publication Date(Web):
DOI:10.1002/anie.201407726
Abstract
The family of polyhalogenated monoterpenes from Plocamium counts over a hundred known members. Using glyceraldehyde acetonide as a chiral-pool precursor, an enantioselective and divergent strategy was developed that provides a blueprint for the synthesis of many of the small yet complex acyclic members of this family. The broad applicability of this approach is demonstrated with the short, eight-step synthesis of four natural products and three analogues. These syntheses are the first of any members of the acyclic polyhalogenated Plocamium monoterpenes and permitted the evaluation of their selectivity against a range of tumor cell lines.
Co-reporter:Yvonne Schmidt ; Jonathan K. Lam ; Hung V. Pham ; K. N. Houk
Journal of the American Chemical Society 2013 Volume 135(Issue 19) pp:7339-7348
Publication Date(Web):May 1, 2013
DOI:10.1021/ja4025963
The unusual intramolecular arene/allene cycloaddition described 30 years ago by Himbert permits rapid access to strained polycyclic compounds that offer great potential for the synthesis of complex scaffolds. To more fully understand the mechanism of this cycloaddition reaction, and to guide efforts to extend its scope to new substrates, quantum mechanical computational methods were employed in concert with laboratory experiments. These studies indicated that the cycloadditions likely proceed via concerted processes; a stepwise biradical mechanism was shown to be higher in energy in the cases studied. The original Himbert cycloaddition chemistry is also extended from heterocyclic to carbocyclic systems, with computational guidance used to predict thermodynamically favorable cases. Complex polycyclic scaffolds result from the combination of the cycloaddition and subsequent ring-rearrangement metathesis reactions.
Co-reporter:Jonathan K. Lam ; Hung V. Pham ; K. N. Houk
Journal of the American Chemical Society 2013 Volume 135(Issue 46) pp:17585-17594
Publication Date(Web):October 10, 2013
DOI:10.1021/ja409618p
Unusual observations in the ring-rearrangement metathesis of Himbert arene/allene cycloadducts to form fused polycylic lactams led to a more in-depth experimental study that yielded conflicting results. Differences in reactivity within related systems and unexpected changes in diastereoselectivity among other similar substrates were not readily explained on the basis of the experimental results. Computational investigations demonstrated substrate-dependent changes in reaction pathways (ring-opening metathesis/ring-closing metathesis [ROM/RCM] cascade vs ring-closing metathesis/ring-opening metathesis [RCM/ROM] cascade). Furthermore, some reactions were judged to be under thermodynamic control and others under kinetic control. The greater understanding of the most likely reaction pathways and their energetics provides a reasonable explanation for the previously irreconcilable results.
Co-reporter:Gregg M. Schwarzwalder, Sarah E. Steinhardt, Hung V. Pham, K. N. Houk, and Christopher D. Vanderwal
Organic Letters 2013 Volume 15(Issue 23) pp:6014-6017
Publication Date(Web):November 12, 2013
DOI:10.1021/ol402905n
A Diels–Alder reaction, a desymmetrizing aldol reaction, and a reductive Heck cyclization are employed in a short synthesis of a tetracycle relevant to exiguaquinol, a potential antibiotic. Ground-state energies of this advanced model system and the natural product rationalize the incorrect hemiaminal configuration experimentally obtained and point to the importance of the sulfonate in dictating the relative configuration of the natural product.
Co-reporter:Dr. Won-jin Chung;Joseph S. Carlson;Dr. D. Karl Bedke; Christopher D. Verwal
Angewandte Chemie International Edition 2013 Volume 52( Issue 38) pp:10052-10055
Publication Date(Web):
DOI:10.1002/anie.201304565
Co-reporter:Dr. Won-jin Chung;Joseph S. Carlson;Dr. D. Karl Bedke; Christopher D. Verwal
Angewandte Chemie 2013 Volume 125( Issue 38) pp:10236-10239
Publication Date(Web):
DOI:10.1002/ange.201304565
Co-reporter:Hung V. Pham, David B. C. Martin, Christopher D. Vanderwal and K. N. Houk
Chemical Science 2012 vol. 3(Issue 5) pp:1650-1655
Publication Date(Web):02 Feb 2012
DOI:10.1039/C2SC01072K
Computational studies show that the base-mediated intramolecular Diels–Alder of tryptamine-derived Zincke aldehydes, used as a key step in the synthesis of the Strychnos alkaloids norfluorocurarine and strychnine, proceeds via a stepwise pathway. The experimentally determined importance of a potassium counterion in the base is explained by its ability to preorganize the Zincke aldehyde diene in an s-cis conformation suitable to bicyclization. Computation also supports the thermodynamic importance of the generation of a stable enolate in the final reaction step. The thermal cycloreversion reaction of the Diels–Alder products is also found to proceed in a stepwise manner.
Co-reporter:Jonathan K. Lam, Yvonne Schmidt, and Christopher D. Vanderwal
Organic Letters 2012 Volume 14(Issue 21) pp:5566-5569
Publication Date(Web):October 15, 2012
DOI:10.1021/ol302680m
The intramolecular arene/allene cycloaddition first described 30 years ago by Himbert and Henn permits rapid access to strained polycyclic compounds. Alkene metathesis processes cleanly rearrange appropriately substituted cycloadducts into complex, functional-group-rich polycyclic lactams of potential utility for natural product synthesis and medicinal chemistry.
Co-reporter:David B. C. Martin, Lucas Q. Nguyen, and Christopher D. Vanderwal
The Journal of Organic Chemistry 2012 Volume 77(Issue 1) pp:17-46
Publication Date(Web):December 14, 2011
DOI:10.1021/jo2020246
A full account of the development of the base-mediated intramolecular Diels–Alder cycloadditions of tryptamine-derived Zincke aldehydes is described. This important complexity-generating transformation provides the tetracyclic core of many indole monoterpene alkaloids in only three steps from commercially available starting materials and played a key role in short syntheses of norfluorocurarine (five steps), dehydrodesacetylretuline (six steps), valparicine (seven steps), and strychnine (six steps). Reasonable mechanistic possibilities for this reaction, a surprisingly facile dimerization of the products, and an unexpected cycloreversion to regenerate Zincke aldehydes under specific conditions are also discussed.
Co-reporter:Dr. Theo D. Michels;Dr. Matthew S. Dowling ;Dr. Christopher D. Verwal
Angewandte Chemie 2012 Volume 124( Issue 30) pp:7690-7694
Publication Date(Web):
DOI:10.1002/ange.201203147
Co-reporter:Dr. Theo D. Michels;Dr. Matthew S. Dowling ;Dr. Christopher D. Verwal
Angewandte Chemie International Edition 2012 Volume 51( Issue 30) pp:7572-7576
Publication Date(Web):
DOI:10.1002/anie.201203147
Co-reporter:Robert S. Paton ; Sarah E. Steinhardt ; Christopher D. Vanderwal ;K. N. Houk
Journal of the American Chemical Society 2011 Volume 133(Issue 11) pp:3895-3905
Publication Date(Web):February 25, 2011
DOI:10.1021/ja107988b
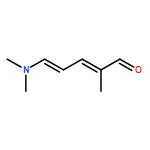
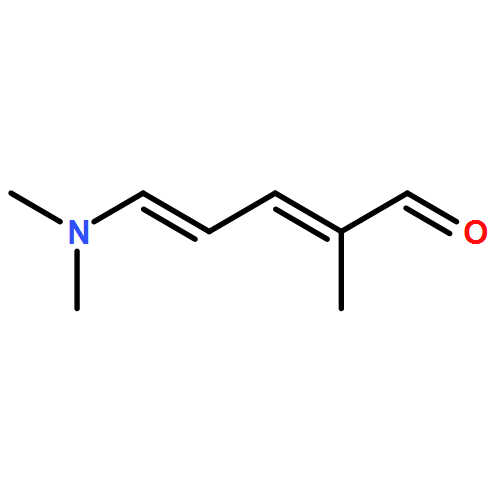
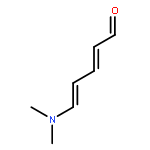
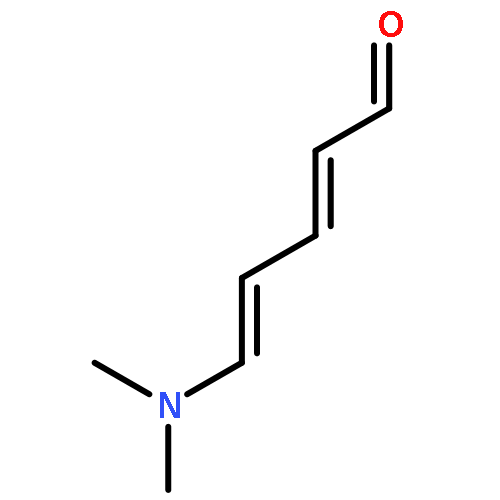
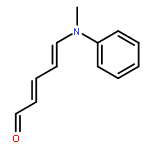
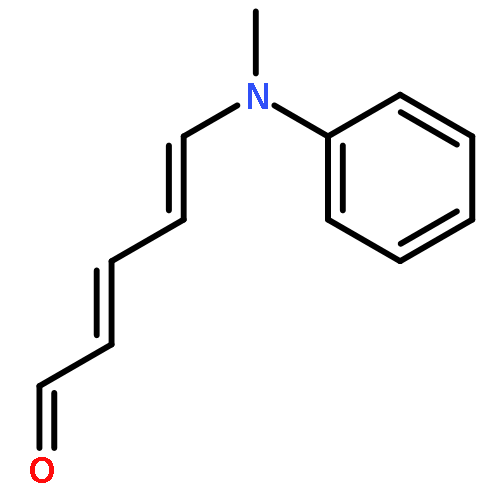
The thermal pericyclic cascade rearrangement of Zincke aldehydes (5-(dialkylamino)-2,4-pentadienals) to afford Z-α,β,γ,δ-unsaturated amides discovered by the Vanderwal group has been studied in depth using quantum mechanical methods. Two mechanistic possibilities that had previously been put forth to explain this internal redox process, one that had been discounted by experiment and the other that had withstood experimental scrutiny, were evaluated. Both of these mechanisms suffered from energetic barriers that appeared too high to allow rearrangement to proceed under the conditions used; however, computational study of a third possibility that implicates the intermediacy of vinylketenes revealed that it is the most likely pathway of rearrangement. Further computational studies accounted for the relative rates of rearrangement in substituted Zincke aldehydes, predicted the feasibility of related processes for other donor−acceptor dienes, and provided insight into the rearrangement of allylamine-derived Zincke aldehydes that provide either dihydropyridones or polycyclic lactams by further pericyclic processes.
Co-reporter:D. Karl Bedke and Christopher D. Vanderwal
Natural Product Reports 2011 vol. 28(Issue 1) pp:15-25
Publication Date(Web):02 Dec 2010
DOI:10.1039/C0NP00044B
Chlorosulfolipids have been isolated from freshwater algae and from toxic mussels. They appear to have a structural role in algal membranes and have been implicated in Diarrhetic Shellfish Poisoning. Further fascinating aspects of these compounds include their stereochemically complex polychlorinated structures and the resulting strong conformational biases, and their poorly understood (yet surely compelling) biosynthesis. Discussions of each of these topics and of efforts in structural and stereochemical elucidation and synthesis are the subject of this Highlight.
Co-reporter:David B. C. Martin and Christopher D. Vanderwal
Chemical Science 2011 vol. 2(Issue 4) pp:649-651
Publication Date(Web):04 Feb 2011
DOI:10.1039/C1SC00009H
Strychnine is synthesized via a longest linear sequence of six steps from commercially available starting materials. Key steps include a base-mediated intramolecular Diels–Alder reaction of a tryptamine-derived Zincke aldehyde, a Ru-catalyzed trans-hydrosilylation of 1,4-butynediol, and a tandem Brook rearrangement/intramolecular conjugate addition reaction that affords the Wieland–Gumlich aldehyde.
Co-reporter:Christopher D. Vanderwal
The Journal of Organic Chemistry 2011 Volume 76(Issue 23) pp:9555-9567
Publication Date(Web):August 30, 2011
DOI:10.1021/jo201625e
The century-old Zincke process for ring-opening of pyridinium salts produces 5-amino-2,4-pentadienals, a type of donor–acceptor dienes known as Zincke aldehydes. Inspired by this reasonably general and often efficient process for dearomatization, my laboratory has used pyridines as a starting point for heterocycle synthesis, which resulted in unusual syntheses of indoles, pyrrolines, and a formal synthesis of the natural product porothramycin A. Furthermore, our study of the reactivity of Zincke aldehydes has led to accidental discoveries of pericyclic cascade reactions that produce Z-α,β-unsaturated amides or polycyclic lactams, depending upon the identity of the substituents on nitrogen. Finally, a base-mediated formal cycloaddition reaction of tryptamine-derived Zincke aldehydes has served as the key step in concise syntheses of the indole alkaloids norfluorocurarine and strychnine.
Co-reporter:D. Karl Bedke ; Grant M. Shibuya ; Alban R. Pereira ; William H. Gerwick
Journal of the American Chemical Society 2010 Volume 132(Issue 8) pp:2542-2543
Publication Date(Web):February 8, 2010
DOI:10.1021/ja910809c
The first enantioselective synthesis of a member of the chlorosulfolipid family of natural products is reported. All of the polar substituents of malhamensilipin A are introduced with high stereoselectivity, and the unique (E)-chlorovinyl sulfate is created by a chemo-, regio-, and stereoselective E2 elimination of HCl in a reaction that likely has a counterpart in the biosynthesis of this fascinating natural product.
Co-reporter:Theo D. Michels, Matthew J. Kier, Aaron M. Kearney and Christopher D. Vanderwal
Organic Letters 2010 Volume 12(Issue 13) pp:3093-3095
Publication Date(Web):June 8, 2010
DOI:10.1021/ol101035p
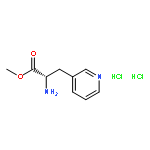
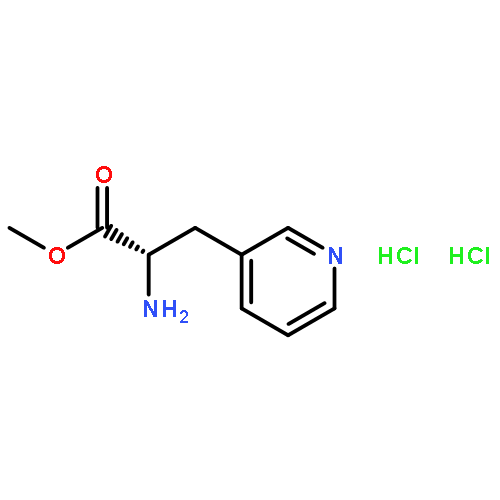
Short formal syntheses of the antitumor antibiotics porothramycins A and B from a commercially available ester of the unnatural amino acid 3-(3-pyridyl)alanine are presented. A rearrangement cascade that presumably involves a Zincke-type pyridinium ring-opening followed by cyclization of a pendant nucleophilic amide generates the salient pyrroline ring of the alkaloids.
Co-reporter:DavidB.C. Martin ;ChristopherD. Verwal
Angewandte Chemie International Edition 2010 Volume 49( Issue 16) pp:2830-2832
Publication Date(Web):
DOI:10.1002/anie.201000045
Co-reporter:Matthew S. Dowling and Christopher D. Vanderwal
The Journal of Organic Chemistry 2010 Volume 75(Issue 20) pp:6908-6922
Publication Date(Web):September 13, 2010
DOI:10.1021/jo101439h
The development of a strategy consisting of allylsilane ring-closing metathesis and subsequent SE′ electrophilic desilylation (allylsilane RCM/SE′) to construct exo-methylidenecycloalkanes is described. Its utility is documented in short syntheses of teucladiol and poitediol. A key transformation in the synthesis of teucladiol is an aldol addition that establishes three stereochemical relationships in one step with ≥10:1 diastereoselectivity and provides a fascinating example of double stereodifferentiation/kinetic resolution with racemic reaction partners in the context of natural product synthesis. The synthesis of (±)-teucladiol required five steps from cyclopentenone and proceeded in 28% overall yield; adaptation of this route to an enantioselective synthesis of (−)-teucladiol enabled the determination of the absolute configuration of this terpene natural product. The use of fluoride-mediated conditions in the final desilylation step preserves the location of the alkene, delivering the natural product (±)-isoteucladiol (five steps and 21% yield from cyclopentenone). The synthesis of poitediol showcases the power of RCM for constructing eight-membered rings and features a highly diastereoselective epoxidation/fluoride-mediated fragmentation sequence for installing the exo-methylidene group with an adjacent hydroxyl-bearing stereocenter. The synthesis of (±)-poitediol required seven steps and proceeded in 18% overall yield. Again, fluoride-mediated desilylation of a late-stage intermediate (with retention of double-bond location) delivered the natural product (±)-dactylol (seven steps and 24% yield). Efforts directed toward incorporating the RCM/SE′ sequence into a synthesis of caryophyllene are also disclosed. While ultimately unsuccessful, these efforts resulted in the identification of a novel metal alkylidene-promoted deallylation reaction of terminal 1,4-dienes. A possible mechanism for this unexpected deallylation reaction of 1,4-dienes is provided.
Co-reporter:DavidB.C. Martin ;ChristopherD. Verwal
Angewandte Chemie 2010 Volume 122( Issue 16) pp:2893-2895
Publication Date(Web):
DOI:10.1002/ange.201000045
Co-reporter:Matthew S. Dowling
Journal of the American Chemical Society 2009 Volume 131(Issue 42) pp:15090-15091
Publication Date(Web):September 29, 2009
DOI:10.1021/ja906241w
A general strategy for the synthesis of exo-methylidenecycloalkanes, which are salient features of many terpenoid natural products, is presented. Ring-closing alkene metathesis of allylsilanes provides intermediates that can be protodesilylated with alkene transposition to afford the exocyclic alkene; alternatively, the reactivity of the cyclic allylsilane intermediate can be harnessed to introduce allylic functionality. These two modes of reactivity are showcased in short syntheses of the sesquiterpene natural products teucladiol and poitediol, respectively.
Co-reporter:D. Karl Bedke ; Grant M. Shibuya ; Alban Pereira ; William H. Gerwick ; Thomas H. Haines
Journal of the American Chemical Society 2009 Volume 131(Issue 22) pp:7570-7572
Publication Date(Web):May 15, 2009
DOI:10.1021/ja902138w
The relative stereochemistry of the major chlorosulfolipid of the chrysophyte alga Ochromonas danica, to which we have given the name “danicalipin A”, is reported. The first synthesis of this lipid, via several stereospecific electrophilic additions to alkenes, serves to corroborate the stereochemical assignment made by NMR spectroscopy. The synthesis strategy described should be applicable to other chlorosulfolipids and should provide access to sufficient material for studies of the lipid’s properties and function in membranes.
Co-reporter:Aaron M. Kearney;Christopher D. Verwal
Angewandte Chemie International Edition 2006 Volume 45(Issue 46) pp:
Publication Date(Web):30 OCT 2006
DOI:10.1002/anie.200602996
The century-old ring-opening reaction of pyridinium salts with tethered nucleophiles has been harnessed for a synthesis of substituted indoles and related nitrogen heterocyles. Extension of this method could lead to oxygen- and sulfur-containing heterocycles and carbocycles, as well as to applications in natural product synthesis and medicinal chemistry.
Co-reporter:Hung V. Pham, David B. C. Martin, Christopher D. Vanderwal and K. N. Houk
Chemical Science (2010-Present) 2012 - vol. 3(Issue 5) pp:NaN1655-1655
Publication Date(Web):2012/02/02
DOI:10.1039/C2SC01072K
Computational studies show that the base-mediated intramolecular Diels–Alder of tryptamine-derived Zincke aldehydes, used as a key step in the synthesis of the Strychnos alkaloids norfluorocurarine and strychnine, proceeds via a stepwise pathway. The experimentally determined importance of a potassium counterion in the base is explained by its ability to preorganize the Zincke aldehyde diene in an s-cis conformation suitable to bicyclization. Computation also supports the thermodynamic importance of the generation of a stable enolate in the final reaction step. The thermal cycloreversion reaction of the Diels–Alder products is also found to proceed in a stepwise manner.
Co-reporter:David B. C. Martin and Christopher D. Vanderwal
Chemical Science (2010-Present) 2011 - vol. 2(Issue 4) pp:NaN651-651
Publication Date(Web):2011/02/04
DOI:10.1039/C1SC00009H
Strychnine is synthesized via a longest linear sequence of six steps from commercially available starting materials. Key steps include a base-mediated intramolecular Diels–Alder reaction of a tryptamine-derived Zincke aldehyde, a Ru-catalyzed trans-hydrosilylation of 1,4-butynediol, and a tandem Brook rearrangement/intramolecular conjugate addition reaction that affords the Wieland–Gumlich aldehyde.
.jpg)










![2-Propenal, 3-(1H-pyrrolo[2,3-b]pyridin-3-yl)-, (2E)-](http://img.cochemist.com/ccimg/923300/923293-12-3.png)
![2-Propenal, 3-(1H-pyrrolo[2,3-b]pyridin-3-yl)-, (2E)-](http://img.cochemist.com/ccimg/923300/923293-12-3_b.png)

![1H-Indole, 5-fluoro-3-[(1E)-3,3,3-trifluoro-1-propen-1-yl]-](http://img.cochemist.com/ccimg/923300/923293-08-7.png)
![1H-Indole, 5-fluoro-3-[(1E)-3,3,3-trifluoro-1-propen-1-yl]-](http://img.cochemist.com/ccimg/923300/923293-08-7_b.png)
![2-Propenal, 3-[6-(trifluoromethyl)-1H-indol-3-yl]-, (2E)-](http://img.cochemist.com/ccimg/923300/923293-07-6.png)
![2-Propenal, 3-[6-(trifluoromethyl)-1H-indol-3-yl]-, (2E)-](http://img.cochemist.com/ccimg/923300/923293-07-6_b.png)
![1H-Indole, 5-methoxy-3-[(1E)-3,3,3-trifluoro-1-propen-1-yl]-](http://img.cochemist.com/ccimg/923300/923293-06-5.png)
![1H-Indole, 5-methoxy-3-[(1E)-3,3,3-trifluoro-1-propen-1-yl]-](http://img.cochemist.com/ccimg/923300/923293-06-5_b.png)
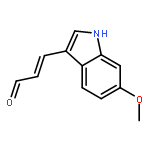
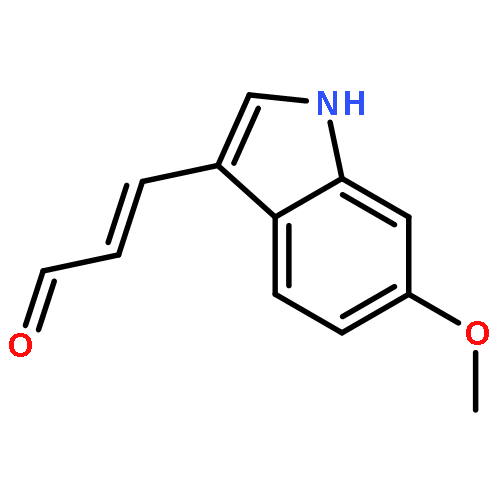
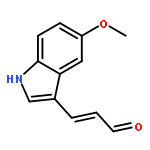
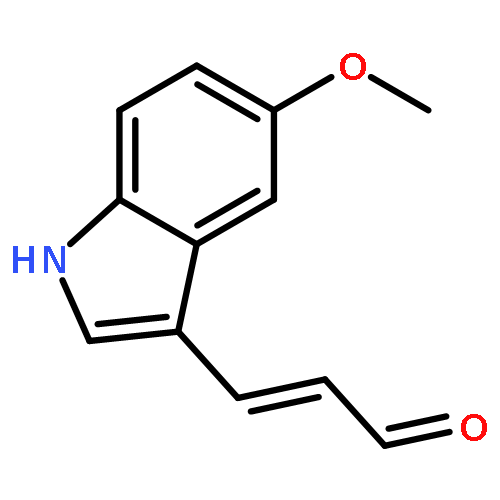


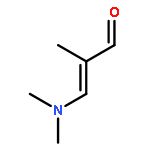
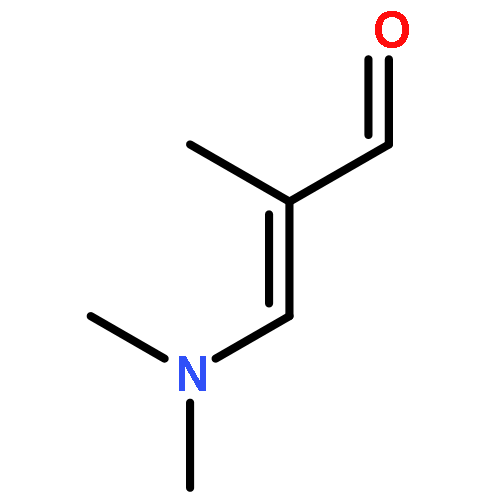


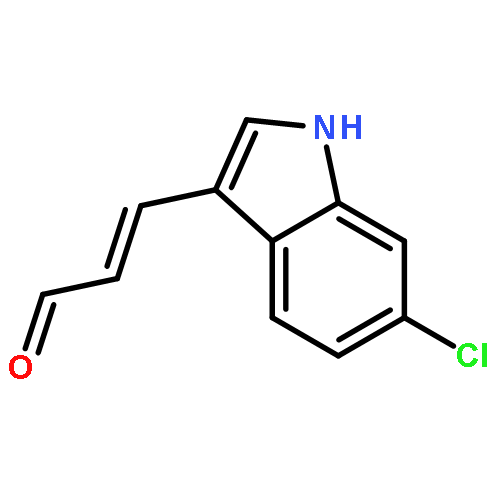
![PROPANEDIOIC ACID, [(1R)-1-(4-FLUOROPHENYL)-2-NITROETHYL]-, DIMETHYL ESTER](http://img.cochemist.com/ccimg/771600/771562-78-8.png)
![PROPANEDIOIC ACID, [(1R)-1-(4-FLUOROPHENYL)-2-NITROETHYL]-, DIMETHYL ESTER](http://img.cochemist.com/ccimg/771600/771562-78-8_b.png)
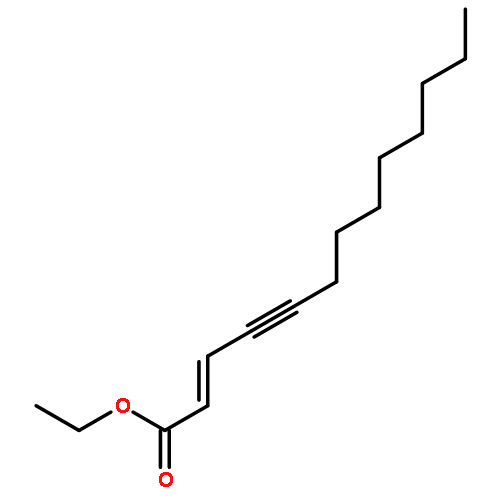


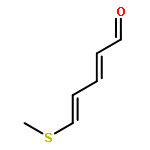

















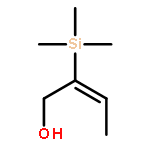
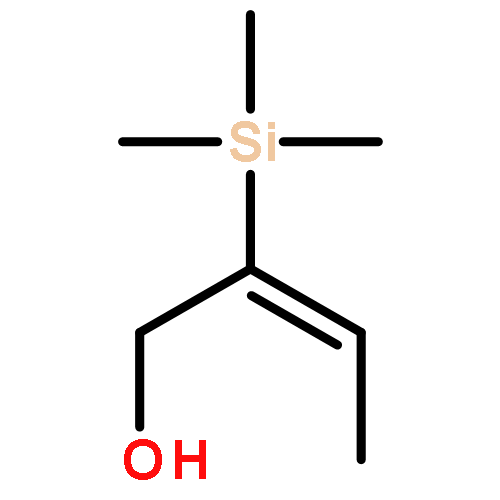
![2,5-Pyrrolidinedione,3-[(1S)-2-[(1R,3S,4aS,7S,8aS)-7-chlorodecahydro-3-hydroxy-5,5,8a-trimethyl-2-methylene-1-naphthalenyl]-1-hydroxyethyl]-,(3R)-](http://img.cochemist.com/ccimg/148800/148717-91-3.png)
![2,5-Pyrrolidinedione,3-[(1S)-2-[(1R,3S,4aS,7S,8aS)-7-chlorodecahydro-3-hydroxy-5,5,8a-trimethyl-2-methylene-1-naphthalenyl]-1-hydroxyethyl]-,(3R)-](http://img.cochemist.com/ccimg/148800/148717-91-3_b.png)
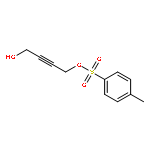

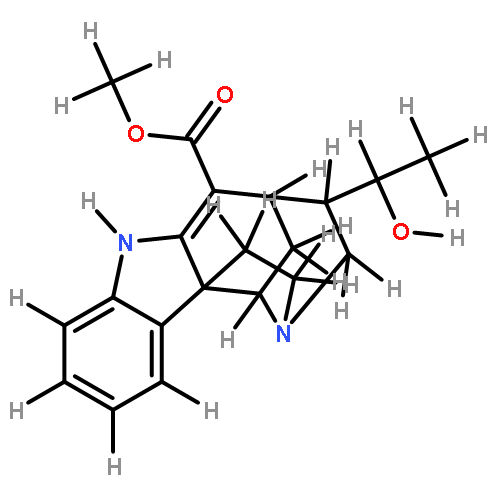
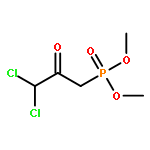
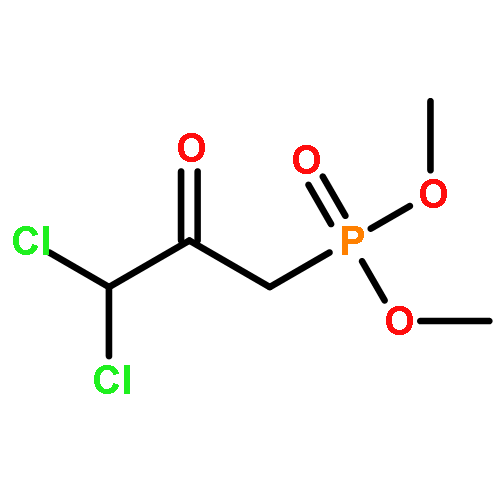
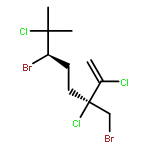
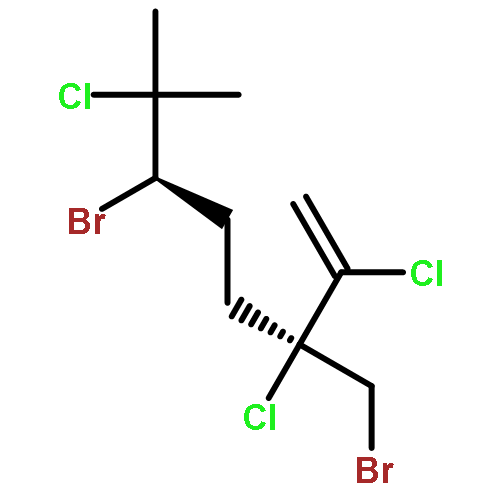


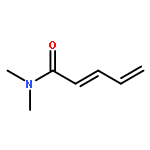
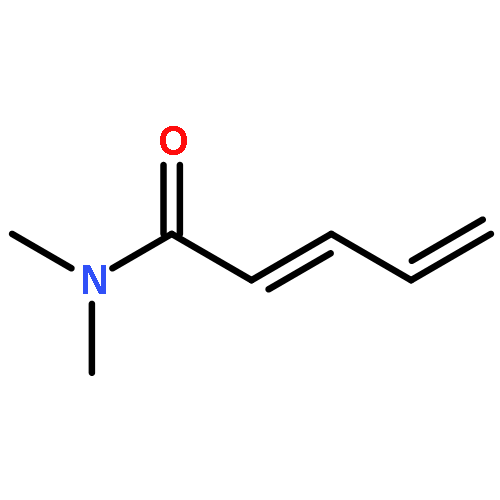
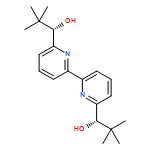




![5-[TERT-BUTYL(DIPHENYL)SILYL]OXYPENT-3-YN-1-OL](http://img.cochemist.com/ccimg/117500/117471-68-8.png)
![5-[TERT-BUTYL(DIPHENYL)SILYL]OXYPENT-3-YN-1-OL](http://img.cochemist.com/ccimg/117500/117471-68-8_b.png)


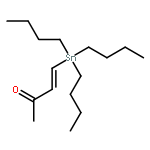
![Cyclohexanone, 2-[(tributylstannyl)methylene]-, (E)-](http://img.cochemist.com/ccimg/112100/112080-42-9.png)
![Cyclohexanone, 2-[(tributylstannyl)methylene]-, (E)-](http://img.cochemist.com/ccimg/112100/112080-42-9_b.png)











![2-Butyn-1-ol, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/86200/86120-46-9.png)
![2-Butyn-1-ol, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/86200/86120-46-9_b.png)
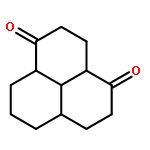
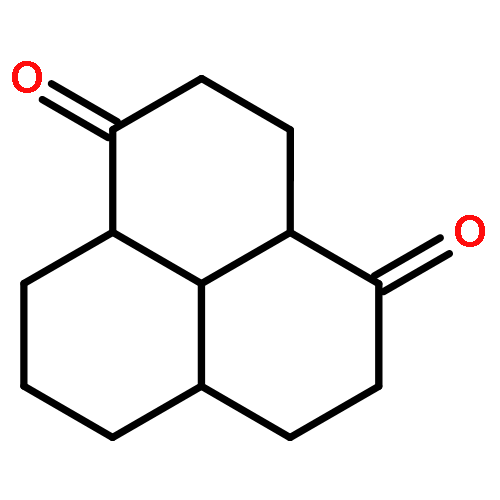
![(2E)-3-[(4S)-2,2-DiMethyl-1,3-dioxolan-4-yl]-2-Methyl-2-propenoic acid ethyl ester](http://img.cochemist.com/ccimg/82000/81997-76-4.png)
![(2E)-3-[(4S)-2,2-DiMethyl-1,3-dioxolan-4-yl]-2-Methyl-2-propenoic acid ethyl ester](http://img.cochemist.com/ccimg/82000/81997-76-4_b.png)
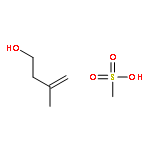
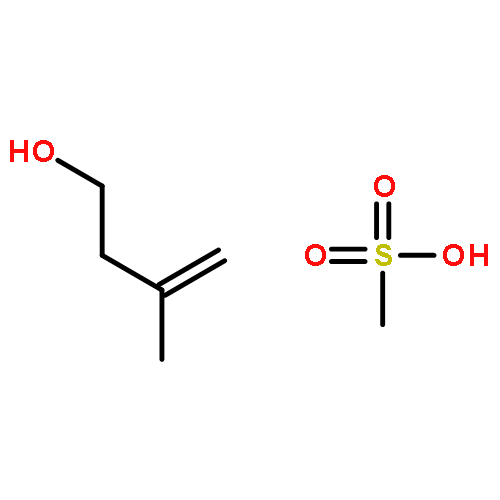



















![2-Cyclopenten-1-one,4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (4R)-](http://img.cochemist.com/ccimg/61400/61305-35-9.png)
![2-Cyclopenten-1-one,4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (4R)-](http://img.cochemist.com/ccimg/61400/61305-35-9_b.png)


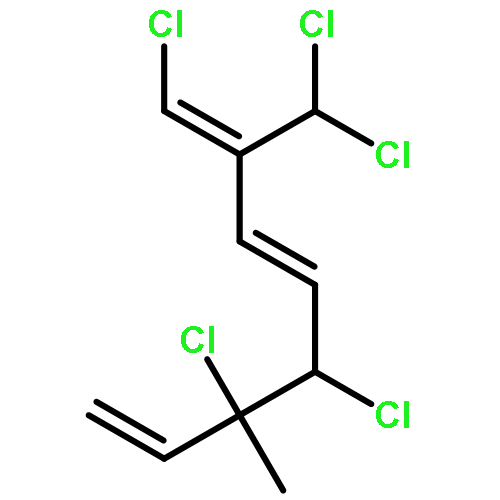

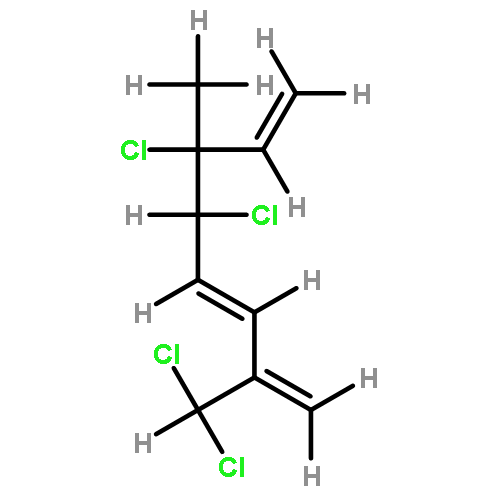
![2-Cyclopenten-1-one, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/56800/56745-67-6.png)
![2-Cyclopenten-1-one, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/56800/56745-67-6_b.png)
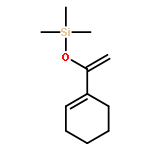
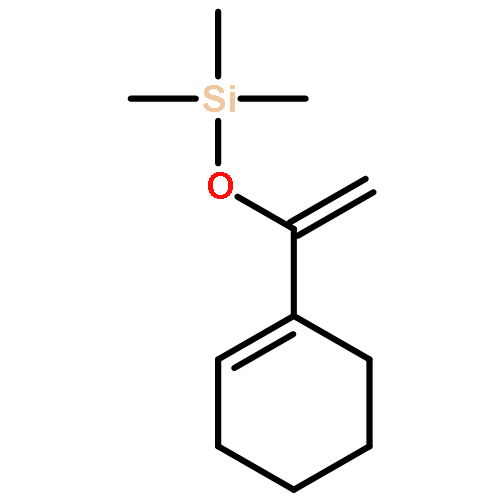













































![Bicyclo[7.2.0]undec-4-ene,4,11,11-trimethyl-8-methylene-](http://img.cochemist.com/ccimg/13900/13877-93-5.png)
![Bicyclo[7.2.0]undec-4-ene,4,11,11-trimethyl-8-methylene-](http://img.cochemist.com/ccimg/13900/13877-93-5_b.png)







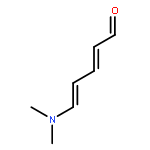
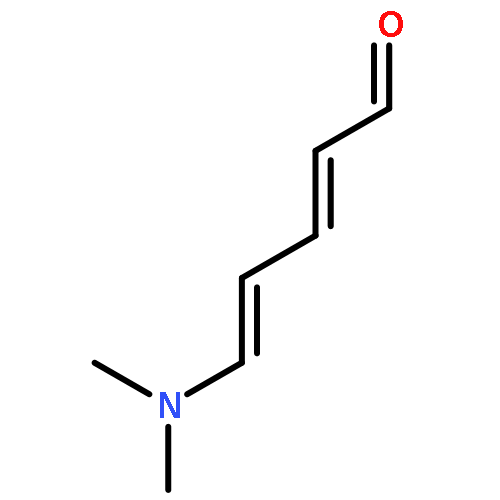
![Cyclohexanone, 2-[(dimethylamino)methylene]-](http://img.cochemist.com/ccimg/6200/6135-19-9.png)
![Cyclohexanone, 2-[(dimethylamino)methylene]-](http://img.cochemist.com/ccimg/6200/6135-19-9_b.png)























![(E)-3-[(6R)-6-HYDROXY-4-METHOXY-11-OXO-5,6,6A,7-TETRAHYDROPYRROLO[2,1-C][1,4]BENZODIAZEPIN-8-YL]-N,N-DIMETHYLPROP-2-ENAMIDE](http://img.cochemist.com/ccimg/110700/110652-73-8.png)
![(E)-3-[(6R)-6-HYDROXY-4-METHOXY-11-OXO-5,6,6A,7-TETRAHYDROPYRROLO[2,1-C][1,4]BENZODIAZEPIN-8-YL]-N,N-DIMETHYLPROP-2-ENAMIDE](http://img.cochemist.com/ccimg/110700/110652-73-8_b.png)
![2-Propenamide,N,N-dimethyl-3-[(11R,11aS)-5,10,11,11a-tetrahydro-9,11-dimethoxy-5-oxo-1H-pyrrolo[2,1-c][1,4]benzodiazepin-2-yl]-,(2E)-](http://img.cochemist.com/ccimg/110700/110652-72-7.png)
![2-Propenamide,N,N-dimethyl-3-[(11R,11aS)-5,10,11,11a-tetrahydro-9,11-dimethoxy-5-oxo-1H-pyrrolo[2,1-c][1,4]benzodiazepin-2-yl]-,(2E)-](http://img.cochemist.com/ccimg/110700/110652-72-7_b.png)



![Silane, [2-(iodomethyl)-2-propenyl]trimethyl-](http://img.cochemist.com/ccimg/80200/80121-73-9.png)
![Silane, [2-(iodomethyl)-2-propenyl]trimethyl-](http://img.cochemist.com/ccimg/80200/80121-73-9_b.png)









![13H-4,6:21,24-Dietheno-8,12-metheno-1H-pyrido[3',2':14,15][1,11]dioxacycloeicosino[2,3,4-ij]isoquinolinium,2,3,13a,14,15,16,25,25a-octahydro-9,19-dihydroxy-18,29-dimethoxy-1,14,14-trimethyl-,(13aR,25aS)-](http://img.cochemist.com/ccimg/100/57-95-4.png)
![13H-4,6:21,24-Dietheno-8,12-metheno-1H-pyrido[3',2':14,15][1,11]dioxacycloeicosino[2,3,4-ij]isoquinolinium,2,3,13a,14,15,16,25,25a-octahydro-9,19-dihydroxy-18,29-dimethoxy-1,14,14-trimethyl-,(13aR,25aS)-](http://img.cochemist.com/ccimg/100/57-95-4_b.png)