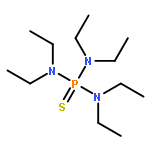
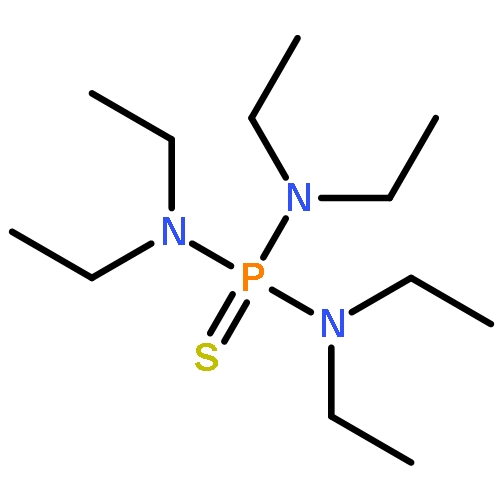
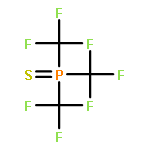
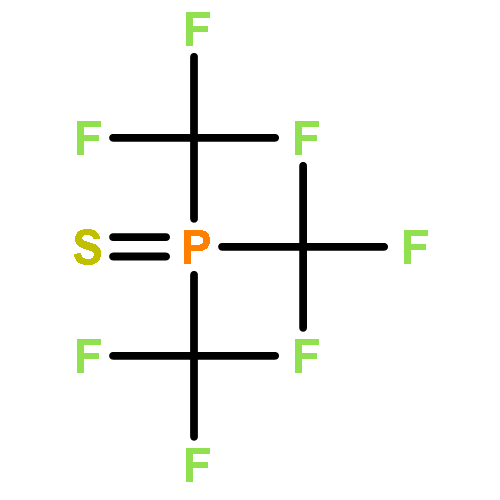
Co-reporter:Bryan A. Rosales, Michael P. Hanrahan, Brett W. Boote, Aaron J. Rossini, Emily A. Smith, and Javier Vela
ACS Energy Letters April 14, 2017 Volume 2(Issue 4) pp:906-906
Publication Date(Web):March 3, 2017
DOI:10.1021/acsenergylett.6b00674
Hybrid lead perovskites containing a mixture of organic and inorganic cations and anions have led to solar cell devices with performance and stability that are better than those of their single-halide analogs. 207Pb solid-state nuclear magnetic resonance and single-particle photoluminescence spectroscopies show that the structure and composition of mixed-halide and likely other hybrid lead perovskites are much more complex than previously thought and are highly dependent on their synthesis. While a majority of reports in the area focus on the construction of photovoltaic devices, this Perspective focuses instead on achieving a better understanding of the fundamental chemistry and photophysics of these materials, because this will aid not only in constructing improved devices but also in generating new uses for these unique materials.
Co-reporter:Chia-Cheng Lin;Shannon J. Tan
Journal of Materials Chemistry A 2017 vol. 5(Issue 38) pp:20351-20358
Publication Date(Web):2017/10/03
DOI:10.1039/C7TA02581E
Transition metal chalcogenide and pnictide nanocrystals are of interest for optoelectronic and catalytic applications. Here, we present a generalized route to the synthesis of these materials from the silylative deoxygenation of metal oxides with trimethylsilyl reagents. Specific nanophases produced in this way include Ni3S2, Ni5Se5, Ni2P, Co9S8, Co3Se4, CoP, Co2P, and heterobimetallic (Ni/Co)9S8. The resulting chalcogenide nanocrystals are hollow, likely due to differential rates of ion diffusion during the interfacial phase transformation reaction (Kirkendall-type effect). In contrast, the phosphide nanocrystals are solid, likely because they form at higher reaction temperatures. In all cases, simultaneous partial decomposition of the deoxygenating silyl reagent produces a coating of amorphous silica around the newly formed nanocrystals, which could impact their stability and recyclability.
Co-reporter:Long Men, Miles A. White, Himashi Andaraarachchi, Bryan A. Rosales, and Javier Vela
Chemistry of Materials 2017 Volume 29(Issue 1) pp:
Publication Date(Web):November 2, 2016
DOI:10.1021/acs.chemmater.6b02906
In this invited paper, we highlight some of our most recent work on the synthesis of low dimensional nanomaterials. Current graduate students and members of our group present four specific case systems: Nowotny–Juza phases, nickel phosphides, germanium-based core/shells, and organolead mixed-halide perovskites. Each system is accompanied by commentary from the student involved, which explains the motivation behind their work, as well as a protocol detailing the key experimental considerations involved in their synthesis. We trust these and similar efforts will help further advance our understanding of the broader field of synthetic nanomaterials chemistry, while, at the same time, highlighting how important this area is to the development of new materials for technologically relevant applications.
Co-reporter:Daniel J. Freppon;Long Men;Sadie J. Burkhow;Jacob W. Petrich;Emily A. Smith
Journal of Materials Chemistry C 2017 vol. 5(Issue 1) pp:118-126
Publication Date(Web):2016/12/22
DOI:10.1039/C6TC03886G
We present the time-correlated luminescence of isolated nanocrystals of five methylammonium lead mixed-halide perovskite compositions (CH3NH3PbBr3−xIx) that were synthesized with varying iodide and bromide anion loading. All analyzed nanocrystals had a spherical morphology with diameters in the range of 2 to 32 nm. The luminescence maxima of CH3NH3PbBr3−xIx nanocrystals were tuned to wavelengths ranging between 498 and 740 nm by varying the halide loading. Both CH3NH3PbI3 and CH3NH3PbBr3 nanocrystals exhibited no luminescence intermittency for more than 90% of the 250 s analysis time, as defined by a luminescence intensity three standard deviations above the background. The mixed halide CH3NH3PbBr0.75I0.25, CH3NH3PbBr0.50I0.50, and CH3NH3PbBr0.25I0.75 nanocrystals exhibited luminescence intermittency in 18%, 4% and 26% of the nanocrystals, respectively. Irrespective of luminescence intermittency, luminescence intensities were classified for each nanocrystal as: (a) constant, (b) multimodal, (c) photobrightening, and (d) photobleaching. Based on their photophysics, the CH3NH3PbBr3−xIx nanocrystals can be expected to be useful in a wide-range of applications where low and non-intermittent luminescence is desirable, for example as imaging probes and in films for energy conversion devices.
Co-reporter:Miles A. White, Gordon J. Miller, and Javier Vela
Journal of the American Chemical Society 2016 Volume 138(Issue 44) pp:14574-14577
Publication Date(Web):October 21, 2016
DOI:10.1021/jacs.6b10054
The first example of polytypism in the I–II–V semiconductors has been demonstrated with the synthesis of cubic LiZnSb by a low-temperature solution-phase method. This phase exhibits a unique coloring pattern that is novel for this class of compounds. The choice of site configuration has a considerable impact on the band structure of these materials, which in turn affects the transport properties. While the hexagonal polytype has been suggested as a promising n-type and extremely poor p-type thermoelectric material, the cubic analogue is calculated to have high efficiencies for both the n- and p-type derivatives (1.64 and 1.43, respectively, at 600 K). Furthermore, the cubic phase is found to be the energetically favored polytype. This surprising result provides a rationale for the lack of success in synthesizing the hexagonal polytype in either stoichiometric or n-type compositions.
Co-reporter:Michelle J. Thompson, Kyle J. Blakeney, Sarah D. Cady, Malinda D. Reichert, Joselyn Del Pilar-Albaladejo, Seth T. White, and Javier Vela
Chemistry of Materials 2016 Volume 28(Issue 6) pp:1668
Publication Date(Web):February 5, 2016
DOI:10.1021/acs.chemmater.5b04411
Because of its useful optoelectronic properties and the relative abundance of its elements, the quaternary semiconductor Cu2ZnSnS4 (CZTS) has garnered considerable interest in recent years. In this work, we dope divalent, high spin transition metal ions (M2+ = Mn2+, Co2+, Ni2+) into the tetrahedral Zn2+ sites of wurtzite CZTS nanorods. The resulting Cu2MxZn1–xSnS4 (CMTS) nanocrystals retain the hexagonal crystalline structure, elongated morphology, and broad visible light absorption profile of the undoped CZTS nanorods. Electron paramagnetic resonance (EPR), X-ray photoelectron spectroscopy (XPS), and infrared (IR) spectroscopy help corroborate the composition and local ion environment of the doped nanocrystals. EPR shows that, similarly to MnxCd1–xSe, washing Cu2MnxZn1–xSnS4 nanocrystals with trioctylphosphine oxide (TOPO) is an efficient way to remove excess Mn2+ ions from the particle surface. XPS and IR of as-isolated and thiol-washed samples show that, in contrast to binary chalcogenides, Cu2MnxZn1–xSnS4 nanocrystals aggregate not through dichalcogenide bonds, but through excess metal ions cross-linking the sulfur-rich surfaces of neighboring particles. Our results may help in expanding the synthetic applicability of CZTS and CMTS materials beyond photovoltaics and into the fields of spintronics and magnetic data storage.
Co-reporter:Bryan A. Rosales, Long Men, Sarah D. Cady, Michael P. Hanrahan, Aaron J. Rossini, and Javier Vela
Chemistry of Materials 2016 Volume 28(Issue 19) pp:6848
Publication Date(Web):July 25, 2016
DOI:10.1021/acs.chemmater.6b01874
Organolead mixed-halide perovskites such as CH3NH3PbX3–aX′a (X, X′ = I, Br, Cl) are interesting semiconductors because of their low cost, high photovoltaic power conversion efficiencies, enhanced moisture stability, and band gap tunability. Using a combination of optical absorption spectroscopy, powder X-ray diffraction (XRD), and, for the first time, 207Pb solid state nuclear magnetic resonance (ssNMR), we probe the extent of alloying and phase segregation in these materials. Because 207Pb ssNMR chemical shifts are highly sensitive to local coordination and electronic structure, and vary linearly with halogen electronegativity and band gap, this technique can provide the true chemical speciation and composition of organolead mixed-halide perovskites. We specifically investigate samples made by three different preparative methods: solution phase synthesis, thermal annealing, and solid phase synthesis. 207Pb ssNMR reveals that nonstoichiometric dopants and semicrystalline phases are prevalent in samples made by solution phase synthesis. We show that these nanodomains are persistent after thermal annealing up to 200 °C. Further, a novel solid phase synthesis that starts from the parent, single-halide perovskites can suppress phase segregation but not the formation of dopants. Our observations are consistent with the presence of miscibility gaps and spontaneous spinodal decomposition of the mixed-halide perovskites at room temperature. This underscores how strongly different synthetic procedures impact the nanostructuring and composition of organolead halide perovskites. Better optoelectronic properties and improved device stability and performance may be achieved through careful manipulation of the different phases and nanodomains present in these materials.
Co-reporter:Miles A. White, Michelle J. Thompson, Gordon J. Miller and Javier Vela
Chemical Communications 2016 vol. 52(Issue 17) pp:3497-3499
Publication Date(Web):28 Jan 2016
DOI:10.1039/C5CC09635A
We report the synthesis and characterization of nanocrystalline LiZnP. The reaction proceeds through a zinc metal intermediate followed by rapid incorporation of lithium and phosphorus. We demonstrate flexibility in the selection of Li, Zn, and P precursors, as well as extension of this method to other half-Heusler phases.
Co-reporter:Himashi P. Andaraarachchi, Michelle J. Thompson, Miles A. White, Hua-Jun Fan, and Javier Vela
Chemistry of Materials 2015 Volume 27(Issue 23) pp:8021
Publication Date(Web):November 11, 2015
DOI:10.1021/acs.chemmater.5b03506
A better understanding of the chemistry of molecular precursors is useful in achieving more predictable and reproducible nanocrystal preparations. Recently, an efficient approach was introduced that consists of fine-tuning the chemical reactivity of the synthetic molecular precursors used, while keeping all other reaction conditions constant. Using nickel phosphides as a research platform, we have studied how the chemical structure and reactivity of a family of commercially available organophosphite precursors (P(OR)3, R = alkyl or aryl) alter the preparation of metallic and metal phosphide nanocrystals. Organophosphites are a versatile addition to the pnictide synthetic toolbox, nicely complementing other available precursors such as elemental phosphorus or trioctylphosphine (TOP). Experimental and computational data show that different organophosphite precursors selectively yield Ni, Ni12P5, and Ni2P and that these phases evolve over time through separate mechanistic pathways. Based on our observations, we propose that nickel phosphide formation requires organophosphite coordination to a nickel precursor, followed by intramolecular rearrangement. We also propose that metallic nickel formation involves outer sphere reduction by uncoordinated organophosphite. These two independent pathways are supported by the fact that preformed Ni nanocrystals do not react with some of the most reactive phosphide-forming organophosphites, failing to evolve into nickel phosphide nanocrystals. Overall, the rate at which organophosphites react with nickel(II) chloride or acetate to form nickel phosphides increases in the order P(OMe)3 < P(OEt)3 < P(OnBu)3 < P(OCH2tBu)3 < P(OiPr)3 < P(OPh)3. Some organophosphites, such as P(OMe)3 or P(OiPr)3, transiently form zerovalent, metallic nickel, while this is the only persistent product observed with the bulky organophosphite P(O-2,4-tBu2C6H4)3. We expect that these results will alleviate the need for time-consuming testing and random optimization of several different reaction conditions, thus enabling a faster development of these and similar pnictide nanomaterials for practical applications.
Co-reporter:Chia-Cheng Lin, Yijun Guo, and Javier Vela
ACS Catalysis 2015 Volume 5(Issue 2) pp:1037
Publication Date(Web):December 24, 2014
DOI:10.1021/cs501650j
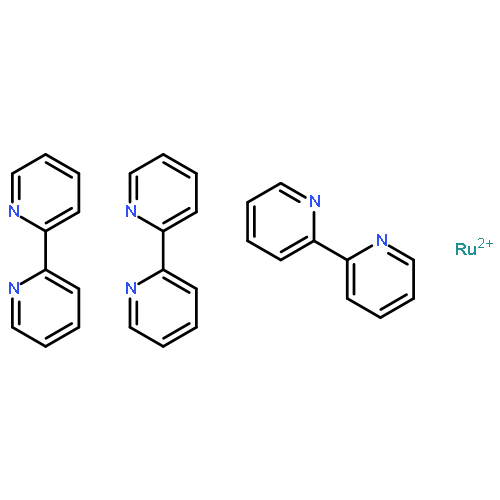
We investigate the effect of microstructuring on the water oxidation (oxygen evolution) activity of two types of Co3O4/porous silica composites: Co3O4/porous SiO2 core/shell nanoparticles with varying shell thicknesses and surface areas, and Co3O4/mesoporous silica nanocomposites with various surface functionalities. Catalytic tests in the presence of Ru(bpy)32+ as a photosensitizer and S2O82– as a sacrificial electron acceptor show that porous silica shells of up to ~20 nm in thickness lead to increased water oxidation activity. We attribute this effect to either (1) a combination of an effective increase in catalyst active area or consequent higher local concentration of Ru(bpy)32+; (2) a decrease in the permittivity of the medium surrounding the catalyst surface and a consequent increase in the rate of charge transfer; or both. Functionalized Co3O4/mesoporous silica nanocomposites show lower water oxidation activity compared with the parent nonfunctionalized catalyst, likely because of partial pore blocking of the silica support upon surface grafting. A more thorough understanding of the effects of microstructure and permittivity on water oxidation ability will enable the construction of next generation catalysts possessing optimal configuration and better efficiency for water splitting.Keywords: Co3O4/SiO2 core/shells; microstructure effects; nanocatalysts; nanocomposites; water oxidation
Co-reporter:Kuangcai Chen, Chia-Cheng Lin, Javier Vela, and Ning Fang
Analytical Chemistry 2015 Volume 87(Issue 8) pp:4096
Publication Date(Web):April 7, 2015
DOI:10.1021/acs.analchem.5b00604
Three-layer core–shell plasmonic nanorods (Au/Ag/SiO2–NRs), consisting of a gold nanorod core, a thin silver shell, and a thin silica layer, were synthesized and used as optical imaging probes under a differential interference contrast microscope for single particle orientation and rotational tracking. The localized surface plasmon resonance modes were enhanced upon the addition of the silver shell, and the anisotropic optical properties of gold nanorods were maintained. The silica coating enables surface functionalization with silane coupling agents and provides enhanced stability and biocompatibility. Taking advantage of the longitudinal LSPR enhancement, the orientation and rotational information of the hybrid nanorods on synthetic lipid bilayers and on live cell membranes were obtained with millisecond temporal resolution using a scientific complementary metal-oxide-semiconductor camera. The results demonstrate that the as-synthesized hybrid nanorods are promising imaging probes with improved sensitivity and good biocompatibility for single plasmonic particle tracking experiments in biological systems.
Co-reporter:Malinda D. Reichert, Miles A. White, Michelle J. Thompson, Gordon J. Miller, and Javier Vela
Inorganic Chemistry 2015 Volume 54(Issue 13) pp:6356-6362
Publication Date(Web):June 19, 2015
DOI:10.1021/acs.inorgchem.5b00679
Low-dimensional cuprous nitride (Cu3N) was synthesized by nitridation (ammonolysis) of cuprous oxide (Cu2O) nanocrystals using either ammonia (NH3) or urea (H2NCONH2) as the nitrogen source. The resulting nanocrystalline Cu3N spontaneously decomposes to nanocrystalline CuO in the presence of both water and oxygen from air at room temperature. Ammonia was produced in 60% chemical yield during Cu3N decomposition, as measured using the colorimetric indophenol method. Because Cu3N decomposition requires H2O and produces substoichiometric amounts of NH3, we conclude that this reaction proceeds through a complex stoichiometry that involves the concomitant release of both N2 and NH3. This is a thermodynamically unfavorable outcome, strongly indicating that H2O (and thus NH3 production) facilitate the kinetics of the reaction by lowering the energy barrier for Cu3N decomposition. The three different Cu2O, Cu3N, and CuO nanocrystalline phases were characterized by a combination of optical absorption, powder X-ray diffraction, transmission electron microscopy, and electronic density of states obtained from electronic structure calculations on the bulk solids. The relative ease of interconversion between these interesting and inexpensive materials bears possible implications for catalytic and optoelectronic applications.
Co-reporter:Samuel R. Alvarado, Ian A. Shortt, Hua-Jun Fan, and Javier Vela
Organometallics 2015 Volume 34(Issue 16) pp:4023-4031
Publication Date(Web):August 10, 2015
DOI:10.1021/acs.organomet.5b00428
Phosphine chalcogenides are useful reagents in chalcogen atom transfer reactions and nanocrystal syntheses. Understanding the strength and electronic structure of these bonds is key to optimizing their use, but a limited number of experimental and computational studies probe these issues. Using density functional theory (DFT), we computationally screen multiple series of trisubstituted phosphine chalcogenide molecules with a variety of phosphorus substituents and examine how these affect the strength of the phosphorus–chalcogen bond. DFT provides valuable data on these compounds including P–E bond dissociation energies, P–E bond order, Löwdin charge on phosphorus and chalcogen atoms, and molecular geometries. Experimentally monitoring the 31P and 77Se NMR chemical shifts and published Hammett constants provides good estimates and confirmation of the relative magnitude of electronic shielding around these nuclei and confirms the predictive value of the computational results.
Co-reporter:Feng Zhu, Long Men, Yijun Guo, Qiaochu Zhu, Ujjal Bhattacharjee, Peter M. Goodwin, Jacob W. Petrich, Emily A. Smith, and Javier Vela
ACS Nano 2015 Volume 9(Issue 3) pp:2948
Publication Date(Web):February 9, 2015
DOI:10.1021/nn507020s
Organometallic halide perovskites CH3NH3PbX3 (X = I, Br, Cl) have quickly become one of the most promising semiconductors for solar cells, with photovoltaics made of these materials reaching power conversion efficiencies of near 20%. Improving our ability to harness the full potential of organometal halide perovskites will require more controllable syntheses that permit a detailed understanding of their fundamental chemistry and photophysics. In this manuscript, we systematically synthesize CH3NH3PbX3 (X = I, Br) nanocrystals with different morphologies (dots, rods, plates or sheets) by using different solvents and capping ligands. CH3NH3PbX3 nanowires and nanorods capped with octylammonium halides show relatively higher photoluminescence (PL) quantum yields and long PL lifetimes. CH3NH3PbI3 nanowires monitored at the single particle level show shape-correlated PL emission across whole particles, with little photobleaching observed and very few off periods. This work highlights the potential of low-dimensional organometal halide perovskite semiconductors in constructing new porous and nanostructured solar cell architectures, as well as in applying these materials to other fields such as light-emitting devices and single particle imaging and tracking.Keywords: morphology control; nanocrystals; organometal halide perovskites; preferred orientation; single particle photoluminescence; size control;
Co-reporter:Samuel R. Alvarado, Yijun Guo, T. Purnima A. Ruberu, Elham Tavasoli, Javier Vela
Coordination Chemistry Reviews 2014 Volumes 263–264() pp:182-196
Publication Date(Web):15 March 2014
DOI:10.1016/j.ccr.2013.09.001
•Molecular precursor reactivity dictates the rate of nucleation of different nanocrystalline phases.•Suppressing homogeneous nucleation enables the growth of nonblinking ‘giant’ nanocrystals.•Metal deposition stabilizes and activates semiconductors in photocatalytic deoxygenation.•Surface ligand doping and organization controls nanocrystal activity and interfacial properties.The optoelectronic and chemical properties of semiconductor nanocrystals heavily depend on their composition, size, shape and internal structure, surface functionality, etc. Available strategies to alter these properties through traditional colloidal syntheses and ligand exchange methods place a premium on specific reaction conditions and surfactant combinations. In this invited review, we apply a molecular-level understanding of chemical precursor reactivity to reliably control the morphology, composition and intimate architecture (core/shell vs. alloyed) of semiconductor nanocrystals. We also describe our work aimed at achieving highly selective, low-temperature photochemical methods for the synthesis of semiconductor–metal and semiconductor–metal oxide photocatalytic nanocomposites. In addition, we describe our work on surface modification of semiconductor nanocrystal quantum dots using new approaches and methods that bypass ligand exchange, retaining the nanocrystal's native ligands and original optical properties, as well as on spectroscopic methods of characterization useful in determining surface ligand organization and chemistry. Using recent examples from our group and collaborators, we demonstrate how these efforts have lead to faster, wider and more systematic application of semiconductor nanocrystal-based materials to biological imaging and tracking, and to photocatalysis of unconventional substrates. We believe techniques and methods borrowed from inorganic chemistry (including coordination, organometallic and solid state chemistry) have much to offer in reaching a better understanding of the synthesis, functionalization and real-life application of such exciting materials as semiconductor nanocrystals (quantum dots, rods, tetrapods, etc.).
Co-reporter:Ji Won Ha ; T. Purnima A. Ruberu ; Rui Han ; Bin Dong ; Javier Vela ;Ning Fang
Journal of the American Chemical Society 2014 Volume 136(Issue 4) pp:1398-1408
Publication Date(Web):January 12, 2014
DOI:10.1021/ja409011y
Metal–semiconductor heterostructures are promising visible light photocatalysts for many chemical reactions. Here, we use high-resolution superlocalization imaging to reveal the nature and photocatalytic properties of the surface reactive sites on single Au–CdS hybrid nanocatalysts. We experimentally reveal two distinct, incident energy-dependent charge separation mechanisms that result in completely opposite photogenerated reactive sites (e– and h+) and divergent energy flows on the hybrid nanocatalysts. We find that plasmon-induced hot electrons in Au are injected into the conduction band of the CdS semiconductor nanorod. The specifically designed Au-tipped CdS heterostructures with a unique geometry (two Au nanoparticles at both ends of each CdS nanorod) provide more convincing high-resolution single-turnover mapping results and clearly prove the two charge separation mechanisms. Engineering the direction of energy flow at the nanoscale can provide an efficient way to overcome important challenges in photocatalysis, such as controlling catalytic activity and selectivity. These results bear enormous potential impact on the development of better visible light photocatalysts for solar-to-chemical energy conversion.
Co-reporter:Malinda D. Reichert, Chia-Cheng Lin, and Javier Vela
Chemistry of Materials 2014 Volume 26(Issue 13) pp:3900
Publication Date(Web):June 2, 2014
DOI:10.1021/cm500896n
Anisotropic II–VI semiconductor nanostructures are important photoactive materials for various energy conversion and optical applications. However, aside from the many available surface chemistry studies and from their ubiquitous photodegradation under continuous illumination, the general chemical reactivity and thermal stability (phase and shape transformations) of these materials are poorly understood. Using CdSe and CdS nanorods as model systems, we have investigated the behavior of II–VI semiconductor nanorods against various conditions of extreme chemical and physical stress (acids, bases, oxidants, reductants, and heat). CdSe nanorods react rapidly with acids, becoming oxidized to Se or SeO2. In contrast, CdSe nanorods remain mostly unreactive when treated with bases or strong oxidants, although bases do partially etch the tips of the nanorods (along their axis). Roasting (heating in air) of CdSe nanorods results in rock-salt CdO, but neither CdSe nor CdO is easily reduced by hydrogen (H2). Another reductant, n-BuLi, reduces CdSe nanorods to metallic Cd. Variable temperature X-ray diffraction experiments show that axial annealing and selective axial melting of the nanorods precede particle coalescence. Furthermore, thermal analysis shows that the axial melting of II–VI nanorods is a ligand-dependent process. In agreement with chemical reactivity and thermal stability observations, silica-coating experiments show that the sharpest (most curved) II–VI surfaces are most active against heterogeneous nucleation of a silica shell. These results provide valuable insights into the fate and possible ways to enhance the stability and improve the use of II–VI semiconductor nanostructures in the fields of optics, magnetism, and energy conversion.
Co-reporter:Patrick S. Dilsaver ; Malinda D. Reichert ; Brittany L. Hallmark ; Michelle J. Thompson
The Journal of Physical Chemistry C 2014 Volume 118(Issue 36) pp:21226-21234
Publication Date(Web):August 19, 2014
DOI:10.1021/jp5062336
Chalcogenide-based semiconductor–metal heterostructures are interesting catalysts for solar-to-chemical energy conversion, but current compositions are impractical due to the relative toxicity and/or scarcity of their constituent elements. To address these concerns, Cu2ZnSnS4 (CZTS) emerged as an interesting alternative to other chalcogenide-based semiconductors; however, the fabrication of CZTS-metal heterostructures remains unexplored. In this paper, we systematically explore four methods of synthesizing CZTS-Au heterostructures, specifically: reaction of CZTS nanorods with either a soluble molecular gold precursor (AuCl3) or preformed gold (Au) nanoparticles, each under thermal (heating in the dark) or photochemical reaction conditions (350 nm lamp illumination at room temperature). We find that using AuCl3 under thermal deposition conditions results in the most well-defined CZTS-Au heterostructures, containing >99% surface-bound 2.1 ± 0.5 nm Au islands along the whole length of the nanorod. These CZTS-Au heterostructures are photocatalytically active, reducing the model compound methylene blue upon irradiation much more effectively than bare CZTS nanorods. We also demonstrate the removal of Au from the CZTS-Au heterostructures by amalgamation. These results open up a new area of greener, CZTS-based photocatalysts for solar-to-chemical energy conversion.
Co-reporter:Yijun Guo, Clare E. Rowland, Richard D. Schaller, and Javier Vela
ACS Nano 2014 Volume 8(Issue 8) pp:8334
Publication Date(Web):July 10, 2014
DOI:10.1021/nn502792m
Ge nanocrystals have a large Bohr radius and a small, size-tunable band gap that may engender direct character via strain or doping. Colloidal Ge nanocrystals are particularly interesting in the development of near-infrared materials for applications in bioimaging, telecommunications and energy conversion. Epitaxial growth of a passivating shell is a common strategy employed in the synthesis of highly luminescent II–VI, III–V and IV–VI semiconductor quantum dots. Here, we use relatively unexplored IV/II–VI epitaxy as a way to enhance the photoluminescence and improve the optical stability of colloidal Ge nanocrystals. Selected on the basis of their relatively small lattice mismatch compared with crystalline Ge, we explore the growth of epitaxial CdS and ZnS shells using the successive ion layer adsorption and reaction method. Powder X-ray diffraction and electron microscopy techniques, including energy dispersive X-ray spectroscopy and selected area electron diffraction, clearly show the controllable growth of as many as 20 epitaxial monolayers of CdS atop Ge cores. In contrast, Ge etching and/or replacement by ZnS result in relatively small Ge/ZnS nanocrystals. The presence of an epitaxial II–VI shell greatly enhances the near-infrared photoluminescence and improves the photoluminescence stability of Ge. Ge/II–VI nanocrystals are reproducibly 1–3 orders of magnitude brighter than the brightest Ge cores. Ge/4.9CdS core/shells show the highest photoluminescence quantum yield and longest radiative recombination lifetime. Thiol ligand exchange easily results in near-infrared active, water-soluble Ge/II–VI nanocrystals. We expect this synthetic IV/II–VI epitaxial approach will lead to further studies into the optoelectronic behavior and practical applications of Si and Ge-based nanomaterials.Keywords: core/shell nanocrystals; germanium; IV/II−VI epitaxy; near-IR photoluminescence; quantum dots
Co-reporter:Yijun Guo, Samuel R. Alvarado, Joshua D. Barclay, and Javier Vela
ACS Nano 2013 Volume 7(Issue 4) pp:3616
Publication Date(Web):March 21, 2013
DOI:10.1021/nn400596e
Dialkyl and diaryl dichalcogenides are highly versatile and modular precursors for the synthesis of colloidal chalcogenide nanocrystals. We have used a series of commercially available dichalcogenide precursors to unveil the molecular basis for the outcome of nanocrystal preparations, more specifically, how precursor molecular structure and reactivity affect the final shape and size of II–VI semiconductor nanocrystals. Dichalcogenide precursors used were diallyl, dibenzyl, di-tert-butyl, diisopropyl, diethyl, dimethyl, and diphenyl disulfides and diethyl, dimethyl, and diphenyl diselenides. We find that the presence of two distinctively reactive C–E and E–E bonds makes the chemistry of these precursors much richer and interesting than that of other conventional precursors such as the more common phosphine chalcogenides. Computational studies (DFT) reveal that the dissociation energy of carbon–chalcogen (C–E) bonds in dichalcogenide precursors (R–E–E–R, E = S or Se) increases in the order (R): diallyl < dibenzyl < di-tert-butyl < diisopropyl < diethyl < dimethyl < diphenyl. The dissociation energy of chalcogen–chalcogen (E–E) bonds remains relatively constant across the series. The only exceptions are diphenyl dichalcogenides, which have a much lower E–E bond dissociation energy. An increase in C–E bond dissociation energy results in a decrease in R–E–E–R precursor reactivity, leading to progressively slower nucleation and higher selectivity for anisotropic growth, all the way from dots to pods to tetrapods. Under identical experimental conditions, we obtain CdS and CdSe nanocrystals with spherical, elongated, or tetrapodal morphology by simply varying the identity and reactivity of the dichalcogenide precursor. Interestingly, we find that precursors with strong C–E and weak E–E bond dissociation energies such as Ph–S–S–Ph serve as a ready source of thiol radicals that appear to stabilize small CdE nuclei, facilitating anisotropic growth. These CdS and CdSe nanocrystals have been characterized using structural and spectroscopic methods. An intimate understanding of how molecular structure affects the chemical reactivity of molecular precursors enables highly predictable and reproducible synthesis of colloidal nanocrystals with specific sizes, shapes, and optoelectronic properties for customized applications.Keywords: anisotropic structures; bond dissociation energies; dichalcogenide precursors; morphology control; selective growth
Co-reporter:Nicholas C. Nelson, T. Purnima A. Ruberu, Malinda D. Reichert, and Javier Vela
The Journal of Physical Chemistry C 2013 Volume 117(Issue 48) pp:25826-25836
Publication Date(Web):November 20, 2013
DOI:10.1021/jp409878a
One-dimensional transition metal nanostructures are of interest in many magnetic and catalytic applications. Using a combination of wet chemical synthesis, optical (infrared), and structural characterization methods (powder X-ray diffraction, scanning and transmission electron microscopy), we have investigated four paths to access 1D nickel nanostructures: (1) direct chemical reduction of a self-assembled nickel–hydrazine coordination complex, (2) thermal decomposition of the silica encapsulated nickel–hydrazine complex, (3) treatment of the silica encapsulated nickel–hydrazine complex with sodium borohydride followed by thermal annealing, and (4) electroless nickel plating using silica encapsulated nickel seed particles. We find that only route 1, which does not require a silica template, results in the formation of nickel nanorods, albeit some particle aggregation is observed. Routes 2 and 3 result in the formation of isotropic nickel structures under a reducing atmosphere. Route 4 results in heterogeneous nucleation and growth of existing particles only when partial etching of the silica capsule occurs. Detailed examination of the encapsulated nickel particles allows studying the effect of silica surface silanols on the oxidation of encapsulated nickel particles, the presence of nanoparticle–silica support interactions, the sintering mechanism of nickel and nickel oxide particles, and the fate of boride impurities. Nickel/silica nanostructures are strongly magnetic at room temperature.
Co-reporter:Michelle J. Thompson, T. Purnima A. Ruberu, Kyle J. Blakeney, Karen V. Torres, Patrick S. Dilsaver, and Javier Vela
The Journal of Physical Chemistry Letters 2013 Volume 4(Issue 22) pp:3918-3923
Publication Date(Web):November 5, 2013
DOI:10.1021/jz402048p
Cu2ZnSnS4 (CZTS) is a promising material for solar energy conversion, but synthesis of phase-pure, anisotropic CZTS nanocrystals remains a challenge. We demonstrate that the initial concentration (loading) of cationic precursors has a dramatic effect on the morphology (aspect ratio) and composition (internal architecture) of hexagonal wurtzite CZTS nanorods. Our experiments strongly indicate that Cu is the most reactive of the metal cations; Zn is next, and Sn is the least reactive. Using this reactivity series, we are able to purposely fine-tune the morphology (dots versus rods) and degree of axial phase segregation of CZTS nanocrystals. These results will improve our ability to fabricate CZTS nanostructures for photovoltaics and photocatalysis.Keywords: composition control; CZTS; precursor loading; shape control;
Co-reporter:Javier Vela
The Journal of Physical Chemistry Letters 2013 Volume 4(Issue 4) pp:653-668
Publication Date(Web):February 1, 2013
DOI:10.1021/jz302100r
Colloidal semiconductor nanocrystals possess unique properties that are unmatched by other chromophores such as organic dyes or transition-metal complexes. These versatile building blocks have generated much scientific interest and found applications in bioimaging, tracking, lighting, lasing, photovoltaics, photocatalysis, thermoelectrics, and spintronics. Despite these advances, important challenges remain, notably how to produce semiconductor nanostructures with predetermined architecture, how to produce metastable semiconductor nanostructures that are hard to isolate by conventional syntheses, and how to control the degree of surface loading or valence per nanocrystal. Molecular chemists are very familiar with these issues and can use their expertise to help solve these challenges. In this Perspective, we present our group’s recent work on bottom-up molecular control of nanoscale composition and morphology, low-temperature photochemical routes to semiconductor heterostructures and metastable phases, solar-to-chemical energy conversion with semiconductor-based photocatalysts, and controlled surface modification of colloidal semiconductors that bypasses ligand exchange.
Co-reporter:Elham Tavasoli, Yijun Guo, Pranaw Kunal, Javier Grajeda, Allison Gerber, and Javier Vela
Chemistry of Materials 2012 Volume 24(Issue 21) pp:4231
Publication Date(Web):October 3, 2012
DOI:10.1021/cm3026957
One remaining challenge in the field of colloidal semiconductor nanocrystal quantum dots is learning to control the degree of functionalization or “valence” per nanocrystal. Current quantum dot surface modification strategies rely heavily on ligand exchange, which consists of replacing the nanocrystal’s native ligands with carboxylate- or amine-terminated thiols, usually added in excess. Removing the nanocrystal’s native ligands can cause etching and introduce surface defects, thus affecting the nanocrystal’s optical properties. More importantly, ligand exchange methods fail to control the extent of surface modification or number of functional groups introduced per nanocrystal. Here, we report a fundamentally new surface ligand modification or “doping” approach aimed at controlling the degree of functionalization or valence per nanocrystal while retaining the nanocrystal’s original colloidal and photostability. We show that surface-doped quantum dots capped with chemically active native ligands can be prepared directly from a mixture of ligands with similar chain lengths. Specifically, vinyl and azide-terminated carboxylic acid ligands survive the high temperatures needed for nanocrystal synthesis. The ratio between chemically active and inactive-terminated ligands is maintained on the nanocrystal surface, allowing to control the extent of surface modification by straightforward organic reactions. Using a combination of optical and structural characterization tools, including IR and 2D NMR, we show that carboxylates bind in a bidentate chelate fashion, forming a single monolayer of ligands that are perpendicular to the nanocrystal surface. Moreover, we show that mixtures of ligands with similar chain lengths homogeneously distribute themselves on the nanocrystal surface. We expect this new surface doping approach will be widely applicable to other nanocrystal compositions and morphologies, as well as to many specific applications in biology and materials science.Keywords: chemical surface modification; loading; quantum dot; valence;
Co-reporter:T. Purnima A. Ruberu, Nicholas C. Nelson, Igor I. Slowing, and Javier Vela
The Journal of Physical Chemistry Letters 2012 Volume 3(Issue 19) pp:2798-2802
Publication Date(Web):September 13, 2012
DOI:10.1021/jz301309d
Photocatalytic conversion of biomass is a potentially transformative concept in renewable energy. Dehydrogenation and hydrogenolysis of biomass-derived alcohols can produce renewable fuels such as H2 and hydrocarbons, respectively. We have successfully used semiconductor-metal heterostructures for sunlight-driven dehydrogenation and hydrogenolysis of benzyl alcohol. The heterostructure composition dictates activity, product distribution, and turnovers. A few metal (M = Pt, Pd) islands on the semiconductor (SC) surface significantly enhance activity and selectivity and also greatly stabilize the SC against photoinduced etching and degradation. Under selected conditions, CdS-Pt favors dehydrogenation (H2) over hydrogenolysis (toluene) 8:1, whereas CdS0.4Se0.6-Pd favors hydrogenolysis over dehydrogenation 3:1. Photochemically generated, surface-adsorbed hydrogen is useful in tandem catalysis, for example, via transfer hydrogenation. We expect this work will lead to new paradigms for sunlight-driven conversions of biomass-relevant substrates.Keywords: alcohol dehydrogenation; biomass; hydrogenolysis; photocatalysis; semiconductor-metal heterostructures;
Co-reporter:Yijun Guo ; Kyle Marchuk ; Siddharth Sampat ; Rachel Abraham ; Ning Fang ; Anton V. Malko
The Journal of Physical Chemistry C 2012 Volume 116(Issue 4) pp:2791-2800
Publication Date(Web):December 23, 2011
DOI:10.1021/jp210949v
Thick-shell CdSe/nCdS (n ≥ 10) nanocrystals were recently reported that show remarkably suppressed fluorescence intermittency or “blinking” at the single-particle level as well as slow rates of Auger decay. Unfortunately, whereas CdSe/nCdS nanocrystal synthesis is well-developed up to n ≤ 6 CdS monolayers (MLs), reproducible syntheses for n ≥ 10 MLs are less understood. Known procedures sometimes result in homogeneous CdS nucleation instead of heterogeneous, epitaxial CdS nucleation on CdSe, leading to broad and multimodal particle size distributions. Critically, obtained core/shell sizes are often below those desired. This article describes synthetic conditions specific to thick-shell growth (n ≥ 10 and n ≥ 20 MLs) on both small (sub2 nm) and large (>4.5 nm) CdSe cores. We find added secondary amine and low concentration of CdSe cores and molecular precursors give desired core/shell sizes. Amine-induced, partial etching of CdSe cores results in apparent shell-thicknesses slightly beyond those desired, especially for very-thick shells (n ≥ 20 MLs). Thermal ripening and fast precursor injection lead to undesired homogeneous CdS nucleation and incomplete shell growth. Core/shells derived from small CdSe (1.9 nm) have longer PL lifetimes and more pronounced blinking at single-particle level compared with those derived from large CdSe (4.7 nm). We expect our new synthetic approach will lead to a larger throughput of these materials, increasing their availability for fundamental studies and applications.
Co-reporter:T. Purnima A. Ruberu, Haley R. Albright, Brandon Callis, Brittney Ward, Joana Cisneros, Hua-Jun Fan, and Javier Vela
ACS Nano 2012 Volume 6(Issue 6) pp:5348
Publication Date(Web):April 22, 2012
DOI:10.1021/nn301182h
We demonstrate molecular control of nanoscale composition, alloying, and morphology (aspect ratio) in CdS–CdSe nanocrystal dots and rods by modulating the chemical reactivity of phosphine–chalcogenide precursors. Specific molecular precursors studied were sulfides and selenides of triphenylphosphite (TPP), diphenylpropylphosphine (DPP), tributylphosphine (TBP), trioctylphosphine (TOP), and hexaethylphosphorustriamide (HPT). Computational (DFT), NMR (31P and 77Se), and high-temperature crossover studies unambiguously confirm a chemical bonding interaction between phosphorus and chalcogen atoms in all precursors. Phosphine–chalcogenide precursor reactivity increases in the order: TPPE < DPPE < TBPE < TOPE < HPTE (E = S, Se). For a given phosphine, the selenide is always more reactive than the sulfide. CdS1–xSex quantum dots were synthesized via single injection of a R3PS–R3PSe mixture to cadmium oleate at 250 °C. X-ray diffraction (XRD), transmission electron microscopy (TEM), and UV/Vis and PL optical spectroscopy reveal that relative R3PS and R3PSe reactivity dictates CdS1–xSex dot chalcogen content and the extent of radial alloying (alloys vs core/shells). CdS, CdSe, and CdS1–xSex quantum rods were synthesized by injection of a single R3PE (E = S or Se) precursor or a R3PS–R3PSe mixture to cadmium–phosphonate at 320 or 250 °C. XRD and TEM reveal that the length-to-diameter aspect ratio of CdS and CdSe nanorods is inversely proportional to R3PE precursor reactivity. Purposely matching or mismatching R3PS–R3PSe precursor reactivity leads to CdS1–xSex nanorods without or with axial composition gradients, respectively. We expect these observations will lead to scalable and highly predictable “bottom-up” programmed syntheses of finely heterostructured nanomaterials with well-defined architectures and properties that are tailored for precise applications.Keywords: molecular control; nanocrystal composition; nanorod aspect ratio; precursor reactivity
Co-reporter:Samuel R. Alvarado, Yijun Guo, T. Purnima A. Ruberu, Andreja Bakac, and Javier Vela
The Journal of Physical Chemistry C 2012 Volume 116(Issue 18) pp:10382-10389
Publication Date(Web):April 18, 2012
DOI:10.1021/jp301459s
Photochemical methods facilitate the generation, isolation, and study of metastable nanomaterials having unusual size, composition, and morphology. These harder-to-isolate and highly reactive phases, inaccessible using conventional high-temperature pyrolysis, are likely to possess enhanced and unprecedented chemical, electromagnetic, and catalytic properties. We report a fast, low-temperature and scalable photochemical route to synthesize very small (∼3 nm) monodisperse cobalt oxyhydroxide (Co(O)OH) nanocrystals. This method uses readily and commercially available pentaamminechlorocobalt(III) chloride, [Co(NH3)5Cl]Cl2, under acidic or neutral pH and proceeds under either near-UV (350 nm) or Vis (575 nm) illumination. Control experiments showed that the reaction proceeds at competent rates only in the presence of light, does not involve a free radical mechanism, is insensitive to O2, and proceeds in two steps: (1) Aquation of [Co(NH3)5Cl]2+ to yield [Co(NH3)5(H2O)]3+, followed by (2) slow photoinduced release of NH3 from the aqua complex. This reaction is slow enough for Co(O)OH to form but fast enough so that nanocrystals are small (ca. 3 nm). The alternative dark thermal reaction proceeds much more slowly and produces much larger (∼250 nm) polydisperse Co(O)OH aggregates. UV–Vis absorption measurements and ab initio calculations yield a Co(O)OH band gap of 1.7 eV. Fast thermal annealing of Co(O)OH nanocrystals leads to Co3O4 nanocrystals with overall retention of nanoparticle size and morphology. Thermogravimetric analysis shows that oxyhydroxide to mixed-oxide phase transition occurs at significantly lower temperatures (up to ΔT = 64 °C) for small nanocrystals compared with the bulk.
Co-reporter:Mussie G. Alemseghed, T. Purnima A. Ruberu, and Javier Vela
Chemistry of Materials 2011 Volume 23(Issue 15) pp:3571
Publication Date(Web):July 13, 2011
DOI:10.1021/cm201513a
Reliable synthesis of semiconductor–metal heterostructures would increase their availability for fundamental studies and applications in catalytic, magnetic, and opto-electronic devices. Here, we demonstrate there are three main pathways for the formation of Pt and Pd nanoparticles on CdS and CdS0.4Se0.6 nanorods. A thermal pathway and photochemical pathway occur when the metal precursor is heated or irradiated directly in the presence of an electron donor, leading to homogeneous nucleation and formation of freestanding metal nanoparticles. A separate photochemical pathway occurs in the presence of semiconductor nanorods, leading to exciton formation and quenching by electron trapping at surface defect sites. The localized electrons act as seeding points, leading to heterogeneous nucleation and formation of surface-bound metal nanoparticles. Careful selection of synthetic conditions allows deposition of Pt and Pd particles on CdS and CdS0.4Se0.6 nanorods with a high degree of selectivity (90–95% surface-bound obtained photochemically) over the formation of freestanding metal particles (70–94% unattached under thermal conditions). In addition, metal photo deposition occurs on specific segments of CdS0.4Se0.6 nanorods with compositional anisotropy by taking advantage of the band gap differential between different nanodomains. Irradiation at short wavelengths favors formation of Pd nanoparticles on the large band gap CdS-rich region of the nanorods (57% and 55% at 350 and 420 nm, respectively), while irradiation at longer wavelengths favors the formation of Pd nanoparticles on the small band gap CdSe-rich region of the nanorods (83% at 575 nm). The ability to tune the spatial composition of these and similar heterostructures will impact the ability to engineer and direct energy flows at the nanoscale.Keywords: heterostructure; hybrid; metal; photo deposition; semiconductor; site selectivity;
Co-reporter:T. Purnima A. Ruberu and Javier Vela
ACS Nano 2011 Volume 5(Issue 7) pp:5775
Publication Date(Web):June 2, 2011
DOI:10.1021/nn201466b
We report the synthesis and characterization of CdS1–xSex nanorods with axial anisotropy. These nanorods were synthesized via single injection of a mixture of trioctylphosphine sulfur and selenium precursors to a cadmium–phosphonate complex at high temperature. Transmission electron microscopy shows nanoparticle morphology changes with relative sulfur and selenium loading. When the synthetic selenium loading is between 5% and 10% of total chalcogenides, the nanorods exhibit pronounced axial anisotropy characterized by a thick “head” and a thin “tail”. The nanorods’ band gap red shifts with increasing selenium loading. X-ray diffraction reveals that CdS1–xSex nanorods have a wurtzite crystal structure with a certain degree of alloying. High-resolution and energy-filtered transmission electron microscopy and energy-dispersive X-ray spectroscopy confirm the head of the anisotropic nanorods is rich in selenium, whereas the tail is rich in sulfur. Time evolution and mechanistic studies confirm the nanorods form by quick growth of the CdSe-rich head, followed by slow growth of the CdS-rich tail. Metal photodeposition reactions with 575 nm irradiation, which is mostly absorbed by the CdSe-rich segment, show effective electronic communication between the nanorod head and tail segments.Keywords: axial anisotropy; cadmium chalcogenide; graded alloy; heterostructure; nanorod
Co-reporter:Miles A. White, Michelle J. Thompson, Gordon J. Miller and Javier Vela
Chemical Communications 2016 - vol. 52(Issue 17) pp:NaN3499-3499
Publication Date(Web):2016/01/28
DOI:10.1039/C5CC09635A
We report the synthesis and characterization of nanocrystalline LiZnP. The reaction proceeds through a zinc metal intermediate followed by rapid incorporation of lithium and phosphorus. We demonstrate flexibility in the selection of Li, Zn, and P precursors, as well as extension of this method to other half-Heusler phases.
Co-reporter:Daniel J. Freppon, Long Men, Sadie J. Burkhow, Jacob W. Petrich, Javier Vela and Emily A. Smith
Journal of Materials Chemistry A 2017 - vol. 5(Issue 1) pp:NaN126-126
Publication Date(Web):2016/11/25
DOI:10.1039/C6TC03886G
We present the time-correlated luminescence of isolated nanocrystals of five methylammonium lead mixed-halide perovskite compositions (CH3NH3PbBr3−xIx) that were synthesized with varying iodide and bromide anion loading. All analyzed nanocrystals had a spherical morphology with diameters in the range of 2 to 32 nm. The luminescence maxima of CH3NH3PbBr3−xIx nanocrystals were tuned to wavelengths ranging between 498 and 740 nm by varying the halide loading. Both CH3NH3PbI3 and CH3NH3PbBr3 nanocrystals exhibited no luminescence intermittency for more than 90% of the 250 s analysis time, as defined by a luminescence intensity three standard deviations above the background. The mixed halide CH3NH3PbBr0.75I0.25, CH3NH3PbBr0.50I0.50, and CH3NH3PbBr0.25I0.75 nanocrystals exhibited luminescence intermittency in 18%, 4% and 26% of the nanocrystals, respectively. Irrespective of luminescence intermittency, luminescence intensities were classified for each nanocrystal as: (a) constant, (b) multimodal, (c) photobrightening, and (d) photobleaching. Based on their photophysics, the CH3NH3PbBr3−xIx nanocrystals can be expected to be useful in a wide-range of applications where low and non-intermittent luminescence is desirable, for example as imaging probes and in films for energy conversion devices.
.jpg)
![(3aR,8R,10aR,10bS,10cR)-8-ethenyl-3-hydroxy-3a,8-dimethyl-2,3,3a,5a,7,8,9,10,10a,10c-decahydro-1H,4H-3,10b-(epoxymethano)phenanthro[10,1-bc]furan-4-one](http://img.cochemist.com/ccimg/51500/51415-08-8.png)
![(3aR,8R,10aR,10bS,10cR)-8-ethenyl-3-hydroxy-3a,8-dimethyl-2,3,3a,5a,7,8,9,10,10a,10c-decahydro-1H,4H-3,10b-(epoxymethano)phenanthro[10,1-bc]furan-4-one](http://img.cochemist.com/ccimg/51500/51415-08-8_b.png)
![1H-Phenanthro[10,1-bc]furan-3,4(2H,3aH)-dione,8-ethenyl-5a,7,8,9,10,10a,10b,10c-octahydro-3a,8,10b-trimethyl-,(3aR,5aR,8R,10aR,10bR,10cR)-](http://img.cochemist.com/ccimg/51500/51415-07-7.png)
![1H-Phenanthro[10,1-bc]furan-3,4(2H,3aH)-dione,8-ethenyl-5a,7,8,9,10,10a,10b,10c-octahydro-3a,8,10b-trimethyl-,(3aR,5aR,8R,10aR,10bR,10cR)-](http://img.cochemist.com/ccimg/51500/51415-07-7_b.png)
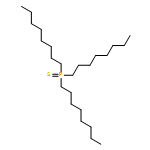
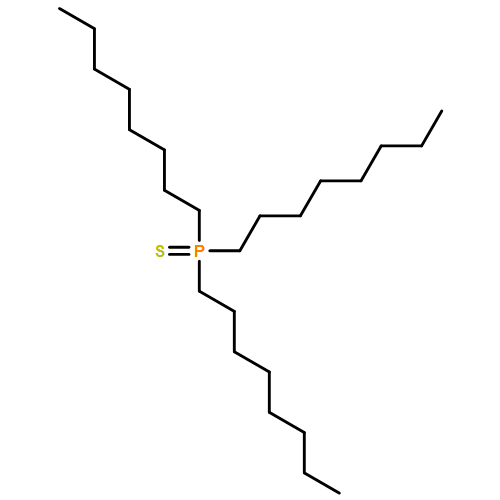
![2,4,6,7-TETRAAZA-1-PHOSPHABICYCLO[2.2.2]OCTANE, 1-SULFIDE](http://img.cochemist.com/ccimg/878300/878292-97-8.png)
![2,4,6,7-TETRAAZA-1-PHOSPHABICYCLO[2.2.2]OCTANE, 1-SULFIDE](http://img.cochemist.com/ccimg/878300/878292-97-8_b.png)

















![5,7-dihydroxy-2-(4-hydroxyphenyl)-3-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-[[(2r,3r,4r,5r,6s)-3,4,5-trihydroxy-6-methyloxan-2-yl]oxymethyl]oxan-2-yl]oxychromen-4-one](http://img.cochemist.com/ccimg/17700/17650-84-9.png)
![5,7-dihydroxy-2-(4-hydroxyphenyl)-3-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-[[(2r,3r,4r,5r,6s)-3,4,5-trihydroxy-6-methyloxan-2-yl]oxymethyl]oxan-2-yl]oxychromen-4-one](http://img.cochemist.com/ccimg/17700/17650-84-9_b.png)
![[(sulfonatoperoxy)sulfonyl]oxidanide](http://img.cochemist.com/ccimg/15100/15092-81-6.png)
![[(sulfonatoperoxy)sulfonyl]oxidanide](http://img.cochemist.com/ccimg/15100/15092-81-6_b.png)






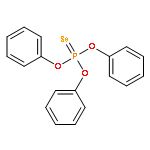
![1-SULFANYLIDENE-2,6,7-TRIOXA-1WEI 5-PHOSPHABICYCLO[2.2.2]OCTANE](http://img.cochemist.com/ccimg/4800/4704-96-5.png)
![1-SULFANYLIDENE-2,6,7-TRIOXA-1WEI 5-PHOSPHABICYCLO[2.2.2]OCTANE](http://img.cochemist.com/ccimg/4800/4704-96-5_b.png)