Co-reporter:Yang Li, Han-Yi Duan, Ming Luo, Ying-Ying Zhang, Xing-Hong Zhang, and Donald J. Darensbourg
Macromolecules November 14, 2017 Volume 50(Issue 21) pp:8426-8426
Publication Date(Web):October 26, 2017
DOI:10.1021/acs.macromol.7b01867
Small quantities of regio-defects in a regio-/stereoregular polymer weaken its tacticity and properties. This work clarified the origin of the regio-defect in the process of synthesizing poly(monothiocarbonate) through the copolymerization of propylene oxide (PO) and carbonyl sulfide (COS) catalyzed by a (salen)CrCl complex accompanied by bis(triphenylphosphoranylidene)ammonium chloride ([PPN]Cl). Quantitative characterization results from the MALDI-TOF-MS and 1H (13C) NMR spectroscopy suggested that the chain transfer reaction resulted in the regio-defect in the final copolymer, i.e., tail-to-tail (T–T) diad and dithiocarbonate (DTC) unit. The chain transferring to water in the reaction system led to the production of a (salen)Cr–OH intermediate, which initiated the copolymerization via either attacking PO first to result in formation of a T–T diad or first activating COS to produce mercapto (−SH) end-capped dormant chains via decarboxylation, thus generating a DTC unit in the final product through another chain transfer reaction and regrowth of the chain. The content of regio-defect in the final copolymer was directly related to the water content in the system. It is essential to reduce the regio-defect for an immortal COS/PO copolymerization reaction by eliminating trace amounts of water. We also demonstrated the application of α-OH, ω-OH poly(propylene monothiocarbonate) for synthesizing a well-defined ABA triblock copolymer, polystyrene-block-poly(propylene monothiocarbonate)-block-polystyrene (PS-b-PPMTC-b-PS), with a Mn of 10 800 g/mol and a PDI of 1.08 via an atom transfer radical polymerization (ATRP) method.
Co-reporter:Jia-Liang Yang;Hai-Lin Wu;Yang Li; Xing-Hong Zhang; Donald J. Darensbourg
Angewandte Chemie 2017 Volume 129(Issue 21) pp:5868-5873
Publication Date(Web):2017/05/15
DOI:10.1002/ange.201701780
AbstractThe preparation of perfectly alternating and regioslective copolymers derived from the copolymerization of carbonyl sulfide (COS) and epoxides by metal-free Lewis pair catalysts composed of a Lewis base (amidine, guanidine, or quaternary onium salts) and a Lewis acid (triethyl borane) is described. Colorless and highly transparent copolymers of poly(monothiocarbonate) were successfully obtained with over 99 % tail-to-head content and high molecular weight (up to 92.5 kg mol−1). In most instances, oxygen–sulfur exchange reactions (O/S ERs), which would generate random thiocarbonate and carbonate units, were effectively suppressed. The turnover frequencies (TOF) of these Lewis pair catalyzed processes were as high as 119 h−1 at ambient temperature.
Co-reporter:Jia-Liang Yang;Hai-Lin Wu;Yang Li; Xing-Hong Zhang; Donald J. Darensbourg
Angewandte Chemie International Edition 2017 Volume 56(Issue 21) pp:5774-5779
Publication Date(Web):2017/05/15
DOI:10.1002/anie.201701780
AbstractThe preparation of perfectly alternating and regioslective copolymers derived from the copolymerization of carbonyl sulfide (COS) and epoxides by metal-free Lewis pair catalysts composed of a Lewis base (amidine, guanidine, or quaternary onium salts) and a Lewis acid (triethyl borane) is described. Colorless and highly transparent copolymers of poly(monothiocarbonate) were successfully obtained with over 99 % tail-to-head content and high molecular weight (up to 92.5 kg mol−1). In most instances, oxygen–sulfur exchange reactions (O/S ERs), which would generate random thiocarbonate and carbonate units, were effectively suppressed. The turnover frequencies (TOF) of these Lewis pair catalyzed processes were as high as 119 h−1 at ambient temperature.
Co-reporter:Ming Luo, Xing-Hong Zhang, and Donald J. Darensbourg
Accounts of Chemical Research 2016 Volume 49(Issue 10) pp:2209
Publication Date(Web):September 27, 2016
DOI:10.1021/acs.accounts.6b00345
Carbonyl sulfide (COS) is an air pollutant that causes acid rain, ozonosphere damage, and carbon dioxide (CO2) generation. It is a heterocumulene and structural analogue of CO2. Relevant to organic synthesis, it is a source of C═O or C═S groups and thus an ideal one-carbon (C1) building block for synthesizing sulfur-containing polymers through the similar route of CO2 copolymerization. In contrast, traditional synthesis of sulfur-containing polymers often involves the condensation of thiols with phosgene and ring-opening polymerization of cyclic thiocarbonates that are generally derived from thiols and phosgene; thus, COS/epoxide copolymerization is a “greener” route to supplement or supplant current processes for the production of sulfur-containing polymers.This Accounts highlights our efforts on the discovery of the selective formation of poly(monothiocarbonate)s from COS with epoxides via heterogeneous zinc–cobalt double metal cyanide complex (Zn–Co(III) DMCC) and homogeneous (salen)CrX complexes. The catalytic activity and selectivity of Zn–Co(III) DMCC for COS/epoxide copolymerization are similar to those for CO2/epoxide copolymerization. (salen)CrX complexes accompanied by onium salts exhibited high activity and selectivity for COS/epoxide copolymerization under mild conditions, affording copolymers with >99% monothiocarbonate units and high tail-to-head content up to 99%. By way of contrast, these catalysts often show moderate or low activity for CO2/epoxide copolymerization. Of note, a specialty of COS/epoxide copolymerization is the occurrence of an oxygen–sulfur exchange reaction (O/S ER), which may produce carbonate and dithiocarbonate units. O/S ER, which are induced by the metal–OH bond regenerated by chain transfer reactions, can be kinetically inhibited by changing the reaction conditions. We provide a thorough mechanistic understanding of the electronic/steric effect of the catalysts on the regioselectivity of COS copolymerization. The regioselectivity of the copolymerization originates from the solely nucleophilic attack of the sulfur anion to methylene of the epoxide, and thus, the chiral configuration of the monosubstituted epoxides is retained.COS-based copolymers are highly transparent sulfur-containing polymers with excellent optical properties, such as high refractive index and Abbe number. Thanks to their good solubility and many available epoxides, COS/epoxide copolymers can potentially be a new applicable optical material. Very recently, crystalline COS-based polymers with or without chiral carbons have been synthesized, which may further expand the scope of application of these new materials.
Co-reporter:Fu-Te Tsai; Yanyan Wang
Journal of the American Chemical Society 2016 Volume 138(Issue 13) pp:4626-4633
Publication Date(Web):March 14, 2016
DOI:10.1021/jacs.6b01327
(S)-3,4-Dihydroxybutyric acid ((S)-3,4-DHBA), an endogenous straight chain fatty acid, is a normal human urinary metabolite and can be obtained as a valuable chiral biomass for synthesizing statin-class drugs. Hence, its epoxide derivatives should serve as promising monomers for producing biocompatible polymers via alternating copolymerization with carbon dioxide. In this report, we demonstrate the production of poly(tert-butyl 3,4-dihydroxybutanoate carbonate) from racemic-tert-butyl 3,4-epoxybutanoate (rac-tBu 3,4-EB) and CO2 using bifunctional cobalt(III) salen catalysts. The copolymer exhibited greater than 99% carbonate linkages, 100% head-to-tail regioselectivity, and a glass-transition temperature (Tg) of 37 °C. By way of comparison, the similarly derived polycarbonate from the sterically less congested monomer, methyl 3,4-epoxybutanoate, displayed 91.8% head-to-tail content and a lower Tg of 18 °C. The tert-butyl protecting group of the pendant carboxylate group was removed using trifluoroacetic acid to afford poly(3,4-dihydroxybutyric acid carbonate). Depolymerization of poly(tert-butyl 3,4-dihydroxybutanoate carbonate) in the presence of strong base results in a stepwise unzipping of the polymer chain to yield the corresponding cyclic carbonate. Furthermore, the full degradation of the acetyl-capped poly(potassium 3,4-dihydroxybutyrate carbonate) resulted in formation of the biomasses, β-hydroxy-γ-butyrolacetone and 3,4-dihydroxybutyrate, in water (pH = 8) at 37 °C. In addition, water-soluble platinum–polymer conjugates were synthesized with platinum loading of 21.3–29.5%, suggesting poly(3,4-dihydroxybutyric acid carbonate) and related derivatives may serve as platinum drug delivery carriers.
Co-reporter:M. Luo, X.-H. Zhang and D. J. Darensbourg
Catalysis Science & Technology 2016 vol. 6(Issue 1) pp:188-192
Publication Date(Web):17 Aug 2015
DOI:10.1039/C5CY00977D
Two guanidine bases were used as organocatalysts for the synthesis of cyclic monothiocarbonates via the coupling reaction of carbonyl sulfide (COS) and epoxides. The systems proved to be efficient single-component, metal-free catalysts for the reaction of simple (propylene oxide, 1,3-butene oxide) or activated epoxides (epichlorohydrin, glycidyl phenyl ether) with COS under solvent-free and mild reaction conditions to selectively afford the corresponding cyclic monothiocarbonates. The yield of this reaction is generally high, thereby providing ready means for pure product isolation.
Co-reporter:Guang-Peng Wu and Donald J. Darensbourg
Macromolecules 2016 Volume 49(Issue 3) pp:807-814
Publication Date(Web):January 14, 2016
DOI:10.1021/acs.macromol.5b02752
The addition of water as a chain transfer reagent during the copolymerization reaction of epoxides and carbon dioxide has been shown as a promising method for producing CO2-based polycarbonate polyols. These polyols can serve as drop-in replacements for petroleum derived polyols for polyurethane production or designer block copolymers. Ironically, during the history of CO2/epoxide coupling development, water was generally considered primarily as an aversion reagent. That is, in its presence, low catalytic activity and high polydispersity was normally observed. Recently, we reported a water-mediated tandem metal-coordination CO2/epoxide copolymerization and organobase catalyzed ring-opening polymerization (ROP) approach for the one-pot synthesis of an ABA CO2-based triblock copolymers. As in previous studies, water was deemed as the chain transfer reagent in this tandem strategy for producing CO2-based polyols. Herein is presented a mechanistic study aimed at determining the intimate role water plays during the metal-catalyzed CO2/epoxide copolymerization process. In this regard, it was observed that under the commonly employed (salen)Co(trifluoroacetate)/onium salt binary catalyst system, water was not the true chain-transfer reagent, but instead reacted initially with the epoxides to afford the corresponding diols which serves as the chain-transfer reagent. The further studies in resultant afforded α,ω-dihydroxyl end-capped polycarbonates were utilized in direct chain extension via ROP of the water-soluble cyclic phosphate monomer, 2-methoxy-2-oxo-1,3,2-dioxaphospholene employing an organocatalyst. These triblock copolymers displayed narrow PDI and were found to provide nanostructure materials which should be of use in biomedical applications.
Co-reporter:Bing Han;Li Zhang;Samuel J. Kyran;Binyuan Liu;Zhongyu Duan
Journal of Polymer Science Part A: Polymer Chemistry 2016 Volume 54( Issue 13) pp:1938-1944
Publication Date(Web):
DOI:10.1002/pola.28052
ABSTRACT
Chromium complexes supported by tetradentate dianionic imine-thioether-bridged bis(phenol) ligands were prepared and employed in the synthesis of poly(cyclohexene carbonate) via the copolymerization of CO2 and cyclohexene oxide. The catalytic activity, product selectivity, and kinetic behaviors of these [ONSO]CrIII complexes have been systematically investigated. Results indicate the presence of electron-withdrawing substituents on the ligands to enhance catalytic activity and polymer selectivity. A turnover frequency of 100 h−1 is observed at a temperature of 110 °C, producing polycarbonate with >60% selectivity. © 2016 Wiley Periodicals, Inc. J. Polym. Sci., Part A: Polym. Chem. 2016, 54, 1938–1944
Co-reporter:Donald J. Darensbourg and Samuel J. Kyran
ACS Catalysis 2015 Volume 5(Issue 9) pp:5421
Publication Date(Web):August 7, 2015
DOI:10.1021/acscatal.5b01375
Poly(1,4-dihydronaphthalene carbonate) has been prepared via the catalytic coupling of carbon dioxide and 1,4-dihydronaphthalene oxide using chromium(III) catalysts. The copolymer formation is found to be greatly dependent on the steric environment around the metal center. Traditional (salen)CrIIIX/cocatalyst systems bearing bulky t-butyl groups hinder the approach of the large monomer, significantly diminishing polymer chain growth and providing the entropically favored cyclic byproduct in excess. In contrast, employing the sterically unencumbered azaannulene-derived catalyst, (tmtaa)CrIIIX/cocatalyst system (tmtaa = tetramethyltetraazaannulene) shows polymer selectivity close to 90% with three times the activity (TOF = 20–30 h–1). With the use of a bifunctional (salen)CrIII catalyst, even higher polymer selectivity (>90%) can be observed. The complete synthesis of a new bifunctional tetraazaannulene ligand for a more effective catalyst is also described herein.Keywords: 1,4-dihydronaphthalene oxide; carbon dioxide; copolymerization; polycarbonate; steric effect
Co-reporter:M. Luo, X.-H. Zhang and D. J. Darensbourg
Polymer Chemistry 2015 vol. 6(Issue 39) pp:6955-6958
Publication Date(Web):18 Aug 2015
DOI:10.1039/C5PY01197C
Polymer structures containing sulfur atoms can provide enhancement of important polymer properties compared to their oxygen-containing counterparts. In this regard, the copolymerization of phenyl glycidyl ether and carbonyl sulfide has been very effectively achieved employing (salen)CrCl in the presence of an onium salt at ambient temperature. The resulting copolymer is shown to be completely alternating and to possess extremely high regioselectivity in the epoxide ring-opening step. That is, the ring-opening step predominantly occurs at the less sterically hindered methylene carbon–oxygen bond leading to a tail-to-head structure (98%) in the copolymer. This observation was further confirmed when using the chiral epoxide monomer, (S)-PGE.
Co-reporter:Donald J. Darensbourg and Yanyan Wang
Polymer Chemistry 2015 vol. 6(Issue 10) pp:1768-1776
Publication Date(Web):19 Dec 2014
DOI:10.1039/C4PY01612B
Terpolymerization reactions of epoxides bearing vinyl groups, propylene oxide and carbon dioxide catalyzed by binary and bifunctional (salen)Co(III) complexes have provided polycarbonates of various compositions depending on the feed ratios of the epoxide monomers. Fineman–Ross analysis of these polymerization reactions revealed reactivity ratios for propylene oxide (PO)/vinyl oxide (VIO)/CO2 of rPO = 3.74 and rVIO = 0.224 at 25 °C which reflect both the binding constants of the epoxides to the cobalt center as well as the rate constants for the ring-opening process. Less discrimination of epoxides was noted when carrying out the process using PO/allyl glycidyl ether (AGE)/CO2, where the reactivity parameters were rPO = 0.755 and rAGE = 0.876 at 25 °C. In order to enhance the polycarbonate's mechanical and thermal properties the terpolymers from PO/AGE/CO2, where the vinyl pendant groups are more randomly distributed in the polymeric material, were cross-linked via thiol–ene chemistry using two different thiols, ethylene glycol bis(3-mercaptoproionate) and pentaerythritol tetrakis(mercaptoacetate). FT-Raman spectroscopy provided evidence that successful cross-linking has occurred. Dynamic mechanical analysis (DMA) measurements on these terpolymers were performed as a function of the cross-link densities. Cross-linked films derived from the tetradentate cross-linker displayed a 50% increase in rubbery modulus between 50% and 100% CC bond conversion; concomitantly, the Tg increased with increasing cross-link densities. In addition, the cross-linked films with 25% CC bond conversion were reacted with N-acetyl-L-cysteine and 2-(Boc-amino)ethanethiol, and confirmed by FTIR and XPS, to provide surface functionalized films containing carboxyl or amine groups for various applications.
Co-reporter:Yanyan Wang;Jingwei Fan ; Donald J. Darensbourg
Angewandte Chemie International Edition 2015 Volume 54( Issue 35) pp:10206-10210
Publication Date(Web):
DOI:10.1002/anie.201505076
Abstract
The construction of amphiphilic polycarbonates through epoxides/CO2 coupling is a challenging aim to provide more diverse CO2-based functional materials. In this report, we demonstrate the facile preparation of diverse and functional nanoparticles derived from a CO2-based triblock polycarbonate system. By the judicious use of water as chain-transfer reagent in the propylene oxide/CO2 polymerization, poly(propylene carbonate (PPC) diols are successfully produced and serve as macroinitiators in the subsequent allyl glycidyl ether/CO2 coupling reaction. The resulting ABA triblock polycarbonate can be further functionalized with various thiols by radical mediated thiol–ene click chemistry, followed by self-assembly in deionized water to construct a versatile and functional nanostructure system. This class of amphiphilic polycarbonates could embody a powerful platform for biomedical applications.
Co-reporter:Yanyan Wang;Jingwei Fan ; Donald J. Darensbourg
Angewandte Chemie 2015 Volume 127( Issue 35) pp:10344-10348
Publication Date(Web):
DOI:10.1002/ange.201505076
Abstract
The construction of amphiphilic polycarbonates through epoxides/CO2 coupling is a challenging aim to provide more diverse CO2-based functional materials. In this report, we demonstrate the facile preparation of diverse and functional nanoparticles derived from a CO2-based triblock polycarbonate system. By the judicious use of water as chain-transfer reagent in the propylene oxide/CO2 polymerization, poly(propylene carbonate (PPC) diols are successfully produced and serve as macroinitiators in the subsequent allyl glycidyl ether/CO2 coupling reaction. The resulting ABA triblock polycarbonate can be further functionalized with various thiols by radical mediated thiol–ene click chemistry, followed by self-assembly in deionized water to construct a versatile and functional nanostructure system. This class of amphiphilic polycarbonates could embody a powerful platform for biomedical applications.
Co-reporter:Donald J. Darensbourg, Wan-Chun Chung, Andrew D. Yeung, and Mireya Luna
Macromolecules 2015 Volume 48(Issue 6) pp:1679-1687
Publication Date(Web):March 5, 2015
DOI:10.1021/acs.macromol.5b00172
The copolymerization of 1,3-cyclohexadiene oxide (1,2-epoxy-3-cyclohexene) with CO2 in the presence of the binary catalyst (salen)CoX or (salen)CrX and onium salts was shown to selectively afford the completely alternating copolymer poly(1,3-cyclohexadiene carbonate) in good yield. In the process catalyzed by the cobalt(III) system, the reaction was 100% selective for copolymer, whereas employing the higher temperature chromium(III) catalyst, the reaction yielded in addition to copolymer a significant quantity of the cis-1,3-cyclohexadiene carbonate. Importantly, no corresponding trans-1,3-cyclohexadiene carbonate was produced. The reactivity of 1,3-cyclohexadiene oxide in coupling reactions with CO2 was strikingly greater than that of 1,4-cyclohexadiene oxide under either catalytic conditions. Authentic samples of the cis-cyclic carbonate and trans-cyclic carbonate were synthesized from epoxide and CO2 using ZnCl2/PPNI catalyzed and trans-diol/ethyl chloroformate routes, respectively. trans-1,3-Cyclohexadiene carbonate was fully characterized by X-ray crystallography. Unlike the copolymer derived from the symmetrical 1,4-cyclohexadiene oxide (1,2-epoxy-4-cyclohexene), deprotonation of poly(1,3-cyclohexadiene carbonate) by a strong base did not lead to depolymerization with formation of the trans-cyclic carbonate. Computational studies revealed the trans-cyclic carbonate was thermodynamically unstable relative to the polycarbonate, with the enthalpy of reaction being +12.8 kcal/mol. The enhanced reactivity of the 1,3-isomeric epoxide versus that of its 1,4-isomer was further demonstrated by the facile terpolymerization reaction of 1,3-cyclohexadiene oxide with propylene oxide and CO2. This latter process is useful for the preparation of cross-linked or functionalized polycarbonates via thiol–ene chemistry.
Co-reporter:Samuel J. Kyran, Sergio G. Sanchez, Christopher J. Arp, and Donald J. Darensbourg
Organometallics 2015 Volume 34(Issue 14) pp:3598-3602
Publication Date(Web):July 9, 2015
DOI:10.1021/acs.organomet.5b00396
Dipyrrolidylmethane, CH2(pyr)2, and dipiperidylmethane, CH2(pip)2, are synthesized via the condensation of their respective secondary amine precursors and dichloromethane at room temperature in the absence of light. Their use as chelating ligands is shown by the isolation and complete characterization of [CH2(pyr)2]Mo(CO)4 and [CH2(pip)2]Mo(CO)4 complexes. X-ray analysis reveals the methylene bis(cycloamines) to possess a sharp bite angle between 61° and 63° and a strong steric impact on the surrounding carbonyl ligands as a result of their ring conformations.
Co-reporter:Donald J. Darensbourg, Wan-Chun Chung, Kecheng Wang, and Hong-Cai Zhou
ACS Catalysis 2014 Volume 4(Issue 5) pp:1511
Publication Date(Web):April 4, 2014
DOI:10.1021/cs500259b
It is presently well-established that the synthesis of polycarbonates or cyclic carbonates from metal-catalyzed reactions of CO2 and oxiranes provides a viable industrial process for the production of these important chemicals. In this study, we have demonstrated that CO2 collected under aerobic conditions at atmospheric pressure over [Cu3(btc)2(H2O)3] (btc = benzene-1,3,5-tricarboxylate) or HKUST-1, a commercially available metal–organic framework material (MOF), can be utilized to synthesize poly(propylene carbonate) from propylene oxide and CO2 catalyzed by Co(III) salen catalysts at optimal pressure. That is, CO2 thermally released from the MOF material selectively affords copolymer in the pressure range that is not rate-limiting. Similar results were noted for the copolymerization of the much less reactive cis-2-butylene oxide monomer with CO2. Comparative studies using CO2 provided directly from a compressed gas source gave similar results. This investigation provides a baseline study for the practical use of atmospheric pressure or below CO2 captured from point sources for the synthesis of useful chemicals without requiring mechanical compression.Keywords: carbon dioxide; copolymerization; epoxides; metal−organic framework; polycarbonates
Co-reporter:Donald J. Darensbourg and Fu-Te Tsai
Macromolecules 2014 Volume 47(Issue 12) pp:3806-3813
Publication Date(Web):June 6, 2014
DOI:10.1021/ma500834r
Common CO2-based polycarbonates are known to be highly hydrophobic, and this “inert” property makes them difficult for the covalent immobilization of bioactive molecules. A practical method for modifying polymers is to introduce various functional groups that permit decoration of polymer chains with bioactive substances. In this report, CO2-based poly(2-vinyloxirane carbonate) (PVIC) with more than 99% carbonate linkages is isolated from the CO2/2-vinyloxirane alternating copolymerization catalyzed by the bifunctional catalyst [(1R,2R)-SalenCo(III)(DNP)2] (1) (DNP = 2,4-dinitrophenolate) bearing a quaternary ammonium salt on the ligand framework. It was also observed that the presence of propylene oxide significantly activates 2-vinyloxirane for incorporation into the polymer chain as well as inhibits the formation of cyclic carbonate in the terpolymerization process. DSC studies demonstrate that the glass transition temperature (Tg) decreases with the increase in the content of vinyl groups in the polycarbonate. By way of thiol–ene coupling, showing mainly “click” characteristics and nearly quantitative yields, amphiphilic polycarbonates (PVIC-OH and PVIC-COOH) with multiple hydroxy or carboxy functionalities have been prepared, providing suitable reactivities for further modifications (ring-opening of l-aspartic acid anhydride hydrochloride salt and deprotonation by aqueous ammonium hydroxide (NH4OH(aq))) to successfully isolate the water-soluble CO2-based polycarbonate PVIC-COONH4, and the PVIC-OH-Asp polymer which shows particles dispersed in water with an average hydrodynamic diameter Dn = 32.2 ± 8.8 nm. It is presumed that this emerging class of amphiphilic/water-soluble polycarbonates could embody a powerful platform for bioconjugation and drug conjugation. In contrast to lower Tgs of PVIC, (PVIC-co-PC), PVIC-OH, and PVIC-COOH, the polycarbonates PVIC-OH-Asp and PVIC-COONH4 show higher Tgs as a consequence of their intrinsic ionic property (ammonium salts).
Co-reporter:Donald J. Darensbourg and Wan-Chun Chung
Macromolecules 2014 Volume 47(Issue 15) pp:4943-4948
Publication Date(Web):July 17, 2014
DOI:10.1021/ma501004w
In this study, we investigate the selectivity for copolymer versus cyclic carbonate production from the coupling of isomeric forms of butene oxide with carbon dioxide in the presence of binary and bifunctional cobalt(III) and chromium(III) salicylaldimine catalysts. Use of the less problematic 1-butene oxide has previously been reported to preferentially afford copolymer from its coupling with CO2. Of the epoxides, cis- and trans-2-butene oxide and isobutene oxide, only cis-2-butene oxide was shown to selectively provide polycarbonate, with both cobalt(III) catalysts being more effective than their chromium(III) analogues. The binary chromium catalyst system produced both cis- and trans-cyclic carbonates from the cycloaddition of CO2 and cis-2-butene oxide, whereas, the corresponding cobalt(III) catalyst selectively yielded 75.4% copolymer at 40 °C with the remaining product being trans-cyclic carbonate. In this instance, the trans-cyclic carbonate results from copolymer degradation, consistent with the observation that depolymerization of the copolymer derived from CO2 and cis-2-butene oxide affords trans-cyclic carbonate exclusively. By way of contrast, both bifunctional catalysts were efficient at producing copolymers with selectivities of 100% (40 °C) and 79% (70 °C) for the cobalt and chromium catalysts, respectively. The glass transition temperature (Tg) of poly(trans-2-butene carbonate) derived from the completely alternating copolymerization of CO2 and cis-2-butene oxide was found to be 68 °C, some 30 deg higher than poly(propylene carbonate). Furthermore, it was shown to have a significantly lower % elongation-to-break value than poly(propylene carbonate).
Co-reporter:Donald J. Darensbourg, Wan-Chun Chung, Christopher J. Arp, Fu-Te Tsai, and Samuel J. Kyran
Macromolecules 2014 Volume 47(Issue 21) pp:7347-7353
Publication Date(Web):October 23, 2014
DOI:10.1021/ma501781k
The coupling reaction of 1,2-epoxy-4-cyclohexene with CO2 in the presence of a ZnCl2/nBu4NI catalyst system was shown to provide the naturally occurring cis-cyclohexadiene carbonate. An alternative synthesis of this compound, which was characterized by X-ray structural analysis, was carried out from the cis-diol and triphosgene. Upon utilizing binary or bifunctional (salen)CrX catalysts, this coupling process resulted in the selective formation of completely alternating copolymer of 1,2-epoxy-4-cyclohexene and carbon dioxide. In the case involving the binary chromium(III)/onium salt catalyst, small quantities of both the cis and trans cyclic carbonates were also produced. The (salen)CoDNP/PPNDNP (DNP = 2,4-dinitrophenolate) catalyst system was most effective at producing high molecular weight copolymer with 100% selectivity. The Tg of this polymer (Mn = 35.9 kDa) was determined to be 123 °C, which is higher than the Tg (116 °C) of the corresponding saturated copolymer. Depolymerization of poly(cyclohexadiene carbonate) to trans-cyclohexadiene carbonate occurred slowly and cleanly at 110 °C following deprotonation of the terminal hydroxyl group. The trans-cyclohexadiene carbonate was independently synthesized via the carbonylation of the trans-diol with ethyl chloroformate. The hydrophobic 1,2-epoxy-4-cyclohexene/CO2 derived copolymer was modified by the quantitative addition of thioglycolic acid by way of the thiol–ene click reaction to afford an amphiphilic copolymer. Upon deprotonation of this functionalized polycarbonate with ammonium hydroxide, the production of a water-soluble polymeric material was achieved which displayed a Tg of 120 °C.
Co-reporter:Donald J. Darensbourg, Wan-Chun Chung, and Stephanie J. Wilson
ACS Catalysis 2013 Volume 3(Issue 12) pp:3050
Publication Date(Web):November 6, 2013
DOI:10.1021/cs4008667
Previously, the polycarbonate produced from the alternating copolymerization of cyclopentene oxide and carbon dioxide has been demonstrated to readily be recyclable to its monomers, hence affording a sustainable route to the production of this polymeric material. This, in turn, necessitates the development of effective synthetic methods for this copolymer. Coupling reactions of the alicyclic epoxides, cyclohexene oxide (CHO) and cyclopentene oxide (CPO), with CO2 were shown to provide grossly different product selectivities utilizing binary (salen)CrX/PPNX catalyst systems. That is, whereas the coupling of CHO/CO2 affords >99% selectivity for completely alternating copolymers, under identical conditions, CPO/CO2 yields >99% selectivity for cis-cyclopentene carbonate. By way of contrast, employing bifunctional (R,R)-(salen)M(III) catalysts (M = Cr and Co) for the coupling reactions of cyclopentene oxide and carbon dioxide results in the highly selective synthesis of well-controllable, narrowly distributed molecular weights copolymers. Although the chromium catalyst was found to be less active than its cobalt analog, it was shown to be thermally more stable, and hence, the reaction could be carried out at higher temperatures with little sacrifice in selectivity for copolymer production. The Tg of the resulting poly(cyclopentene carbonate) was determined to be 84.5 °C.Keywords: alternating copolymerization; carbon dioxide; chromium; cobalt; cyclopentene oxide
Co-reporter:Donald J. Darensbourg, Andrew D. Yeung and Sheng-Hsuan Wei
Green Chemistry 2013 vol. 15(Issue 6) pp:1578-1583
Publication Date(Web):09 Apr 2013
DOI:10.1039/C3GC40475G
High-accuracy CBS-QB3(+) calculations were used to obtain the free energy barriers for several polycarbonates of interest to undergo alkoxide back-biting to give the corresponding epoxide and carbon dioxide. Free energy barriers to epoxide formation were modest for most polymeric alkoxides (12.7–17.4 kcal mol−1), and they were higher than for the same starting material to give cyclic carbonate (10.7–14.6 kcal mol−1). Poly(cyclopentene carbonate) differs: epoxide formation has a lower free energy barrier (13.3 kcal mol−1) than cyclic carbonate formation (19.9 kcal mol−1). These results explain why poly(cyclopentene carbonate) depolymerizes to cyclopentene oxide when treated with a strong base, whereas propylene and styrene polycarbonates depolymerize to their respective cyclic carbonates. Recycling via regeneration of the monomer represents the ideal method for producing material of the highest quality.
Co-reporter:Donald J. Darensbourg, Stephanie J. Wilson, and Andrew D. Yeung
Macromolecules 2013 Volume 46(Issue 20) pp:8102-8110
Publication Date(Web):October 3, 2013
DOI:10.1021/ma4015438
The catalytic coupling of cyclopentene oxide with carbon disulfide has been investigated utilizing (salen)CrCl in the presence of added onium salts. Both polymeric and cyclic materials were produced, with oxygen/sulfur atom scrambling observed in both instances. This atom redistribution process was found to require (salen)CrCl and excess epoxide, though an increase in the rate of atom scrambling was noted upon the addition of the onium salt. Cyclopentene sulfide was observed as a side product of the coupling reaction and was found to be unreactive toward both CS2 and CO2, instead undergoing desulfurization to cyclopentene under the conditions of the reaction. Of the 12 cyclic cyclopentene [thio]carbonates possibly produced by this coupling reaction, eight were observed, and the crystal structure of trans-cyclopentene trithiocarbonate is reported herein. Computational studies reveal that the cis- cyclic materials are more stable than their trans-counterparts by >5 kcal/mol of enthalpy, and there is a 10–25 kcal/mol preference for the formation of a C═O vs C═S double bond. When trans-cyclohexene trithiocarbonate was exposed to the catalyst system in the presence of excess cyclopentene oxide, mixed-species scrambling was observed, whereby cyclic [thio]carbonate compounds displaying both cyclopentyl and cyclohexyl backbones were produced. A proposed mechanistic pathway for atom scrambling involves nucleophilic alkoxide attack at a [thio]carbonyl center to induce oxygen/sulfur atom exchange.
Co-reporter:Dr. Donald J. Darensbourg;Dr. Guang-Peng Wu
Angewandte Chemie International Edition 2013 Volume 52( Issue 40) pp:10602-10606
Publication Date(Web):
DOI:10.1002/anie.201304778
Co-reporter:Dr. Donald J. Darensbourg;Dr. Guang-Peng Wu
Angewandte Chemie 2013 Volume 125( Issue 40) pp:10796-10800
Publication Date(Web):
DOI:10.1002/ange.201304778
Co-reporter:Donald J. Darensbourg and Andrew D. Yeung
Macromolecules 2013 Volume 46(Issue 1) pp:
Publication Date(Web):December 12, 2012
DOI:10.1021/ma3021823
The copolymerization reactions of carbon dioxide and epoxides to give polycarbonates were examined by density functional theory (DFT), and chemically accurate thermochemical data (benchmarked to experimental values) were obtained via composite ab initio methods. All of the examples studied, i.e., formation of poly(ethylene carbonate), poly(propylene carbonate), poly(chloropropylene carbonate), poly(styrene carbonate), poly(cyclohexene carbonate), and poly(indene carbonate), exhibited enthalpies of polymerization of 21–23 kcal/mol, with the exception of poly(cyclopentene carbonate) (15.8 kcal/mol) which suffers both ring strain and intramolecular steric repulsion caused by the cyclopentane ring fused to the polymer chain. The metal-free carbonate backbiting reaction by a free anionic polycarbonate strand is inhibited by bulky groups at the methine carbon but is accelerated by resonance stabilization of the pentavalent transition state in the case involving poly(styrene carbonate). Nucleophilic attack at the methylene carbon of a substituted epoxide has a lower barrier than for the corresponding reaction involving ethylene oxide due to charges being distributed onto the pendant groups. The undesired backbiting reaction to afford cyclic organic carbonates observed under polymerization conditions for many systems due to the low activation barrier (ΔG‡ = 18–25 kcal/mol) was negligible for poly(cyclohexene carbonate) because, in this instance, it must overcome an additional endergonic conformational change (ΔG = 4.7 kcal/mol) before traversing the activation barrier (ΔG‡ = 21.1 kcal/mol) to cyclization. Backbiting from an alkoxide chain end is proposed to proceed via a tetrahedral alkoxide intermediate, where formation of this intermediate is barrierless. Further reaction of this intermediate to the cyclic carbonate has a free energy barrier 10 kcal/mol less than the carbonate chain end backbiting reaction.
Co-reporter:Donald J. Darensbourg and Stephanie J. Wilson
Macromolecules 2013 Volume 46(Issue 15) pp:5929-5934
Publication Date(Web):August 1, 2013
DOI:10.1021/ma4013779
The copolymerization of carbon dioxide and indene oxide to yield poly(indene carbonate) has been achieved through the use of bifunctional cobalt(III) catalysts. When compared to our earlier studies utilizing the traditional binary (salen)Co(III)X/cocatalyst system, the bifunctional catalysts display large increases in activity and selectivity for polymer while maintaining good control (PDI < 1.2). The copolymerization reactions can proceed at 25 °C while maintaining >99% selectivity for poly(indene carbonate) production. Polymer samples have been achieved with Mns and Tgs of up to 9700 g/mol and 138 °C, respectively. This represents the highest Tg yet observed for polycarbonates produced from the coupling of CO2 and epoxides. Additionally, the activation energy for the direct coupling of indene oxide and CO2 to yield cis-indene carbonate employing the (salen)CrCl/n-Bu4NCl catalyst system was determined to be 114.4 ± 5.7 kJ/mol utilizing in situ ATR-FTIR.
Co-reporter:Donald J. Darensbourg, Sheng-Hsuan Wei, Andrew D. Yeung, and W. Chadwick Ellis
Macromolecules 2013 Volume 46(Issue 15) pp:5850-5855
Publication Date(Web):August 1, 2013
DOI:10.1021/ma401286x
The hydroxyl-terminated copolymer, poly(cyclopentene carbonate), derived from carbon dioxide and cyclopentene oxide was deprotonated by the strong base sodium bis(trimethylsilyl)amide (NaHMDS) in toluene and shown to undergo depolymerization to cyclopentene oxide and cis-cyclopentene carbonate. The degradation process was demonstrated to be retarded under a 0.7 MPa pressure of CO2, with the product distribution being enhanced in favor of cis-cyclopentene carbonate. Unlike the related copolymer, poly(indene carbonate), the degradation pathway was found to be insensitive to either light or the radical trap, TEMPO. The depolymerization process was further shown to be catalyzed by (salen)CrCl/n-Bu4NN3, with the major product being cyclopentene oxide. Although the reaction rate in the presence of metal catalyst was inhibited by an added pressure of CO2, the product distribution still highly favored epoxide production. On the other hand, upon reducing the pressure above the polymer solution during the depolymerization reaction, the rate of reaction was accelerated and was more selective for cyclopentene oxide formation. Preliminary studies were investigated to optimize the efficient recycling of poly(cyclopentene carbonate) to its monomers, cyclopentene oxide and CO2. Computational studies have been performed on depolymerization reaction of polycarbonates derived from CO2 and epoxides which strongly support these experimental findings.
Co-reporter:Donald J Darensbourg, Jason C Yarbrough
Journal of Organometallic Chemistry 2000 Volumes 614–615() pp:305-308
Publication Date(Web):8 December 2000
DOI:10.1016/S0022-328X(00)00594-5
Our attempts to synthesize [H2B(3-Phpz)2]Zn(acetate) from K[H2B(3-Phpz)2] and anhydrous zinc acetate has instead resulted in the formation of the 2:1 complex, [H2B(3-Phpz)2]2Zn. This derivative was isolated and crystallized from THF at −10°C. Its structure has been defined as a distorted tetrahedron with an average Zn–N bond distance of 2.011(1) Å as revealed by X-ray crystallography. As anticipated, this complex is inactive at catalyzing the coupling of cyclohexene oxide and carbon dioxide.
Co-reporter:M. Luo, X.-H. Zhang and D. J. Darensbourg
Catalysis Science & Technology (2011-Present) 2016 - vol. 6(Issue 1) pp:NaN192-192
Publication Date(Web):2015/08/17
DOI:10.1039/C5CY00977D
Two guanidine bases were used as organocatalysts for the synthesis of cyclic monothiocarbonates via the coupling reaction of carbonyl sulfide (COS) and epoxides. The systems proved to be efficient single-component, metal-free catalysts for the reaction of simple (propylene oxide, 1,3-butene oxide) or activated epoxides (epichlorohydrin, glycidyl phenyl ether) with COS under solvent-free and mild reaction conditions to selectively afford the corresponding cyclic monothiocarbonates. The yield of this reaction is generally high, thereby providing ready means for pure product isolation.
.jpg)


![6H-Indeno[1,2-b]oxirene,1a,6a-dihydro-, (1aR,6aS)-](http://img.cochemist.com/ccimg/85400/85354-35-4.png)
![6H-Indeno[1,2-b]oxirene,1a,6a-dihydro-, (1aR,6aS)-](http://img.cochemist.com/ccimg/85400/85354-35-4_b.png)


![(1as,6ar)-6,6a-dihydro-1ah-indeno[1,2-b]oxirene](http://img.cochemist.com/ccimg/67600/67528-26-1.png)
![(1as,6ar)-6,6a-dihydro-1ah-indeno[1,2-b]oxirene](http://img.cochemist.com/ccimg/67600/67528-26-1_b.png)
![Poly[oxy(phenyl-1,2-ethanediyl)oxy[(2Z)-1,4-dioxo-2-butene-1,4-diyl]]](http://img.cochemist.com/ccimg/66000/65988-55-8.png)
![Poly[oxy(phenyl-1,2-ethanediyl)oxy[(2Z)-1,4-dioxo-2-butene-1,4-diyl]]](http://img.cochemist.com/ccimg/66000/65988-55-8_b.png)
![Poly[oxy(phenyl-1,2-ethanediyl)oxycarbonyl-1,2-phenylenecarbonyl]](http://img.cochemist.com/ccimg/66000/65988-53-6.png)
![Poly[oxy(phenyl-1,2-ethanediyl)oxycarbonyl-1,2-phenylenecarbonyl]](http://img.cochemist.com/ccimg/66000/65988-53-6_b.png)


![7-methyl-1,3,5-triaza-7-phosphoniatricyclo[3.3.1.1~3,7~]decane](http://img.cochemist.com/ccimg/64500/64452-02-4.png)
![7-methyl-1,3,5-triaza-7-phosphoniatricyclo[3.3.1.1~3,7~]decane](http://img.cochemist.com/ccimg/64500/64452-02-4_b.png)
![2,4-ditert-butyl-6-[[2-[(3,5-ditert-butyl-2-hydroxyphenyl)methylamino]ethylamino]methyl]phenol](http://img.cochemist.com/ccimg/63600/63551-08-6.png)
![2,4-ditert-butyl-6-[[2-[(3,5-ditert-butyl-2-hydroxyphenyl)methylamino]ethylamino]methyl]phenol](http://img.cochemist.com/ccimg/63600/63551-08-6_b.png)
![1-(3-ACETYL-5-OXO-1,3,7-TRIAZA-5WEI 5-PHOSPHABICYCLO[3.3.1]NONAN-7-YL)ETHANONE](http://img.cochemist.com/ccimg/63300/63250-00-0.png)
![1-(3-ACETYL-5-OXO-1,3,7-TRIAZA-5WEI 5-PHOSPHABICYCLO[3.3.1]NONAN-7-YL)ETHANONE](http://img.cochemist.com/ccimg/63300/63250-00-0_b.png)
![2-Thia-1,3,5-triaza-7-phosphatricyclo[3.3.1.13,7]decane, 2,2,7-trioxide](http://img.cochemist.com/ccimg/55800/55776-70-0.png)
![2-Thia-1,3,5-triaza-7-phosphatricyclo[3.3.1.13,7]decane, 2,2,7-trioxide](http://img.cochemist.com/ccimg/55800/55776-70-0_b.png)
![2-THIA-1,3,5-TRIAZA-7-PHOSPHATRICYCLO[3.3.1.13,7]DECANE, 2,2-DIOXIDE](http://img.cochemist.com/ccimg/55800/55776-69-7.png)
![2-THIA-1,3,5-TRIAZA-7-PHOSPHATRICYCLO[3.3.1.13,7]DECANE, 2,2-DIOXIDE](http://img.cochemist.com/ccimg/55800/55776-69-7_b.png)
![Ferrate(4-), chloro[[4,4',4'',4'''-(21H,23H-porphine-5,10,15,20-tetrayl-κN21,κN22,κN23,κN24)tetrakis[benzoato]](6-)]-, hydrogen (1:4), (SP-5-12)-](/data/chemimg/99300/55266-17-6.png)
![Ferrate(4-), chloro[[4,4',4'',4'''-(21H,23H-porphine-5,10,15,20-tetrayl-κN21,κN22,κN23,κN24)tetrakis[benzoato]](6-)]-, hydrogen (1:4), (SP-5-12)-](/data/chemimg/99300/55266-17-6_b.png)
![L-Asparticacid, N-[(phenylmethoxy)carbonyl]-, 1,4-bis(1,1-dimethylethyl) ester](http://img.cochemist.com/ccimg/42500/42417-76-5.png)
![L-Asparticacid, N-[(phenylmethoxy)carbonyl]-, 1,4-bis(1,1-dimethylethyl) ester](http://img.cochemist.com/ccimg/42500/42417-76-5_b.png)






![3-METHYL-3,4-DIHYDRO-1H-BENZO[E][1,4]DIAZEPINE-2,5-DIONE](http://img.cochemist.com/ccimg/30900/30860-31-2.png)
![3-METHYL-3,4-DIHYDRO-1H-BENZO[E][1,4]DIAZEPINE-2,5-DIONE](http://img.cochemist.com/ccimg/30900/30860-31-2_b.png)
![[1,2-BIS(DIPHENYLPHOSPHINO)ETHANE]TETRACARBONYLTUNGSTEN](http://img.cochemist.com/ccimg/29900/29890-05-9.png)
![[1,2-BIS(DIPHENYLPHOSPHINO)ETHANE]TETRACARBONYLTUNGSTEN](http://img.cochemist.com/ccimg/29900/29890-05-9_b.png)


![POLY[OXY[(1R)-1-METHYL-2-OXO-1,2-ETHANEDIYL]]](http://img.cochemist.com/ccimg/27000/26917-25-9.png)
![POLY[OXY[(1R)-1-METHYL-2-OXO-1,2-ETHANEDIYL]]](http://img.cochemist.com/ccimg/27000/26917-25-9_b.png)
![Poly[oxy(1-methyl-3-oxo-1,3-propanediyl)]](http://img.cochemist.com/ccimg/26800/26744-04-7.png)
![Poly[oxy(1-methyl-3-oxo-1,3-propanediyl)]](http://img.cochemist.com/ccimg/26800/26744-04-7_b.png)












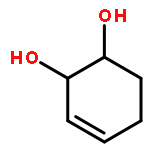
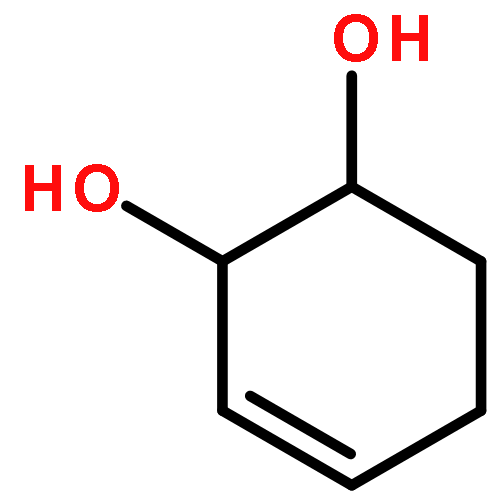
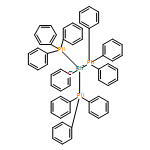
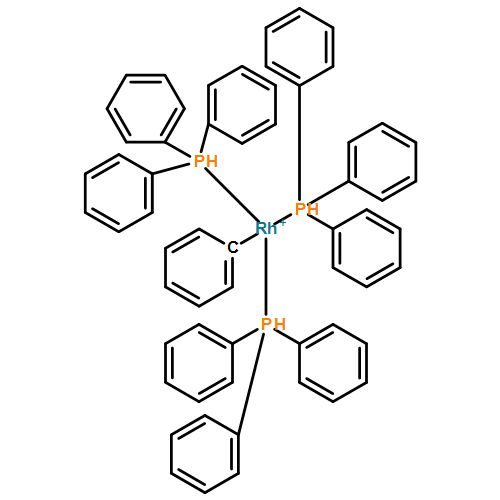










![Iron,tetracarbonyl[(2,3-h)-methyl 2-propenoate]-](http://img.cochemist.com/ccimg/12300/12287-67-1.png)
![Iron,tetracarbonyl[(2,3-h)-methyl 2-propenoate]-](http://img.cochemist.com/ccimg/12300/12287-67-1_b.png)






![4-[2-(4-OXOPENTAN-2-YLIDENEAMINO)ETHYLIMINO]PENTAN-2-ONE](http://img.cochemist.com/ccimg/6400/6310-76-5.png)
![4-[2-(4-OXOPENTAN-2-YLIDENEAMINO)ETHYLIMINO]PENTAN-2-ONE](http://img.cochemist.com/ccimg/6400/6310-76-5_b.png)












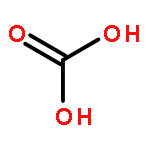
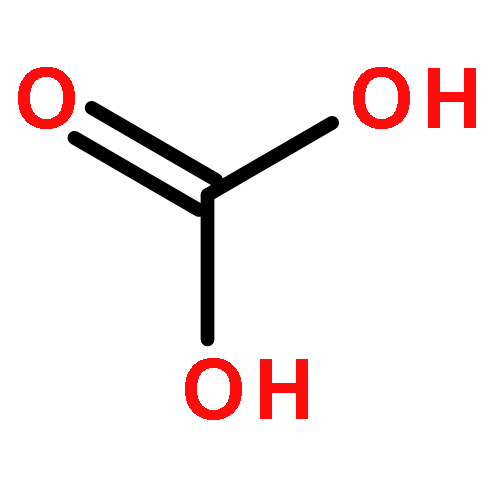
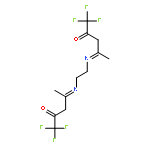
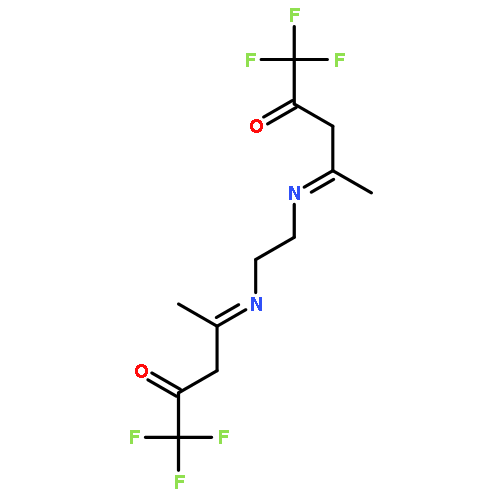
![6-thiabicyclo[3.1.0]hexane](http://img.cochemist.com/ccimg/300/285-75-6.png)
![6-thiabicyclo[3.1.0]hexane](http://img.cochemist.com/ccimg/300/285-75-6_b.png)


![Phenol, 2,4-bis(1,1-dimethylethyl)-6-[(8-quinolinylimino)methyl]-](http://img.cochemist.com/ccimg/252100/252048-83-2.png)
![Phenol, 2,4-bis(1,1-dimethylethyl)-6-[(8-quinolinylimino)methyl]-](http://img.cochemist.com/ccimg/252100/252048-83-2_b.png)
![Phenol, 2,4-bis(1,1-dimethylethyl)-6-[[(2-pyridinylmethyl)imino]methyl]-](http://img.cochemist.com/ccimg/252100/252048-82-1.png)
![Phenol, 2,4-bis(1,1-dimethylethyl)-6-[[(2-pyridinylmethyl)imino]methyl]-](http://img.cochemist.com/ccimg/252100/252048-82-1_b.png)
![Phenol, 2-[[[2,6-bis(1-methylethyl)phenyl]imino]methyl]-4,6-dichloro-](http://img.cochemist.com/ccimg/210900/210882-40-9.png)
![Phenol, 2-[[[2,6-bis(1-methylethyl)phenyl]imino]methyl]-4,6-dichloro-](http://img.cochemist.com/ccimg/210900/210882-40-9_b.png)
![Naphtho[2,3-d]-1,3-dioxol-2-one, 3a,4,9,9a-tetrahydro-, (3aR,9aS)-rel-](http://img.cochemist.com/ccimg/127600/127541-95-1.png)
![Naphtho[2,3-d]-1,3-dioxol-2-one, 3a,4,9,9a-tetrahydro-, (3aR,9aS)-rel-](http://img.cochemist.com/ccimg/127600/127541-95-1_b.png)
![Phenol,2,2'-[1,2-phenylenebis(nitrilomethylidyne)]bis[4,6-bis(1,1-dimethylethyl)-](http://img.cochemist.com/ccimg/103600/103595-82-0.png)
![Phenol,2,2'-[1,2-phenylenebis(nitrilomethylidyne)]bis[4,6-bis(1,1-dimethylethyl)-](http://img.cochemist.com/ccimg/103600/103595-82-0_b.png)
![1-methyl-3,5-diaza-1-azonia-7-phosphatricyclo[3.3.1.1~3,7~]decane](http://img.cochemist.com/ccimg/53600/53597-71-0.png)
![1-methyl-3,5-diaza-1-azonia-7-phosphatricyclo[3.3.1.1~3,7~]decane](http://img.cochemist.com/ccimg/53600/53597-71-0_b.png)




![Poly[oxy(1-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/25300/25248-42-4.png)
![Poly[oxy(1-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/25300/25248-42-4_b.png)









![7-oxabicyclo[4.1.0]hept-3-ene](http://img.cochemist.com/ccimg/6300/6253-27-6.png)
![7-oxabicyclo[4.1.0]hept-3-ene](http://img.cochemist.com/ccimg/6300/6253-27-6_b.png)












![Naphth[2,3-b]oxirene,1a,2,7,7a-tetrahydro-](http://img.cochemist.com/ccimg/2500/2461-35-0.png)
![Naphth[2,3-b]oxirene,1a,2,7,7a-tetrahydro-](http://img.cochemist.com/ccimg/2500/2461-35-0_b.png)


![Poly[oxycarbonyloxy(phenyl-1,2-ethanediyl)]](http://img.cochemist.com/ccimg/70700/70694-67-6.png)
![Poly[oxycarbonyloxy(phenyl-1,2-ethanediyl)]](http://img.cochemist.com/ccimg/70700/70694-67-6_b.png)
![Poly[oxy[(1S)-1-methyl-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26200/26161-42-2.png)
![Poly[oxy[(1S)-1-methyl-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26200/26161-42-2_b.png)






![[1,1':3',1''-Terphenyl]-2'-ol](http://img.cochemist.com/ccimg/2500/2432-11-3.png)
![[1,1':3',1''-Terphenyl]-2'-ol](http://img.cochemist.com/ccimg/2500/2432-11-3_b.png)


![Poly[oxycarbonyloxy(methyl-1,2-ethanediyl)]](http://img.cochemist.com/ccimg/37300/37248-85-4.png)
![Poly[oxycarbonyloxy(methyl-1,2-ethanediyl)]](http://img.cochemist.com/ccimg/37300/37248-85-4_b.png)




![Chromium,chloro[[2,2'-[(1R,2R)-1,2-cyclohexanediylbis[(nitrilo-kN)methylidyne]]bis[4,6-bis(1,1-dimethylethyl)phenolato-kO]](2-)]-, (SP-5-13)-](http://img.cochemist.com/ccimg/165000/164931-83-3.png)
![Chromium,chloro[[2,2'-[(1R,2R)-1,2-cyclohexanediylbis[(nitrilo-kN)methylidyne]]bis[4,6-bis(1,1-dimethylethyl)phenolato-kO]](2-)]-, (SP-5-13)-](http://img.cochemist.com/ccimg/165000/164931-83-3_b.png)
![1,3,5-Triaza-7-phosphatricyclo[3.3.1.13,7]decane](http://img.cochemist.com/ccimg/53600/53597-69-6.png)
![1,3,5-Triaza-7-phosphatricyclo[3.3.1.13,7]decane](http://img.cochemist.com/ccimg/53600/53597-69-6_b.png)