Co-reporter:Fengying Zhang, Xinyu Song, and Yuxiang Bu
The Journal of Physical Chemistry C August 17, 2017 Volume 121(Issue 32) pp:17160-17160
Publication Date(Web):August 1, 2017
DOI:10.1021/acs.jpcc.7b06429
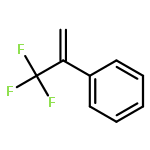
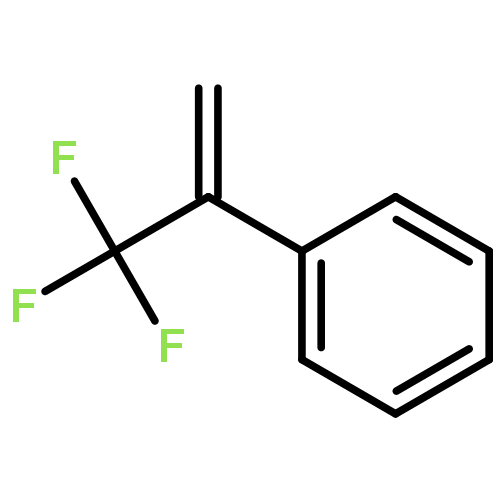
Proton-induced magnetic enhancement in an organic diradical is an appealing phenomenon. Here, taking two nitroxide groups as spin sources, we predict the magnetic properties of the trans and cis forms of azobenzene (AB)-bridged diradicals in which the central −N═N– unit can undergo single protonation to convert to its protonated counterpart or vice versa. The calculated results for these two pairs of diradicals (protonated versus unprotonated trans and cis forms) indicate that the signs of their magnetic coupling constants J do not change, but the magnitudes remarkably increase after protonation from −716.4 to −1787.1 cm–1 for the trans form and from −388.1 to −1227.9 cm–1 for the cis form, respectively. Such noticeable magnetic enhancements induced by protonation are mainly attributed to the strong mediating role of the coupler AB between two radical groups through its lowest unoccupied molecular orbital (LUMO) with a lower energy level after protonation. The planar structure for the protonated trans diradical and two reduced CCNN torsional angles due to protonation for the cis one are responsible for the significant magnetic enhancements. Protonation not only supports the development of π conjugation among the spin groups and coupler but also creates a very favorable condition for spin transmission through the coupler AB LUMO by lowering the LUMO energy level and improving spin polarization and charge delocalization and thus enhances the spin coupling effectively. In addition, different spin sources and linking modes of the radical groups are also considered to confirm our conclusions, and the possibilities of protonation of such diradical systems are further discussed. The studied diradicals could be the promising candidates for the rational design of magnetic molecular switches.
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 4) pp:2816-2825
Publication Date(Web):2017/01/25
DOI:10.1039/C6CP07628A
In this work, the effect of diffuse function types (atom-centered diffuse functions versus floating functions and s-type versus p-type diffuse functions) on the structures and properties of three representative water cluster anions featuring a surface-bound excess electron is studied and we find that an effective combination of such two kinds of diffuse functions can not only reduce the computational cost but also, most importantly, considerably improve the accuracy of results and even avoid incorrect predictions of spectra and the EE shape. Our results indicate that (a) simple augmentation of atom-centered diffuse functions is beneficial for the vertical detachment energy convergence, but it leads to very poor descriptions for the singly occupied molecular orbital (SOMO) and lowest unoccupied molecular orbital (LUMO) distributions of the water cluster anions featuring a surface-bound excess electron and thus a significant ultraviolet spectrum redshift; (b) the ghost-atom-based floating diffuse functions can not only contribute to accurate electronic calculations of the ground state but also avoid poor and even incorrect descriptions of the SOMO and the LUMO induced by excessive augmentation of atom-centered diffuse functions; (c) the floating functions can be realized by ghost atoms and their positions could be determined through an optimization routine along the dipole moment vector direction. In addition, both the s- and p-type floating functions are necessary to supplement in the basis set which are responsible for the ground (s-type character) and excited (p-type character) states of the surface-bound excess electron, respectively. The exponents of the diffuse functions should also be determined to make the diffuse functions cover the main region of the excess electron distribution. Note that excessive augmentation of such diffuse functions is redundant and even can lead to unreasonable LUMO characteristics.
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 8) pp:5932-5943
Publication Date(Web):2017/02/23
DOI:10.1039/C6CP08201G
While the conductance behavior of carbon-based couplers has been successfully investigated, insight into the magnetic properties of such carbon-based molecule coupled diradical systems is still scarce, and especially the structural effect of such couplers on the magnetic properties is poorly understood. The present work reports three different interference effects on the magnetic properties of carbon-based molecule coupled nitroxide diradicals: twisting, sideways group, and position effects. DFT calculations reveal that (i) torsion does not change their broken-symmetry singlet ground state and antiferromagnetic coupling, but decreases their magnetism; (ii) different linkages of two radical moieties result in different ground states and thus different magnetisms, depending on a combination of meta-sites and para-sites; (iii) the antiferromagnetic coupling with a broken-symmetry singlet ground state is not changed by adding sideways groups, but the coupling magnitude can be tuned by modifying the side-bridge. Discussions on geometries, magnetic properties, SOMO–SOMO splittings, and spin density distributions are made to clarify relevant magnetic behaviors. Clearly, the findings concerning the regulation of the diradicalized material molecules through modifying the carbon-based bridges provide a comprehensive understanding of the magnetism of such carbon-based diradicals and new prospects for the design of building blocks of magnetic functional molecular materials.
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 21) pp:13807-13818
Publication Date(Web):2017/05/31
DOI:10.1039/C7CP01847A
In this work, we present an ab initio molecular dynamics simulation study on the interaction of an excess electron (EE) with histidine in its aqueous solution. Two different configurations of histidine (imidazole group protonated or not) are considered to reflect its different existing forms in neutral or slightly acidic surroundings. The simulation results indicate that localizations of EEs in different aqueous histidine solutions are quite different and are strongly affected by protonation of the side chain imidazole group and are thus pH-controlled. In neutral aqueous histidine solution, an EE localizes onto the carboxyl anionic group of the amino acid backbone after a relatively lengthy diffuse state, performing just like in an aliphatic amino acid solution. But in weakly acidic solution in which the side chain imidazole group is protonated, an EE undergoes a short lifetime diffuse state and finally localizes on the protonated imidazole group. We carefully examine these two different localization dynamics processes and analyze the competition between different dominating groups in their corresponding electron localization mechanisms. To explain the difference, we investigate the frontier molecular orbitals of these two systems and find that their energy levels and compositions are important to determine these differences. These findings can provide helpful information to understand the interaction mechanisms of low energy EEs with amino acids and even oligopeptides, especially with aromatic rings.
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 28) pp:18868-18879
Publication Date(Web):14 Jun 2016
DOI:10.1039/C6CP03552C
In this work, we conduct ab initio molecular dynamics simulations on the localization dynamics of an excess electron (EE) in acetamide/Ca2+ aqueous solutions with three different interaction modes of Ca2+ with acetamide: tight contact, solvent-shared state, and separated interaction. The simulated results reveal that an EE could exhibit two different localization behaviors in these acetamide/Ca2+ aqueous solutions depending on different amide⋯Ca2+ interactions featuring different contact distances. For the tight contact and solvent-shared state of amide⋯Ca2+ solutions, vertically injected diffuse EEs follow different mechanisms with different dynamics, forming a cavity-shaped hydrated electron or a hydrated amide anion, respectively. Meanwhile, for the separated state, only one localization pattern of a vertically injected diffuse EE towards the formation of hydrated amide anion is observed. The hindrance of hydrated Ca2+ and the attraction of the hydrated amide group originating from its polarity and low energy π* orbital are the main driving forces. Additionally, different EE localization modes have different effects on the interaction between the amide group and Ca2+ in turn. This work provides an important basis for further understanding the mechanisms and dynamics of localizations/transfers of radiation-produced EEs and associated EE-induced lesions and damage to biological species in real biological environments or other aqueous solutions.
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 4) pp:2913-2923
Publication Date(Web):21 Dec 2015
DOI:10.1039/C5CP06133D
Novel DNA base pair derivatives (A2CunU, A2CunC, G3CunU, and G3CunC) are designed by aromatic expansion of pyrimidine bases with four kinds of hetero-rings (denoted by nC and nU, n = 1, 2, 3, and 4) and metal-decoration through Cu replacement of hydrogens in the Watson–Crick hydrogen bond region. Their structures and properties are calculated for examining the cooperative effects of the two modification ways. The calculated results reveal that multiple Cu decoration makes up the deficiencies of size-expansion, and exhibits not only increase of structural stability and reduction of ionization potentials, but also ideal shrink of the HOMO–LUMO gaps, notable enhancement of interbase coupling as well as remarkable redshifts of π → π* transitions for all M-x modified base pairs. The decrease extents of the gaps and ionization potentials follow the same order G3CunU > G3CunC > A2CunU > A2CunC, and in each series (denoted by different n), the gaps, ionization potentials and first π → π* transition energies have an order of 4 < 1 < 2 < 3. The Cu d orbitals function as bridges for π electron delocalization on the conjugated aromatic rings of two bases, leading to an enhancement of transverse electronic communication, as verified by spin density delocalization, orbital composition changes, redshift of the π → π* transition and also advocated by the electron-sharing indexes such as delocalization index, Mayer bond orders and multicenter bonding. Electron localization function ELF-π isosurfaces above the molecular plane further suggested that effective longitudinal conduction is closely relevant with the bicyclic domain involving good electron delocalization and strong π–π stacking between layers. This work presents theoretical evidence for the cooperative effects of metal decoration and ring-expansion modifications on the electronic properties of the modified base pairs and also proves that the base pairs designed here could be competent building blocks for the DNA-based nanowires with improved electron activity and excellent conductivity.
The Journal of Physical Chemistry C 2016 Volume 120(Issue 19) pp:10215-10226
Publication Date(Web):May 3, 2016
DOI:10.1021/acs.jpcc.6b03891
While the photoelectrochemical behavior of azapentacene has been investigated successfully, insight into the dynamic electronic properties of azapentacene triggered by different energy pulses is very scarce. The present work reports a fascinating phenomenon about potential diradical character governed by structural vibrations in hexazapentacene. In complete contrast to the static equilibrium configuration of hexazapentacene without diradical character, due to the vibration-based structural perturbation, DFT calculations show that some of the transient configurations possess diradical character and thus magnetism, which exhibit different periodic pulse behavior in time evolution. Since each vibrational mode refers to two distortion ways (positive/negative distortions from equilibrium configuration), 7 different possibilities are observed for the vibration-induced diradical character for all vibrational modes (e.g., a combination of nonradical, singlet diradical, or triplet diradical for positive distortion and those of for negative distortion for each vibrational mode). This intriguing diradical character is rationalized by structural distortions with considerable changes of some energy quantities. The structural distortions cause the HOMO energy raising and LUMO energy lowering and thus an efficient reduction of the HOMO–LUMO energy gap and singlet–triplet gap of the system, which are favorable to the formation of the broken-symmetry open-shell singlet or triplet states. The periodic pulsing behavior is attributed to persistent molecular vibrations and is thus vibrational mode controlled. Compared with pentacene, the remarked effects of nitrogen substitution on the diradical properties and their pulsing behaviors are mainly due to the decreases of both the HOMO and the LUMO energies and considerable narrowing of their gaps in the vibrations-distorted configurations. This intriguing potential diradical character and its different dynamic behavior suggest hexazapentacene potential applications as promising building blocks in the rational design of novel electromagnetic materials because of its controllable magnetism through energy pulses. This work provides comprehensive understanding of the nature of dynamic variations of the electronic structures and properties of the nitrogen-rich acene derivatives and other materials molecules.
The role of adenine (A) derivatives in DNA damage is scarcely studied due to the low electron affinity of base A. Experimental studies demonstrate that low-energy electron (LEE) attachment to adenine derivatives complexed with amino acids induces barrier-free proton transfer producing the neutral N7-hydrogenated adenine radicals rather than conventional anionic species. To explore possible DNA lesions at the A sites under physiological conditions, probable bond ruptures in two models—N7-hydrogenated 2′-deoxyadenosine-3′-monophosphate (3′-dA(N7H)MPH) and 2′-deoxyadenosine-5′-monophosphate (5′-dA(N7H)MPH), without and with LEE attachment—are studied by DFT. In the neutral cases, DNA backbone breakage and base release resulting from C3′−O3′ and N9−C1′ bond ruptures, respectively, by an intramolecular hydrogen-transfer mechanism are impossible due to the ultrahigh activation energies. On LEE attachment, the respective C3′−O3′ and N9−C1′ bond ruptures in [3′-dA(N7H)MPH]− and [5′-dA(N7H)MPH]− anions via a pathway of intramolecular proton transfer (PT) from the C2′ site of 2′-deoxyribose to the C8 atom of the base moiety become effective, and this indicates that substantial DNA backbone breaks and base release can occur at non-3′-end A sites and the 3′-end A site of a single-stranded DNA in the physiological environment, respectively. In particular, compared to the results of previous theoretical studies, not only are the electron affinities of 3′-dA(N7H)MPH and 5′-dA(N7H)MPH comparable to those of hydrogenated pyrimidine derivatives, but also the lowest energy requirements for the C3′−O3′ and N9-glycosidic bond ruptures in [3′-dA(N7H)MPH]− and [5′-dA(N7H)MPH]− anions, respectively, are comparable to those for the C3′−O3′ and N1-glycosidic bond cleavages in corresponding anionic hydrogenated pyrimidine derivatives. Thus, it can be concluded that the role of adenine derivatives in single-stranded DNA damage is equally important to that of pyrimidine derivatives in an irradiated cellular environment.
Co-reporter:Shoushan Wang, Peiwen Zhao, Changzhe Zhang, and Yuxiang Bu
The Journal of Physical Chemistry B 2016 Volume 120(Issue 10) pp:2649-2657
Publication Date(Web):February 25, 2016
DOI:10.1021/acs.jpcb.5b11432
Experimental studies showed that high energy radiation induced base release and DNA backbone breaks mainly occur at the neighboring 5′ nucleotide when a single-stranded DNA is modified by radiosensitizing 5-halogenated deoxyuridines. However, no mechanism can be used to interpret these experimental observations. To better understand the radiosensitivity of 5-halogenated deoxyuridines, mechanisms involving hydrogen abstraction by the uracil-5-yl radical from the C2′ and C3′ positions of an adjacent nucleotide separately followed by the C3′–O3′ or N–glycosidic bond rupture and the P–O3′ bond breakage are investigated in the DNA sequence 5′-TU•-3′ employing density functional theory calculations in the present study. It is found that hydrogen abstractions from both positions are comparable with the one from the C2′ site slightly more favorable. The N–glycosidic bond cleavage in the neighboring 5′ nucleotide following the internucleotide C2′–Ha abstraction is estimated to have the lowest activation free energies, indicating that the adjacent 5′ base release dominates electron induced damage to single-stranded DNA incorporated by 5-halogenated deoxyuridines. Relative to the P–O3′ bond breakage after the internucleotide C3′-H abstraction, the C3′–O3′ bond rupture in the neighboring 5′ nucleotide following the internucleotide C2′-Ha abstraction is predicted to have a lower activation free energy, implying that single-stranded DNA backbone breaks are prone to occur at the C3′–O3′ bond site. The 5′-TU•-3′ species has substantial electron affinity and can even capture a hydrated electron, forming the 5′-TU–-3′ anion. However, the electron induced C3′–O3′ bond rupture in 5′-TU–-3′ anion via a pathway of internucleotide proton abstraction is only minor in both the gas phase and aqueous solution. The present theoretical predictions can interpret rationally experimental observations, thereby demonstrating that the mechanisms proposed here are responsible for high energy radiation induced damage to single-stranded DNA incorporated by radiosensitizing 5-halogenated deoxyuridines. By comparing with previous results, our work proves that the radiosensitizing action of 5-bromo-2-deoxyuridine is not weaker but stronger than its isomer 6-bromo-2-deoxyuridine on the basis of the available data.
Co-reporter:Xiuxiu Wu, Liang Gao, Jinxiang Liu, Hongfang Yang, Shoushan Wang and Yuxiang Bu
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 40) pp:26854-26863
Publication Date(Web):15 Sep 2015
DOI:10.1039/C5CP03720D
Studies on the structure, states, and reactivity of excess electrons (EEs) in biological media are of great significance. Although there is information about EE interaction with desolvated biological molecules, solution effects are hardly explored. In this work, we present an ab initio molecular dynamics simulation study on the interaction and reactivity of an EE with glycine in solution. Our simulations reveal two striking results. Firstly, a pre-solvated EE partially localizes on the negatively charged –COO− group of the zwitterionic glycine and the remaining part delocalizes over solvent water molecules, forming an anion-centered quasi-localized structure, due to relative alignment of the lowest unoccupied molecular orbital energy levels of potential sites for EE residence in the aqueous solution. Secondly, after a period of anion-centered localization of an EE, the zwitterionic glycine is induced to spontaneously fragment through the cleavage of the N–Cα bond, losing ammonia (deamination), and leaving a ˙CH2–COO− anion radical, in good agreement with experimental observations. Introduction of the same groups (–COO− or –NH3+) in the side chain (taking lysine and aspartic acid as examples) can affect EE localization, with the fragmentation of the backbone part of these amino acids dependent on the properties of the side chain groups. These findings provide insights into EE interaction mechanisms with the backbone parts of amino acids and low energy EE induced fragmentation of amino acids and even peptides and proteins.
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 30) pp:19797-19805
Publication Date(Web):24 Jun 2015
DOI:10.1039/C5CP02693H
Radiation-generated secondary electrons can induce resonance processes in a target molecule and fragment it via different pathways. Although the associating electronic resonant states at equilibrium geometry have been well studied for many target molecules in the gas phase, vibrational resonance contributions and the solvent effect are still poorly understood for relevant processes in solution. Taking a radiosensitive drug, 5-bromopyrimidine (5-BrPy), as an example, we here present a combined ab initio molecular dynamics simulation and time-dependent wave packet study with an emphasis on vibrational resonance and solvation effects on excess electron interaction with 5-BrPy in solution. The gaseous results reveal two primary channels for the electron induced C–Br bond cleavage: the highest vibrational resonance on vertical potential energy curve via a tunneling mechanism , and auto-dissociation along repulsive relaxed potential energy curve , which account for the two peaks at 0.2 and 0 eV observed in Modelli's experiment. However, a strong solvation effect modifies the mechanism and dynamics of the dissociation of the electron⋯5-BrPy system. On one hand, the spontaneous dissociation becomes unfavorable due to a barrier on the relaxed free energy surface created by the coupling between the π* and σ* states. Seven vibrational resonances (v = 0–6) are identified for the solution process and only the high-level v = 5, 6 with non-negligible quantum tunneling coefficient can cause the dissociation . On the other hand, protonation is also observed at the N sites of the hydrated 5-BrPy anion , and this inhibits the dissociation along the C–Br bond, suggesting a competing pathway against C–Br bond cleavage. Clearly, this work provides a combination strategy using an ab initio molecular dynamics technique and time-dependent wave packet method to explore the effects of vibrational resonances and solvation on the interaction of radio-generated excess electrons with target biological molecules in complicated solution surroundings.
Co-reporter:Shoushan Wang, Changzhe Zhang, Peiwen Zhao, and Yuxiang Bu
The Journal of Physical Chemistry B 2015 Volume 119(Issue 44) pp:13971-13979
Publication Date(Web):October 6, 2015
DOI:10.1021/acs.jpcb.5b06195
Possible electron-induced ruptures of C3′–O3′, C5′–O5′, and N1–C1′ bonds in O4-hydrogenated 2′-deoxythymidine-3′-monophosphate (3′-dT(O4H)MPH) and 2′-deoxythymidine-5′-monophosphate (5′-dT(O4H)MPH) are investigated using density functional theory calculations, and efficient pathways are proposed. Electron attachment causes remarkable structural relaxation in the thymine C6 site. A concerted process of intramolecular proton transfer (IPT) from the C2′ site of 2′-deoxyribose to the C6 site and the C3′–O3′ bond rupture is observed in [3′-dT(O4H)MPH]−. A low activation barrier (9.32 kcal/mol) indicates that this pathway is the most efficient one as compared to other known pathways leading to backbone breaks of a single strand DNA at the non-3′-end thymine, which prevents the N1–C1′ bond cleavage in [3′-dT(O4H)MPH]−. However, essentially spontaneous N1–C1′ bond cleavage following similar IPT is predicted in [5′-dT(O4H)MPH]−. A moderate activation barrier (13.02 kcal/mol) for the rate-controlling IPT step suggests that base release from the N1–C1′ cleavage arises readily at the 3′-end of single strand DNA with the strand ended by a thymine. The C5′–O5′ bond has only an insignificant change in the IPT process. Solvent effects are found to increase slightly the energy requirements for either bond ruptures (11.23 kcal/mol (C3′–O3′) vs 16.18 kcal/mol (N1–C1′)), but not change their relative efficiencies.
Co-reporter:Ru Zhang;Dr. Jinxiang Liu;Dr. Hongfang Yang;Shoushan Wang;Meng Zhang ; Dr. Yuxiang Bu
ChemPhysChem 2015 Volume 16( Issue 2) pp:436-446
Publication Date(Web):
DOI:10.1002/cphc.201402657
Abstract
Density functional theory calculations suggest that β-turn peptide segments can act as a novel dual-relay elements to facilitate long-range charge hopping transport in proteins, with the N terminus relaying electron hopping transfer and the C terminus relaying hole hopping migration. The electron- or hole-binding ability of such a β-turn is subject to the conformations of oligopeptides and lengths of its linking strands. On the one hand, strand extension at the C-terminal end of a β-turn considerably enhances the electron-binding of the β-turn N terminus, due to its unique electropositivity in the macro-dipole, but does not enhance hole-forming of the β-turn C terminus because of competition from other sites within the β-strand. On the other hand, strand extension at the N terminal end of the β-turn greatly enhances hole-binding of the β-turn C terminus, due to its distinct electronegativity in the macro-dipole, but does not considerably enhance electron-binding ability of the N terminus because of the shared responsibility of other sites in the β-strand. Thus, in the β-hairpin structures, electron- or hole-binding abilities of both termini of the β-turn motif degenerate compared with those of the two hook structures, due to the decreased macro-dipole polarity caused by the extending the two terminal strands. In general, the high polarity of a macro-dipole always plays a principal role in determining charge-relay properties through modifying the components and energies of the highest occupied and lowest unoccupied molecular orbitals of the β-turn motif, whereas local dipoles with low polarity only play a cooperative assisting role. Further exploration is needed to identify other factors that influence relay properties in these protein motifs.
We present an ab initio molecular dynamics (AIMD) simulation study into the transfer dynamics of an excess electron from its cavity-shaped hydrated electron state to a hydrated nucleobase (NB)-bound state. In contrast to the traditional view that electron localization at NBs (G/A/C/T), which is the first step for electron-induced DNA damage, is related only to dry or prehydrated electrons, and a fully hydrated electron no longer transfers to NBs, our AIMD simulations indicate that a fully hydrated electron can still transfer to NBs. We monitored the transfer dynamics of fully hydrated electrons towards hydrated NBs in aqueous solutions by using AIMD simulations and found that due to solution-structure fluctuation and attraction of NBs, a fully hydrated electron can transfer to a NB gradually over time. Concurrently, the hydrated electron cavity gradually reorganizes, distorts, and even breaks. The transfer could be completed in about 120–200 fs in four aqueous NB solutions, depending on the electron-binding ability of hydrated NBs and the structural fluctuation of the solution. The transferring electron resides in the π*-type lowest unoccupied molecular orbital of the NB, which leads to a hydrated NB anion. Clearly, the observed transfer of hydrated electrons can be attributed to the strong electron-binding ability of hydrated NBs over the hydrated electron cavity, which is the driving force, and the transfer dynamics is structure-fluctuation controlled. This work provides new insights into the evolution dynamics of hydrated electrons and provides some helpful information for understanding the DNA-damage mechanism in solution.
Co-reporter:Jinxiang Liu, Robert I. Cukier, Yuxiang Bu, and Yuan Shang
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 10) pp:4189-4197
Publication Date(Web):August 23, 2014
DOI:10.1021/ct500238k
Ab initio molecular dynamics simulations reveal that an excess electron (EE) can be more efficiently localized as a cavity-shaped state in aqueous glucose solution (AGS) than in water. Compared with that (∼1.5 ps) in water, the localization time is shortened by ∼0.7–1.2 ps in three AGSs (0.56, 1.12, and 2.87 M). Although the radii of gyration of the solvated EEs are all close to 2.6 Å in the four solutions, the solvated EE cavities in the AGSs become more compact and can localize ∼80% of an EE, which is considerably larger than that (∼40–60% and occasionally ∼80%) in water. These observations are attributed to a modification of the hydrogen-bonded network by the introduction of glucose molecules into water. The water acts as a promoter and stabilizer, by forming voids around glucose molecules and, in this fashion, favoring the localization of an EE with high efficiency. This study provides important information about EEs in physiological AGSs and suggests a new strategy to efficiently localize an EE in a stable cavity for further exploration of biological function.
The Journal of Physical Chemistry C 2014 Volume 118(Issue 33) pp:18861-18867
Publication Date(Web):July 30, 2014
DOI:10.1021/jp504041n
Amino fragments (−NH2) are well-known to exist widely in biological systems and their protonated forms are inclined to trap electrons and form Rydberg radicals (−NH3•) in the electron-excess systems. Taking CH3–NH3+ as a mimicking group of the protonated alkylamine side-chain of lysine, ab initio calculations indicate that the proton/electron cooperatively transfer from CH3NH3 to CH3NH2 via a single-proton-coupled Rydberg-state electron transfer (ET) mechanism with an Rydberg-orbital channel for ET outside the −NHn hydrogens and a N–H+ → N proton migrating pathway. Besides, in big amine clusters, CH3NH3·(NH3)n·NH2CH3 (n = 1–3), the proton/electron transfer along an amine wire is stepwise and every step takes place via the similar single-proton-coupled Rydberg-state ET mechanism with low energy barrier (<4.0 kcal/mol). When a water chain, (H2O)n (n = 1–3), lies between CH3NH3 and NH2CH3 as a bridge, the energy barriers (8.5–15.0 kcal/mol) of proton/electron cooperatively transfer between CH3NH3 and NH2CH3 are raised significantly as compared to these of the pure amine wires (<4.0 kcal/mol). We attribute this fact to the combined effects of the proton binding energies and electron affinities of CH3NH2 and H2O. Interestingly, different from the amine-wire case, movement of the solvated electron along the water-wire can promote two or three protons synchronously moving at the same direction. This process can be described in terms of a multi-proton-coupled solvated-ET mechanism.
Co-reporter:Shoushan Wang, Jinxiang Liu, Changzhe Zhang, Li Guo, and Yuxiang Bu
The Journal of Physical Chemistry A 2014 Volume 118(Issue 39) pp:9212-9219
Publication Date(Web):May 15, 2014
DOI:10.1021/jp5030284
We present an ab initio molecular dynamics simulation study of a CH3CN–(H2O)40 cluster with an excess electron (EE) injected vertically in this work. Instead of surface bound or internally solvated electron, a hydrated CH3CN– is first formed as the CN transient after geometrical relaxation. The driving forces for the formation of CH3CN– are bending vibration of ∠CCN angle, which initiates transfer of an extra charge to the CH3CN LUMO, and hydration effect of the immediate water molecules, which plays a stabilizing role. Solvent thermal fluctuation can lead to different resonances (the quasi-C2-resonance versus quasi-N-resonance) from the CN transient and further cause the hydrated CH3CN– system to evolve via two distinctly different pathways featuring spontaneous proton transfer to the central C and N sites, producing two different protonation products, respectively. The solvent thermal fluctuation induced formation of hydrogen bonding with the corresponding sites (C2 versus N) is responsible for the quasi-resonances and interconversion between three resonant structures and further proton transfers featuring spontaneous transfer of a proton to C2 or to N from its interacting water molecule. The duration of CH3CN– for either of the two proton transfer processes is less than 200 fs. On the basis of experimental ESR results in which only the CH3CHN radical was found and present theoretical calculations, it is suggested that the trans-CH3CNH radical can be further converted to the CH3CHN radical via a water-mediated hydrogen atom transfer path.
Co-reporter:Jinxiang Liu, Robert I. Cukier, and Yuxiang Bu
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 11) pp:4727-4734
Publication Date(Web):October 15, 2013
DOI:10.1021/ct4002174
We report an ab initio molecular dynamics simulation study of the solvation and dynamics of an excess electron in liquid acetonitrile (ACN). Four families of states are observed: a diffusely solvated state and three ACN core-localized states with monomer core, quasi-dimer (π*-Rydberg mode) core, and dual-core/dimer core (a coupled dual-core). These core localized states cannot be simply described as the corresponding anions because only a part of the excess electron resides in the core molecule(s). The quasi-dimer core state actually is a mixture that features cooperative excess electron capture by the π* and Rydberg orbitals of two ACNs. Well-defined dimer anion and solvated electron cavity were not observed in the 5–10 ps simulations, which may be attributed to slow dynamics of the formation of the dimer anion and difficulty of the formation of a cavity in such a fluxional medium. All of the above observed states have near-IR absorptions and thus can be regarded as the solvated electron states but with different structures, which can interpret the experimentally observed IR band. These states undergo continuous conversions via a combination of long-lasting breathing oscillation and core switching, characterized by highly cooperative oscillations of the electron cloud volume and vertical detachment energy. The quasi-dimer core and diffusely solvated states dominate the time evolution, with the monomer core and dual-core/dimer core states occurring occasionally during the breathing and core switching processes, respectively. All these oscillations and core switchings are governed by a combination of the electron-impacted bending vibration of the core ACN molecule(s) and thermal fluctuations.
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 42) pp:18453-18463
Publication Date(Web):02 Sep 2013
DOI:10.1039/C3CP52745J
We present a computational study of the double-electron oxidized guanine–cytosine base pair as well as its deprotonated derivatives, focusing on their structural and electronic properties. Some novel electromagnetic characteristics are found. A hydrazine-like (N–N) cross-linked structure between the G and C radical moieties is the lowest-energy one for the [GC]2+ complexes. Double-electron oxidation can considerably destabilize the GC unit and leads to a barrier-hindered dissociation channel with negative dissociation energy. This channel is governed by a balance between electrostatic repulsion and attractive hydrogen-bonding interaction co-existing between G˙+ and C˙+. The proton/electron transfer reactions in the double-electron oxidized Watson–Crick base pair occur through a proton transfer induced charge migration mechanism. For the deprotonated [GC]2+ derivative, the [G(–H+)C]+ series prefers to accompany by transfer of an electron from the G to C moiety when the G+ is deprotonated, and its highest-doubly occupied molecular orbital mainly localizes over the C moiety with a π-bonding character. For the diradical G˙+C(–H)˙ series in which the C moiety is deprotonated, the two unpaired electrons reside one on each moiety in the π system. The diradical base pairs possess open-shell broken symmetry singlet states, and their magnetic coupling interactions are controlled by both intra- and inter-molecular interactions. The double-electron oxidized Watson–Crick base pair shows strong antiferromagnetic coupling, whereas the magnetic interactions of other diradical derivatives are relatively weak. This study highlights the crucial role of H-bonding in determining the magnetic interactions.
Computational and Theoretical Chemistry 2013 Volume 1016() pp:54-61
Publication Date(Web):15 July 2013
DOI:10.1016/j.comptc.2013.04.014
•We explored the electron-binding properties of the hydrated protonated imidazole.•Hydration strongly influences the excess electron binding of the imidazole group.•Electron distribution depends on the number of water molecules and hydration mode.•Excess electron occupies the Rydberg-like orbital or the imidazole π* LUMO.•Geometry relaxation can result in conversion of the states of an excess electron.An excess electron in condensed phase models of hydrated protonated histidine residue (HisH+) in proteins has been investigated using ab initio calculations and molecular dynamics simulations, which focuses on electron-binding mode and evolution mechanism of the captured electron in the HisH+ side chain hydrated clusters. Results indicate that distribution of an excess electron is highly associated with the number of water molecules and the geometric configurations of the hydrated clusters. An electron can stably localize in three cases depending on the size of the hydrated clusters. First, an excess electron always occupies the π* orbital of the imidazole ring not only after vertical binding but also in relaxation process. Second, an excess electron firstly vertically localizes in a Rydberg-like orbital, and then relaxes to reside in the π* LUMO localizing in the imidazole ring. Third, an electron always occupies the Rydberg-like orbital both after vertical binding and after relaxation. Furthermore, molecular dynamics simulations reveal that the captured excess electron prefers to localize around HisH+ no mater how it distributes at the initially bound states (localized versus delocalized), and the evolution time for the solvated electron from the initially bound state to the finally localized state at HisH+ is about 220 fs, as demonstrated from an excess electron-bound HisH+(H2O)40 model cluster.
Density functional theory calculations were employed to study the stabilization process of the guanine radical cation through amino acid interactions as well as to understand the protection mechanisms. On the basis of our calculations, several protection mechanisms are proposed in this work subject to the type of the amino acid. Our results indicate that a series of three-electron bonds can be formed between the amino acids and the guanine radical cation which may serve as relay stations supporting hole transport. In the three-electron-bonded, π–π-stacked, and H-bonded modes, amino acids can protect guanine from oxidation or radiation damage by sharing the hole, while amino acids with reducing properties can repair the guanine radical cation through proton-coupled electron transfer or electron transfer. Another important finding is that positively charged amino acids (ArgH+, LysH+, and HisH+) can inhibit ionization of guanine through raising its ionization potential. In this situation, a negative dissociation energy for hydrogen bonds in the hole-trapped and positively charged amino acid–Guanine dimer is observed, which explains the low hole-trapping efficiency. We hope that this work provides valuable information on how to protect DNA from oxidation- or radiation-induced damages in biological systems.
Co-reporter:Meng Zhang, Jing Zhao, Hongfang Yang, Ping Liu, and Yuxiang Bu
The Journal of Physical Chemistry B 2013 Volume 117(Issue 21) pp:6385-6393
Publication Date(Web):May 8, 2013
DOI:10.1021/jp4012526
We present a density functional calculational study to clarify that a 310-helix peptide can serve as a novel dual-relay element to mediate long-range charge migrations via its C- and N-termini in proteins. The ionization potential of the 310-helix C-terminus correlates inversely with the helix length, HOMO energy, and dipole moment. In particular, it decreases considerably with the increase in the number of peptide units, even to a smaller value than that of the easily oxidized amino acid residue, which implies the possibility of releasing an electron and forming a hole at the 310-helical C-terminus. On the other hand, the electron affinity of the 310-helical N-terminus correlates positively with the helix length and dipole moment but inversely with the LUMO energy. Clearly, the increasing positive electron-binding energy with the increase in the number of peptide units implies that the 310-helical N-terminus can capture an excess electron and play an electron-relaying role. The relaying ability of the 310-helical C-terminus and N-terminus not only depends on the helix length but also varies subject to the capping effect, the collaboration and competition of proximal groups, and solvent environments. In contrast to the known hole relays such as the side chains of Tyr and Trp and electron relays such as the side chains of protonated Lys and Arg, either the hole relay (the 310-helix C-terminus) or the electron relay (the 310-helix N-terminus) is property-tunable and could apply to different proteins in assisting or mediating long-range charge migrations.
Co-reporter:Jing Zhao, Mei Wang, Hongfang Yang, Meng Zhang, Ping Liu, and Yuxiang Bu
The Journal of Physical Chemistry B 2013 Volume 117(Issue 37) pp:10698-10710
Publication Date(Web):August 21, 2013
DOI:10.1021/jp4042149
We present here a theoretical investigation of the structural and electronic properties of di-ionized GG base pairs (G•+G•+,G(-H1)•G•+, and G(-H1)•G(-H1)•) consisting of the guanine cation radical (G•+) and/or dehydrogenated guanine radical (G(-H1)•) using density functional theory calculations. Different coupling modes (Watson–Crick/WC, Hoogsteen/Hoog, and minor groove/min hydrogen bonding, and π–π stacking modes) are considered. We infer that a series of G•+G•+ complexes can be formed by the high-energy radiation. On the basis of density functional theory and complete active space self-consistent (CASSCF) calculations, we reveal that in the H-bonded and N–N cross-linked modes, (G•+G•+)WC, (G(-H1)•G(-H1)•)WC, (G(-H1)•G(-H1)•)minI, and (G(-H1)•G(-H1)•)minIII have the triplet ground states; (G•+G•+)HoogI, (G(-H1)•G•+)WC, (G(-H1)•G•+)HoogI, (G(-H1)•G•+)minI, (G(-H1)•G•+)minII, and (G(-H1)•G(-H1)•)minII possess open-shell broken-symmetry diradical-characterized singlet ground states; and (G•+G•+)HoogII, (G•+G•+)minI, (G•+G•+)minII, (G•+G•+)minIII, (G•+G•+)HoHo, (G(-H1)•G•+)minIII, (G(-H1)•G•+)HoHo, and (G(-H1)•G(-H1)•)HoHo are the closed-shell systems. For these H-bonded diradical complexes, the magnetic interactions are weak, especially in the diradical G•+G•+ series and G(-H1)•G(-H1)• series. The magnetic coupling interactions of the diradical systems are controlled by intermolecular interactions (H-bond, electrostatic repulsion, and radical coupling). The radical–radical interaction in the π–π stacked di-ionized GG base pairs ((G•+G•+)ππ, (G(-H1)•G•+)ππ, and (G(-H1)•G(-H1)•)ππ) are also considered, and the magnetic coupling interactions in these π–π stacked base pairs are large. This is the first theoretical prediction that some di-ionized GG base pairs possess diradical characters with variable degrees of ferromagnetic and antiferromagnetic characteristics, depending on the dehydrogenation characters of the bases and their interaction modes. Hopefully, this work provides some helpful information for the understanding of different structures and properties of the di-ionized GG base pairs.
Co-reporter:Boran Han, Xiaohua Chen, Jing Zhao and Yuxiang Bu
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 45) pp:15849-15859
Publication Date(Web):01 Oct 2012
DOI:10.1039/C2CP41566F
This work presents a density functional theory calculational study for clarifying that peptide loops (–[peptide]n–) including the N-terminal and the C-terminal oligopeptides and the α-helix N-terminal can serve as an intriguing kind of relay elements, as an addition to the known relay stations served by aromatic amino acids for electron hopping migration. For these protein motifs, an excess electron generally prefers to reside at the –NH3+ group in a Rydberg state for the N-terminal peptides, or at the –COOH group in a dipole-bound state for the C-terminal peptides, and at the N-terminal in a dipole-bound π*-orbital state for the peptide loops and α-helices. The electron binding ability can be effectively enhanced by elongation for the α-helix N-terminal, and by bending, twisting, and even β-turning for the peptide chains. The relay property is determined by the local dipole instead of the total dipole of the peptide chains. Although no direct experiment supports this hypothesis, a series of recent studies regarding charge hopping migration associated with the peptide chains and helices could be viewed as strong evidence. But, further studies are still needed by considering the effects from relative redox potential between the donor and acceptor sites, protein environment, and structure water molecules.
Co-reporter:Hongfang Yang, Qisheng Song, Wenchao Li, Xinyu Song, and Yuxiang Bu
The Journal of Physical Chemistry C 2012 Volume 116(Issue 9) pp:5900-5907
Publication Date(Web):February 1, 2012
DOI:10.1021/jp2107654
Two classes of multi-Zn-expanded oligoacenes from benzene to pentacene are computationally designed through introducing a Zn array into acene rings in two ways: acene-chain axial versus single-ring quasi-transversal direction. Combined density functional theory, CASSCF, and CCSD calculations predict that all these multi-Zn-expanded oligoacenes have the open-shell singlet diradical ground states, in contrast with the common fact that their parent oligoacenes are closed-shell systems or may have a triplet ground state and only acenes larger than octacene have open-shell singlet diradical ground states. These results offer the first theoretical prediction that the multi-Zn introduction into the acene ring(s), forming the Zn-expanded oligoacenes, can lead them to diradical structures. The diradical character of the ground states of these molecules arises from the Zn-participation-induced disjoint nature of the nonbonding molecular orbitals that are singly occupied in the diradicals. This work provides a strategy to design perfect and stable singlet diradicals from oligoacenes or their derivatives.
Co-reporter:Li Han, Hongfang Yang, Jing Zhao, and Yuxiang Bu
The Journal of Physical Chemistry C 2012 Volume 116(Issue 44) pp:23214-23223
Publication Date(Web):October 23, 2012
DOI:10.1021/jp306099m
In the present work, cyclopentadienyl radicals are introduced to nucleobases to gain the building blocks of DNA-based molecular wires with novel electromagnetic characteristics. Calculations reveal that the radicalized DNA bases exist stably because their extended π-conjugated structures are beneficial to spin delocalization, diradical base pairs possess open-shell singlet ground states, and magnetic coupling interactions of the multiradical systems are controlled by both intra- and intermolecular interactions. For the designed base pairs, the intra-base-pair magnetic interactions are weak, especially in the diradical rA–rT base pair; as for the inter-base-pair magnetic interactions, different cases are observed depending on the relative position of the radicalized bases. The overlap-stacking diradical helices manifest variable degrees of ferromagnetic and antiferromagnetic characteristics, whereas the magnetic coupling interactions in the cross-stacking diradical helices are generally weak. The latter is attributed to the long spatial distances between the two spin centers. Thus, for the tetraradical helices, their magnetic characteristics can be viewed as a combination of two overlap-stacking diradical base pairs, and mostly are antiferromagnetic. This work provides a reasonable strategy of designing magnetic building blocks for the magnetic DNA molecular wires or DNA molecular magnets.
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 13) pp:5906-5914
Publication Date(Web):18 Feb 2011
DOI:10.1039/C0CP02297G
Motivated by a promising expansion of the genetic alphabet and a successful design of conductive DNA bases justified from the hetero-ring-expanded purine base (G and A) analogs, we extend our hetero-ring expansion scheme to the pyrimidine bases (C and T) to examine the ring-expansion effects on various properties of these single-ring bases with a comparison with those in the double-ring purine case. Four kinds of the hetero-rings are considered to expand C and T, forming the C and T analogs (nC and nT), respectively. The relevant structures and properties were investigated by means of quantum calculations and molecular dynamics simulations. The results reveal that all the modified bases can form base pairs specifically with their natural counterparts and assemble duplex helices which have comparable stability to native ones. The HOMO–LUMO gaps of G–nC and A–nT are smaller than those of the natural pairs, and the assembled duplex helices ((G–nC)12 and (A–nT)12) are diameter-enlarged but with smaller rise and twist, both of which favor DNA-conduction, as confirmed by ionization potentials and spin density distributions. In addition, the hetero-ring expansion can lower the activation barriers and reduce the reaction heats of the inter-base double proton transfers. In particular, as evidenced by NMR parameters and the excited states, the hetero-ring expansion leads to an enhancement of the transverse electronic communication between two pairing bases, clearly facilitating the conduction along the helices. Furthermore, the hetero-ring expansion effect on the pyrimidine bases is larger than that on the purine bases. In summary, this work presents clear theoretical evidence for the possibility of hetero-ring expanded pyrimidine bases as promising candidates for the motifs of the genetic alphabet and DNA nanowires.
Co-reporter:Genqin Li ; Haiying Liu ; Xiaohua Chen ; Laibin Zhang
The Journal of Physical Chemistry C 2011 Volume 115(Issue 6) pp:2855-2864
Publication Date(Web):January 24, 2011
DOI:10.1021/jp107605k
Transition-metal-mediated base pairs are under intense research because of their potential application in nanoscale molecular devices. To pursue suitable building blocks for DNA-based molecular wires, a three-copper-mediated guanine−cytosine (G3CuC) and a two-copper-mediated adenine−thymine (A2CuT) base pair were designed by equi-stoichiometric H-by-Cu replacements in this Article. Their structural and electronic properties were examined by theoretical methods. Geometrically, G3CuC and A2CuT have great resemblances to the natural GC and AT with a size-expansion of about 1.0 Å due to the larger radii of Cu(I). Their significantly larger binding energies promise them to be structurally suitable for DNA helix construction. Electronically, the equi-number H-by-Cu replacement not only leads to considerable reductions of the HOMO−LUMO gaps and ionization potentials, but also enhances transverse electronic communication within isolated G3CuC and A2CuT pairs, revealed by the charge-transfer transitions in the UV absorption spectra of G3CuC and A2CuT. To further examine the effect of H-by-Cu substitution on conductivity, three-layer-stacked G3CuC and A2CuT of repeat and cross sequences were studied with positive results obtained. It can be reasonably concluded that the multi-Cu-mediated G3CuC and A2CuT pairs are promising candidates for building blocks of the Cum−DNA nanowires. This work would open a new prospective for rational design of the DNA-based molecular wires by multimetal incorporation.
Co-reporter:Haiying Liu ; Genqin Li ; Hongqi Ai ; Jilai Li
The Journal of Physical Chemistry C 2011 Volume 115(Issue 45) pp:22547-22556
Publication Date(Web):October 10, 2011
DOI:10.1021/jp2070198
The effect of the new designed multicopper modification of base pairs on the conductivity of DNA was investigated by the nonequilibrium Green’s function method combined with density functional theory. Electronic transport calculations revealed that the equi-number H-by-Cu replacement can significantly enhance the conductivity of DNA from two aspects: transverse base-to-base communication along the hydrogen-bond direction and longitudinal transport along the DNA duplex. Furthermore, the enhancement effect on the longitudinal direction is more notable than that on the transverse. A tunneling mechanism is suggested for the short DNA segments. The decay factor of conductance in Cu-DNA decreases by half compared with the native DNA, thus making it more promising for constructing nanowires. In addition, Cu-DNA may prefer electron migration to hole transport with the lengthening of DNA segments. This work will shed some light on the design of promising DNA-based molecular wires.
Co-reporter:Wenming Sun and Yuxiang Bu , Yixuan Wang
The Journal of Physical Chemistry C 2011 Volume 115(Issue 8) pp:3220-3228
Publication Date(Web):February 9, 2011
DOI:10.1021/jp108812z
The major objective of this paper is to address a controversial binding sequence between nucleic acid bases (NABs) and C60 by investigating adsorptions of NABs and their cations on C60 fullerene with a variety of density functional theories including two novel hybrid meta-GGA functionals, M05-2x and M06-2x, as well as a dispersion-corrected density functional, PBE-D. The M05-2x/6-311++G** provides the same binding sequence as previously reported, guanine (G) > cytosine (C) > adenine (A) > thymine (T); however, M06-2x switches the binding strengths of A and C, and PBE-D eventually results in the following sequence, G > A >T > C, which is the same as the widely accepted hierarchy for the stacking of NABs on other carbon nanomaterials such as single-walled carbon nanotube and graphite. The results indicate that the questionable relative binding strength is due to insufficient electron correlation treatment with the M05-2x or even the M06-2x method. The binding energy of G@C60 obtained with the M06-2x/6-311++G(d,p) and the PBE-D/cc-pVDZ is −7.10 and −8.07 kcal/mol, respectively, and the latter is only slightly weaker than that predicted by the MP2/6-31G(d,p) (−8.10 kca/mol). Thus, the PDE-D performs better than the M06-2x for the observed NAB@C60 π-stacked complexes. To discuss whether C60 could prevent NABs from radiation-induced damage, ionization potentials of NABs and C60 and frontier molecular orbitals of the complexes NABs@C60 and (NABs@C60)+ are also extensively investigated. These results revealed that when an electron escapes from the complexes, a hole was preferentially created in C60 for T and C complexes, while for G and A the hole delocalizes over the entire complex, rather than a localization on the C60 moiety. The interesting finding might open a new strategy for protecting DNA from radiation-induced damage and offer a new idea for designing C60-based antiradiation drugs.
Co-reporter:Ping Li, Zhitao Shen, Weihua Wang, Zhiying Ma, Siwei Bi, Haitao Sun and Yuxiang Bu
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 20) pp:5256-5267
Publication Date(Web):01 Apr 2010
DOI:10.1039/B924058F
To gain a better understanding of the antioxidation behaviors of vitamin C, the reactions between vitamin C (monoanionic form, AAH−) and two radicals, ˙H and ˙OH, have been investigated employing the B3LYP and BHandHLYP methods in combination with the atoms in molecules (AIM) theory and energy decomposition analyses (EDA). Both the radical additions to the five-membered ring of AAH− and H-abstraction reactions are explored. The reaction profiles of various reactions have been obtained. The most favorable active site to be attacked by radical addition has been confirmed to be the C2 site of AAH−, which is different from that of the C3 site in the neutral vitamin C. The ˙OH addition reactions are essentially diffusion-controlled processes, which is in contrast to the previous reports. A new source for the formation of the principal anion free radical (AFR) of AAH− has been observed in the ˙OH attack process, i.e., AFR can be formed mainly from the H13 abstraction reaction involving two types of concerted proton–electron transfer (CPET) mechanisms. Moreover, the binding characters and formation mechanisms of the stable reaction complex formed during the formation of AFR have been systematically investigated.
Co-reporter:Jun Wang, Huifang Li, Liang Zhang and Yuxiang Bu
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 40) pp:13099-13106
Publication Date(Web):08 Sep 2010
DOI:10.1039/B927202J
We present here a theoretical investigation of the electronic and energetic properties of Na+GC, a DNA motif bound to a sodium ion (Na+) at the N7 and O6 sites of guanine (G), and its hole-trapped derivative [Na+GC]+ using density functional theory calculations. Normally, Na+GC has positive dissociation energies along various dissociation channels. However, hole-trapping of the Na+GC motif can lead to an unusual energetic phenomenon. Hole-trapping can reduce not only the dissociation barrier by destabilizing the Na+GC motif to a metastable state, but also the dissociation energy of the Na+⋯N7/O6 bond with an unexpected change from a positive to a negative value (61.51 versus −16.18 kcal mol−1). This unexpected negative dissociation energy phenomenon implies that this motif can store energy (∼16 kcal mol−1) in the Na+⋯N7/O6 bond zone due to hole-trapping. The topological properties of electron densities and the Laplacian values at the bond critical points indicate that this energetic phenomenon mainly originates from additional electrostatic repulsions between two moieties linked via a high-energy bond (Na+⋯N7/O6). Proton transfer from G induced by hole-trapping can expand the negative dissociation energy zone to both Na+⋯N7/O6 and Watson–Crick (WC) H-bond zones. Similar phenomena can be observed for the Na+ binding at the minor groove. Solvation of the hole-trapped Na+GC motif can change the negative dissociation energies by varying degrees, depending on the solvent-binding sites and the polarity of the solvents.
Co-reporter:Zhiping Wang, Liang Zhang, Robert I. Cukier and Yuxiang Bu
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 8) pp:1854-1861
Publication Date(Web):07 Jan 2010
DOI:10.1039/B921104G
The structural and electronic properties of an excess electron (EE) in the ionic liquid (IL) 1-methylpyridinium chloride were explored using ab initio molecular dynamics simulations and quantum chemical calculations to give an overall understanding of the solvation and transport behavior of an EE in this IL. The results show that the EE resides in cation π*-type orbitals and that the electronic states can be characterized by the alternating appearance of localized and delocalized states during the time evolution. The characters of the EE electronic states are determined by the number of cations contributing to the LUMO of the IL. In a localized state one or two cations contribute to the LUMO of the bulk ionic liquid, while in the delocalized state the IL LUMO is composed of π*-type orbitals spanning nearly all the cations in the cell. The arrangement and fluctuation-induced changes of the orbital components in the empty band produce an alternation of different states and leads to the migration of the excess electron. These findings can be attributed to the special features of the electronic structures and geometries of the IL, and they can be used to explain similarities and differences between pyridinium-based and imidazolium-based ILs in mediating electron migration.
Co-reporter:Ping Li Dr.;Zhi-Ying Ma;Wei-Hua Wang Dr.;Zhi-Tao Shen;Si-Wei Bi Dr.;Hai-Tao Sun ;Yu-Xiang Bu Dr.
ChemPhysChem 2010 Volume 11( Issue 3) pp:696-705
Publication Date(Web):
DOI:10.1002/cphc.200900781
Abstract
Radical–molecule complexes associated with the hydroperoxyl radical (HOO) play an important role in atmospheric chemistry. Herein, the nature of the coupling interactions between sulfurous acid (H2SO3) and the HOO radical is systematically investigated at the B3LYP/6-311++G(3df,3pd) level of theory in combination with the atoms in molecules (AIM) theory, the natural bond orbital (NBO) method, and energy decomposition analyses (EDA). Eight stable stationary points possessing double H-bonding features were located on the H2SO3⋅⋅⋅HOO potential energy surface. The largest binding energies of −12.27 and −11.72 kcal mol−1 are observed for the two most stable complexes, where both of them possess strong double intermolecular H-bonds of partially covalence. Moreover, the characteristics of the IR spectra for the two most stable complexes are discussed to provide some help for their possible experimental identification.
The Journal of Physical Chemistry B 2010 Volume 114(Issue 2) pp:1144-1147
Publication Date(Web):December 10, 2009
DOI:10.1021/jp9100637
Ab initio calculations reveal an unknown energetic phenomenon for H-bonds in the hole-trapping triplex Cp•G◦C motif observed experimentally in hole migration which can explain the lower but really available oxidization possibility in Cp•G◦C site. Hole trapping can considerably destabilize the Cp•G◦C unit and lead to an unexpected barrier-hindered channel with a negative dissociation energy. This channel is governed by a balance between electrostatic repulsion and H-bonding attraction in the two associated moieties and different attenuations of two opposite interactions with respect to the H-bond distance. This Cp•G◦C unit can be viewed as a high-energy node in a DNA wire which modulates migration of a hole into or through it via its unusual energetics. It provides useful information for understanding of an unknown type of the complicated intermolecular interactions, a novel type of “high-energy” bond, and can be applied further to interpret the hidden transport properties and the energy conversion/transfer mechanisms in the related fields.
Co-reporter:Laibin Zhang, Xiaohua Chen, Haiying Liu, Li Han, Robert I. Cukier and Yuxiang Bu
The Journal of Physical Chemistry B 2010 Volume 114(Issue 10) pp:3726-3734
Publication Date(Web):February 22, 2010
DOI:10.1021/jp9117503
We present the results of a detailed and systematic computational investigation into the excited-state properties of the fluorescent cytosine analogue x-cytosine (xC). Also examined were the influences of hydration, linking to deoxyribose, base pairing with guanine (G), and base stacking on its absorption and emission processes. The calculated excitation and emission energies agree well with the experimentally measured data. It was found that hydration, linking to deoxyribose, and base pairing with G have a hyperchromic effect on the excitation maximum of xC. The linking sugar will red-shift the fluorescence emission of xC by 7 nm, while hydration and base pairing with G, on the contrary, results in a blue-shift of the fluorescence emission by 8 and 9 nm, respectively. In addition, hydration of xCG will further blue-shift the fluorescence emission of xC by about 17 nm. Furthermore, the fluorescence quantum yield of xC would be increased after hydration, linking to deoxyribose, and base pairing with G. When sandwiched by two identical natural bases, a significant decrease of the oscillator strength as well as a red-shift of the dipole-allowed transition with respect to free xC is observed in all cases. The fluorescence quantum yield of xC was expected to be lowered in the stacked complexes due to a static quenching mechanism.
Co-reporter:Dianxiang Xing, Xuejie Tan, Changhua Zhang, Yuxiang Bu
Journal of Molecular Structure: THEOCHEM 2010 Volume 939(1–3) pp:82-90
Publication Date(Web):15 January 2010
DOI:10.1016/j.theochem.2009.09.044
The interactions of N7,N9-dimethylguaninium with different anions (fluoride, bromide or tetrafluoroborate) and the related electronic properties are reported. The calculations show that the cation interactions with one to three different anion(s) give 31 stable structures. Among these structures, the cation interactions with two anions, especially with N1H (or/and N2Ha) and C8H of guanine, are the most favorable, while with one anion are the least favorable. Moreover, with the increase of the anionic radius, the interaction energies are decreased. NBO analyses reveal that the interactions between the F anion(s) with the cation mainly occur through the formation of FH/C covalent bond besides Flp→σC/NH∗ or/and Flp→πC8N7∗ interaction(s). However, with the increase of the anionic radius, the designed pairs are more favorable for the ionic bond, which may suggest that N7,N9-dimethylguaninium fluoride is not suitable to be selected as an ionic liquid.
Co-reporter:Xiaohua Chen, Laibin Zhang, Liang Zhang, Wenming Sun, Zhenwei Zhang, Haiying Liu, Yuxiang Bu and Robert I. Cukier
The Journal of Physical Chemistry Letters 2010 Volume 1(Issue 10) pp:1637-1641
Publication Date(Web):May 6, 2010
DOI:10.1021/jz100210t
Ab initio calculations suggest that the C-terminus of an α-helix can serve as a novel relay element for long-range charge migration in proteins, as an addition or alternative to the known relay properties of aromatic and S-containing amino acids. The relay ability of an α-helical C-terminus varies with helix length, capping, proximal group competition, and helix twisting/bending. The vertical ionization potential (IPV) is a suitable indicator of hole relay ability and correlates positively with the HOMO energies and inversely with helix length and dipole moment. Different capping groups can yield different effects on the IPV but hardly change the IPV−helix length dependence. A series of recent experimental observations regarding charge migrations by hopping along helices or across different helices and the distance dependence of the charge-transfer rates through helices could be viewed as strong evidence for this prediction that also provides the impetus for further experimental tests and continued theoretical exploration.Keywords (keywords): DFT calculations; hole migration relay; peptide helices; peptide size dependence of ionization energies; spin density distribution;
Frontiers of Chemistry in China 2010 Volume 5( Issue 3) pp:309-324
Publication Date(Web):2010 September
DOI:10.1007/s11458-010-0217-2
This feature article addresses several novel aspects regarding the peptide-mediated charge migrations, including: i) radical exchanges with tunable radical types (σ-radical versus π-radical) and electron-transfer (ET)-channel-tunable cooperative proton-coupled ET (PCET) mechanism, including hydrogen-atom transfer (HAT), single ET-channel PCET, double ET channel PCET, and channel-type-tunable (σ-channel versus π-channel) PCET; ii) hole hopping migration between the active groups in the side-chains and its controllability; iii) hole hopping through stepping-stones via a solvated “hole” form; and iv) electron hopping through positively charged groups as stepping-stones via a solvated electron state. In particular, the controllability of the ET channels (pathways and types) and solvated-“hole”/“electron”-based relay mechanisms are mainly mentioned. Clearly, this is an important addition to the well-documented mechanisms for charge migration in proteins. In view of the complexity of protein charge migration, further exploration on details of the stepping-stone-based relay mechanisms, by considering the properties and structures of the redox active centers, their intercalators, and the real surroundings, is still needed.
Co-reporter:Laibin Zhang, Huifang Li, Xiaohua Chen, Robert I. Cukier and Yuxiang Bu
The Journal of Physical Chemistry B 2009 Volume 113(Issue 4) pp:1173-1181
Publication Date(Web):January 7, 2009
DOI:10.1021/jp8094648
We present an ab initio study of the optical absorption and emission spectra of size-expanded nucleic acid base analogues (yA, yT, yT-m, yG, yG-t2, and yC) obtained by benzo homologation (see Krueger, A. T.; Lu, H.; Lee, A. H. F.; Kool, E. T. Acc. Chem. Res. 2007, 40, 141 and references therein). Also examined were the effects of linking to deoxyribose and hydrogen bonding to their natural complementary bases (T, A, C, and G, respectively). The calculated excitation and emission energies are in good agreement with the measured data where experimental results are available. The geometries corresponding to the first excited singlet state of yA and yT are found to be quasi-planar, while those for yG and yC are nonplanar. In general, binding to deoxyribose will red shift the absorbance and fluorescence emission maxima of the y-bases. The ground-state geometries of the Watson−Crick analog base pairs (yAT, yTA, yGC, and yCG) are found to be planar, and the calculated interaction energies are very close to those of natural base pairs, indicating that the y-bases can pair with their natural complementary partners to generate stable base pairs. The base pairing has no significant effects on the fluorescence emission of yA, yC, and yT, but blue shifts the fluorescence emission of yG by 22 nm.
Co-reporter:Li Han, Huifang Li, Robert I. Cukier and Yuxiang Bu
The Journal of Physical Chemistry B 2009 Volume 113(Issue 13) pp:4407-4412
Publication Date(Web):February 26, 2009
DOI:10.1021/jp8088726
The design of new DNA motifs is at present a very interesting topic. Recent progress indicates that the hetero-ring-expanded guanine (G) analogues possess enhanced properties compared with natural guanine. In this work, a series of hetero-ring-expanded adenine (A) analogues are designed, and their structures and electronic properties are investigated by means of density functional calculations and molecular dynamics simulations. The results indicate that the designed A-analogues can form stable base pairs with natural counterpart, and the pairing energetics for the Watson−Crick hydrogen-bonded dimers between the expanded A-analogues and natural T exhibit similarity to natural AT. Their tautomeric preferences are close to natural A, too. Furthermore, compared with natural ones, most size-expanded adenines and corresponding base pairs have smaller ionization potentials. In particular, several designed A analogues have ionization potentials even lower than natural G. The electron affinities of these modified A are comparable with that of natural A. The HOMO−LUMO gaps also behave with sensible trends. Most of A-analogues and their interrelated base pairs possess smaller gaps than the corresponding natural base and base pairs. Further, molecular dynamics simulations show the sufficient stabilities of the DNA analogues (dnA·dT)12 (where nA represents the size-expanded A-analogues designed here) when forming duplexes as the natural one does. Clearly, these observations imply their promising applications as molecular wires and new DNA motifs.
Co-reporter:Zhiping Wang, Liang Zhang, Xiaohua Chen, Robert I. Cukier and Yuxiang Bu
The Journal of Physical Chemistry B 2009 Volume 113(Issue 24) pp:8222-8226
Publication Date(Web):May 27, 2009
DOI:10.1021/jp902575s
We present the first approach to the excess electron solvation in a novel medium, room-temperature ionic liquid, using ab initio molecular dynamics simulation techniques in this work. Results indicate that an excess electron can be solvated in the [dmim]+Cl− IL as long-lived delocalized states and two short-lifetime localized states, one a single-cation-residence parasitical type and the other a double-cation-based solvated type state. The presence of a low-lying π*-LUMO as the site of excess electron residence in the cation moiety disables the C−H unit as a H-bond donor, while the aromaticity requirement of the rings and the effect of the counterion Cl−’s make the resulting ion pairs a weak stabilizer for an excess electron. Although no large solvent reorganization in IL was found at the picosecond scale, the IL fluctuations sufficiently modify the relative energy levels of the excess electron states to permit facile state-to-state conversion and adiabatic migration. The binding energy of the excess electron is only ∼0.2 eV, further indicating that it is in a quasi-free state, with a large drift mobility, suggesting that ILs are unreactive and promising mediators for transport of excess electrons, in agreement with the experimental findings. The present study provides insight into the novel electron solvation character in a new class of promising media for physical and chemical processes, which are fundamental for understanding of electron migration mechanisms in IL-based applications.
Co-reporter:Xiaohua Chen, Laibin Zhang, Liang Zhang, Jun Wang, Haiying Liu and Yuxiang Bu
The Journal of Physical Chemistry B 2009 Volume 113(Issue 52) pp:16681-16688
Publication Date(Web):November 19, 2009
DOI:10.1021/jp9077689
Charge transfer between tyrosine and tryptophan residues in proteins is continuously a hot topic because of its important biological implication. On the basis of DFT calculations and ab initio molecular dynamics simulations, several possible proton/electron cooperative transfer mechanisms from tyrosine to a tryptophan radical (or cation) without or with the assistence of a base in proteins are proposed in this work which range from direct proton-coupled π-electron π-channel/σ-channel transfers (PCπEπT versus PCπEσT) to proton-coupled long-range hopping mechanisms depending on the peptide conformations. In general, because of a smaller ionization potential, tryptophan readily behaves as two oxidized states: dehydrogenated neutral radical and ionized radical cation. For the neutral radical, the proton/electron transfers between a tyrosine and a tryptophan radical prefer a cooperative direct coupling mode through the PCπEπT or PCπEσT mechanism if two residues are proximal or can approach each other, while they cannot take place without assistence if two residues are far apart. The tryptophan radical cation prefers to form a complex with tyrosine to stabilize the hole when two residues are proximal or can approach each other. However, the electron transfer from tyrosine to tryptophan could occur via a hopping mechanism but is regulated by a base as a proton acceptor in the vicinity of tyrosine when two residues are separated. The dynamics properties and characters for these transfer events are also presented. The energetics comparison indicates that the energy barriers for the direct PCET (PCπEπT or PCπEσT) mechanisms are higher than those of the base-assisting hopping mechanism (6.8−12.5 versus ≤4.9 kcal/mol), implying the latter is a favorable way for the proton/electron cooperative transfers. Hopefully, this work provides some helpful information for understanding the mechanisms of physiologically important electron transfer reactions.
Co-reporter:Huichun Liu, Yuxiang Bu, Yunjie Mi, Yixuan Wang
Journal of Molecular Structure: THEOCHEM 2009 Volume 901(1–3) pp:163-168
Publication Date(Web):15 May 2009
DOI:10.1016/j.theochem.2009.01.021
A novel hybrid density functional theory, MPWB1K, was firstly employed to investigate static adsorptions of a nifedipine on a (10, 10) type of single-walled carbon nanotube (SWCNT), which was modeled by C200H40 and C280, respectively. For both SWCNT models the internal adsorption is more stable than the external adsorption in a range of 5.3–7.8 kcal/mol, which indicates that a nifedipine has a preference to internally adsorb on the (10, 10) SWCNT. Molecular dynamic simulations were then used to predict the dynamic behaviors of a nifedipine and the (10, 10) SWCNT system in both gas phase and aqueous solution. The classical MD simulations show that for both cases a nifedipine could spontaneously encapsulate into the SWCNT and migrate in a surprising oscillation behavior inside the SWCNT; however, both phenomena are significantly delayed in the presence of water molecules. The present study suggests that the nanotube network may be used as an efficient tool for transporting this kind of calcium channel antagonists.
The influences of thioketo substitution on the properties of uracil monomer and dimer and their interactions with Zn2+ have been systematically investigated at the B3LYP/6-311+G*level of theory. Those properties include the structural characteristics, acidities, ionization potentials, and singlet–triplet energy gaps of SU monomers and their dimers, where SU=2-thiouracil, 4-thiouracil, and 2,4-dithiouracil, respectively. Computational results suggest that thioketo substitution leads to an increase in the acidities of the N-H groups for both uracil and its dimer, where the N1–H group is still the most acidic site relative to that of N3–H group. However, the opposite behaviors are true for the ionization potentials and the singlet–triplet energy gaps of uracil monomer and its dimer, suggesting that thiouracils are more susceptible to radiation damage relative to the unsubstituted uracil. For uracil and 2-thiouracil, the corresponding triplet excited-state geometries are predicted to be highly nonplanar compared with the planar geometries of the ground state as well as 4-thiouracil and 2,4-dithiouracil upon triplet excitation. As a rule, the intermolecular H-bonds involving the sulfur atom directly have been influenced more significant than those the oxygen atom directly involved for U::U and SU::SU base pairs upon ionization and excitation. Additionally, Zn2+ binding is expected to lead to an increase in the stability of U::U and SU::SU base pairs.
Co-reporter:Huifang Li, Robert I. Cukier and Yuxiang Bu
The Journal of Physical Chemistry B 2008 Volume 112(Issue 30) pp:9174-9181
Publication Date(Web):July 4, 2008
DOI:10.1021/jp8030545
The effects of metal ion binding on the 2hJNN-coupling and δ(1H)/Δδ(15N) chemical shifts of N−H···N H-bond units in internucleotide base pairs were explored by a combination of density functional theory calculations and molecular dynamics (MD) simulations. Results indicate that the NMR parameters vary considerably upon cation binding to the natural GC or AT base pairs, and thus can be used to identify the status of the base pairs, if cation-perturbed. The basic trend is that cation perturbation causes 2hJNN to increase, Δδ(15N) to decrease, and δ(1H) to shift upfield for GC, and in the opposite directions for AT. The magnitudes of variation are closely related to the Lewis acidity of the metal ions. For both base pair series (Mz+GC and Mz+AT), these NMR parameters are linearly correlated among themselves. Their values depend strongly on the energy gaps (ΔELP→σ*) and the second-order interaction energies (E(2)) between the donor N lone pair (LPN) and the acceptor σ*N−H localized NBO orbitals. In addition, the 2hJNN changes are also sensitive to the amount of σ charge transfer from LPN to σ*N−H NBOs or from the purine to the pyrimidine moieties. The different trends are a consequence of the different H-bond patterns combined with the polarization effect of the metal ions in the cationized Mz+AT series, Mz+ ← A → T, and the cationized GC series, Mz+ ← G ← C. The predicted cation-induced systematic trends of 2hJNN and δ(15N,1H) in N−H···N H-bond units may provide a new approach to the determination of H-bond structure and strength in Watson−Crick base pairs, and provide an alternative probe of the heterogeneity of DNA sequences.
The Journal of Physical Chemistry B 2008 Volume 112(Issue 34) pp:10723-10731
Publication Date(Web):July 31, 2008
DOI:10.1021/jp802556a
We present the results of the CIS and TDB3LYP calculations of the optical absorption and emission spectra of some newly designed guanine (G) analogues and their Watson−Crick base pairs. Compared with natural G, the onset absorption peaks of these newly designed analogues are red-shifted, while the fluorescence peaks are blue-shifted. In general, the first excited singlet states (ππ*) of these analogues are nonplanar for all bases considered here. But, the Stokes shifts for the designed G-analogues are much smaller than that of natural G, suggesting that they have stronger molecular rigidity and higher fluorescence quantum yields than those of natural G. The first excited states of these Watson−Crick base pairs essentially originate from those of their isolated purine moieties, as demonstrated from the S1 geometries of their Watson−Crick base pairs. For G and its analogues, A1 and A2 (they are ring-expanded with one-bond intercalation at the C5 site), the pairing with cytosine reduces the oscillator strengths of both the first absorption peak (by 27%−60%) and the fluorescent emission (by 19%−23%), while for the analogues A3, A4, and xG in which G is ring-expanded with a two-bond intercalation at the C5 site, the pairing, in contrast, increases the oscillator strengths of both the first absorption peak (by 11%−15%) and the fluorescent emission (by 3%−20%). These observations indicate that the pairing with cytosine can quench the fluorescence for G, A1, and A2 but enhance the fluorescence quantum yields for A3, A4, and xG. The significant shifts induced by ring-expansion in the ring-expanded G with a two-bond intercalation at the C5 site reveal a possibility for their fluorescent detections.
Co-reporter:Wenming Sun and Yuxiang Bu, Yixuan Wang
The Journal of Physical Chemistry B 2008 Volume 112(Issue 48) pp:15442-15449
Publication Date(Web):November 11, 2008
DOI:10.1021/jp804965x
The adsorptions of a glycine molecule as well as dehydrogenated radicals on the side walls of both intrinsic and boron-doped (B-doped) single-walled (8,0) carbon nanotubes (SWCNTs) were investigated by a density functional theory. A glycine molecule tends to physically adsorb on intrinsic SWCNTs yet chemically adsorb on B-doped SWCNTs as a result of a somewhat chemical bond between the electron-rich nitrogen atom of the glycine molecule and the electron-scarce boron atom of the doped SWCNT. Opposite to the previous report (J. Phys. Chem. B 2006, 110, 6048−6050), it is found in the present study that both the N-centered and C-centered glycine radicals can form quite stable complexes with intrinsic as well as B-doped (8,0) SWCNTs. When the B-doped SWCNT interacts with glycine radicals, although there is a competition between B and the neighbor C in the nanotube axis direction, glycine radicals preferentially bind to the C site. The encapsulations of a glycine molecule into SWCNTs with various diameters are also discussed. We find that the encapsulation process is endothermic for (8,0) and (9,0) SWCNTs, while it is exothermic for (10,0) SWCNTs, indicating that the critical diameter of the zigzag SWCNT for the encapsulation is 7.83 Å, the diameter of (10,0).
Co-reporter:Dianxiang Xing, Xiaohua Chen and Yuxiang Bu
New Journal of Chemistry 2007 vol. 31(Issue 8) pp:1514-1524
Publication Date(Web):29 May 2007
DOI:10.1039/B702640D
The pairing strength, proton properties associated with proton transfer and deprotonation as well as relevant structural perturbations of 7,9-dimethyl GC ([mGC]+) and 7,9-dimethyl AT ([mAT]+) base pairs have been investigated. Energies related to proton transfer and deprotonation have also been predicted. It is found that the guanine/adenine N7 methylation improves the stability of GC/AT base pair. The proton transfer between the guanine N1 and the cytosine N3 is observed in [mGC]+ with an out-of-plane transition state. However, no proton-transfer reaction occurs for [mAT]+. For the deprotonation of [mGC]+ and [mAT]+, guanine C8 and N1 and adenine C8 are the most favorable sites. Deprotonation from the pyrimidine N1 site generates the most stable deprotonated base pairs, and the dissociation energies surprisingly amount to ∼100.00 and ∼65.49 kcal mol−1, respectively, much higher than those of their neutral base pairs. Deprotonation from the other sites of pyrimidine exhibits the most significant structural changes and gives the most interesting deprotonated base pairs. This process is accompanied by a barrier-free proton transfer (BFPT) from guanine N1 to cytosine N3 or adenine N6 to thymine N3 or O4 site. In this way, two rare imino tautomers of cytosine (trans-C* or cis-C*) are easily generated by removing a proton from the N4 site of cytosine in [mGC]+. In addition, the reason for BFPT from guanine N1 to cytosine N3 has been explored.
Co-reporter:Shihai Yan Dr.;Liang Zhang Dr.;Robert I. Cukier
ChemPhysChem 2007 Volume 8(Issue 6) pp:944-954
Publication Date(Web):23 MAR 2007
DOI:10.1002/cphc.200600674
Proton transfer along a single-file hydrogen-bonded water chain is elucidated with a special emphasis on the investigation of chain length, side water, and solvent effects, as well as the temperature and pressure dependences. The number of water molecules in the chain varies from one to nine. The proton can be transported to the acceptor fragment through the single-file hydrogen-bonded water wire which contains at most five water molecules. If the number of water molecule is more than five, the proton is trapped by the chain in the hydroxyl-centered H7O3+ state. The farthest water molecule involved in the formation of H7O3+ is the fifth one away from the donor fragment. These phenomena reappear in the molecular dynamics simulations. The energy of the system is reduced along with the proton conduction. The proton transfer mechanism can be altered by excess proton. The augmentation of the solvent dielectric constant weakens the stability of the system, but favors the proton transfer. NMR spin–spin coupling constants can be used as a criterion in judging whether the proton is transferred or not. The enhancement of temperature increases the thermal motion of the molecule, augments the internal energy of the system, and favors the proton transfer. The lengthening of the water wire increases the entropy of the system, concomitantly, the temperature dependence of the Gibbs free energy increases. The most favorable condition for the proton transfer along the H-bonded water wire is the four-water contained chain with side water attached near to the acceptor fragment in polar solvent under higher temperature.
Co-reporter:Huichun Liu Dr.;Liang Zhang Dr.;Robert I. Cukier ;Ping Li Dr.
ChemPhysChem 2007 Volume 8(Issue 2) pp:304-314
Publication Date(Web):19 DEC 2006
DOI:10.1002/cphc.200600499
A comprehensive study is carried out using quantum chemical computation and molecular dynamics (MD) simulations to gain insight into the interaction between Ca2 ions and the most important class of calcium channel antagonists—nifedipine. First, the chelating structures and energetic characters of nifedipine–Ca2 in the gas phase are explored, and 25 isomers are found. The most favorable chelating mode is a tridentate one, that is, Ca2 binds to two carbonyl O atoms and one nitryl O atom, where Ca2 is above the plane of the three O atoms to form a pyramidal structure. Accurate geometric structures, relative stabilities, vertical and adiabatic binding energies, and charge distributions are discussed. The differences in the geometries and energies among these isomers are analyzed from the contributions of chelating sites, electrostatics and polarizations, steric repulsions, and charge distributions. The interconversions among isomers with similar geometries and energies are also investigated because of the importance of the geometric transformation in the biological system. Furthermore, certain numbers of water molecules are added to the nifedipine–Ca2 system to probe the effect of water. A detailed study is performed on the hydrated geometries on the basis of the most stable isomer 1. Stepwise hydration can weaken the nifedipine–Ca2 interaction, and the chelating sites of nifedipine are gradually replaced by the added water molecules. Hexacoordination is found to be the most favorable geometry no matter how many water molecules were added, which can be verified by the MD simulations. The transfer of water molecules from the inner shell to the outer shell is also supported by MD simulations of the hexahydrated complexes.
Co-reporter:Feng Xiang, Ping Li, Shihai Yan, Lixiang Sun, Robert I. Cukier and Yuxiang Bu
New Journal of Chemistry 2006 vol. 30(Issue 6) pp:890-900
Publication Date(Web):21 Apr 2006
DOI:10.1039/B518408H
The stepwise hydration effect on the glutamic acid–Ca2+ (GC) complexes in the gas phase has been investigated by density functional theory (DFT) calculations. The thermodynamics parameters for the hydration reactions, the stepwise hydration energies and accurate geometries have been explored. To elucidate the Ca2+–ligand interaction, the charge transfer, bonding analysis and IR spectroscopic characteristics have also been investigated. The correlating data have shown that all of the stepwise hydration reactions are enthalpy-driven because of the relatively small value of ΔS, but the number of coordinated water molecules in the first shell of Ca2+ is not limitless. In our study, the optimal coordination number (CN) of Ca2+ in the first shell is 6 or 7; the former value agrees well with the data reported in the Protein Data Bank (PDB), and the latter is the reflection of the most frequent Ca2+-binding motif, EF-hand, in soluble proteins. Furthermore, the self-consistent reaction field (SCRF) and higher-level MP2 calculations have confirmed our conclusions. Additionally and very importantly, the stepwise hydration in either the first or second coordination shell can weaken the glutamic acid–Ca2+ interaction gradually till the glutamic acid ligand is replaced by the added water molecules, resulting in the conversion of coordination mode of the glutamic acid to Ca2+ from the inner-sphere one to a peripheral interaction mode, just like the ligand exchange process in the Ca2+ release channel existing in the real biological system. Finally, the similarities and discrepancies between our model and the Ca2+-channel in vivo have been compared.
Co-reporter:Fangfang Liu, Peng Qian, Shihai Yan, Yuxiang Bu
Journal of Molecular Structure: THEOCHEM 2006 Volume 760(1–3) pp:209-217
Publication Date(Web):28 February 2006
DOI:10.1016/j.theochem.2005.11.033
The coupling characteristics and the proton transfer mechanisms of guanine–Na+ monohydrate are determined in this investigation after the implementation of the geometry optimization and the harmonic vibrational frequency calculations. There are two elementary coupling modes: the interaction of monohydrated sodium ion with two heteroatoms which form a ringed coupling, and hydrogen-bond involved coupling mode. Two potential reaction pathways, coupling mode and hydration have been taken into account, and the accurate values of binding energy are corrected for basis set superposition error (BSSE) and zero-point vibrational energy (ZPVE). Relative energies of the hydrated guanine–sodium ion complexes indicate that the ringed-coupling complexes are predominant geometries with much lower energies. Monohydrated sodium ion coupling with O6 and N7 generates the most stable geometry with a five-member cycle. Sodium ion plays an important role in the tautomerization for guanine–sodium ion complexes. This investigation indicates that the stable cation-π complexes cannot be optimized for guanine–sodium ion monohydrate. Amino-involved coupling often gives rise to a twisted four-membered cycle with unrealistic distribution of positive charge and higher energies. The rotation of amino group is likely to lead to the redistribution of the base pair hydration bonding. Effective distribution of the positive charge is an important factor in the stabilization of biological systems and binding energies for the monohydrated guanine–sodium ion complexes. The enolic coupling complex has the higher energy than the keto type due to the hindrance for the positive charge.
Co-reporter:Hongqi Ai, Yuxiang Bu, Ping Li and Chong Zhang
New Journal of Chemistry 2005 vol. 29(Issue 12) pp:1540-1548
Publication Date(Web):24 Oct 2005
DOI:10.1039/B509496H
The regulatory roles of metal ions (M+/2+
= Li+, Na+, K+, Be2+, Mg2+, and Ca2+) and water molecules in stabilizing the zwitterionic form of glycine derivatives are discussed in the present paper. The involved glycine conformers are (1) the glycine zwitterion (OO), which can bind an ion to form the salt-bridge-form derivative, and (2) two neutral forms (NO and OH), which can bind the ion with both the carboxyl oxygen and the amino nitrogen (NO), or both the carboxyl oxygen and the hydroxyl oxygen (OH) to form charge-solvated structures. Results show that four water molecules can make the OO-mode glycine–Li+/Na+/K+ derivatives more stable than each of the corresponding NO-mode counterparts. Similarly, two water molecules can make the OO-mode glycine–Be2+ more stable than its corresponding NO-mode counterpart, while the OO-mode glycine–Mg2+/Ca2+ are always more stable than their corresponding NO counterparts whether there is a water molecule(s) attached or not. No OH-mode glycine–Be2+/Mg2+/Ca2+ hydrates are observed because the large electrostatic effect of these divalent metal ions makes the OH-mode hydrates degenerate into the OO-mode ones. For the non-hydrated or monohydrated glycine–Li+/Na+ isomers, the global minimum derives from the NO-mode structure. Their (di–tetra)hydrates, however, prefer the OH-mode to the other two. For all these different glycine–K+ hydrates, each OH-mode structure is always the global minimum among the corresponding isomers, as for the non-hydrated reactant (glycine–K+). These predictions are consistent with other theoretical and experimental results. Most of the relative binding strengths, between a metal ion and its corresponding ligand, of the three modes are in good agreement with the relative stabilities, i.e., the larger the binding energy is, the more stable the structure would be. Some exceptions to the agreement are explained. A comparison of binding strengths shows that the smaller the ion radius is, the greater its binding strength would be. For instance, Li–NO·W (−50.5 kcal mol−1) > Na–NO·W (−36.4 kcal mol−1) > K–NO·W (−25.4 kcal mol−1).
Co-reporter:Qiao Sun, Mei Qin, Yuxiang Bu, Keli Han
Journal of Molecular Structure: THEOCHEM 2005 Volume 714(2–3) pp:165-174
Publication Date(Web):14 February 2005
DOI:10.1016/j.theochem.2004.09.043
The geometries and vibrational frequencies of NH3, NH3+, NF3, NF3+ as well as their encounter complexes ((NH3⋯NH3)+ and (NF3⋯NF3)+) are calculated using DFT and ab initio methods at 6-311++G** basis set level. This paper also discusses the inapplicability of DFT methods in predicting the dissociation energy curves, especially for the systems at large contact distance, due to the ‘inverse symmetry breaking’ problem. Finally, the contact distance dependence of the activation energy, the coupling matrix element and the electron-transfer rate has been analyzed at MP2/6-311++G** level. For the NX3/NX3+ (X=H, F) coupling systems, electron transfer occurs chiefly over a range of contact distance, 2.00 Å
Different morphologies of BaSO4 nanocrystals including nanoparticles and short rods and long fibers were prepared via the static organic/aqueous biphase boundary at the room temperature. The organic phase was a containing primary amine sulfate solution, while the aqueous phase was a solution of Ba2+. The effect of [SO42−]/[Ba2+] molar ratios on the BaSO4 growth process was studied principally by TEM. In addition, it was found that the diluent polarity can also affect self-assembly process of BaSO4 crystals at the interface. The possible mechanism of nanocrystalline BaSO4 self-assembly is discussed.
Chemical Physics Letters 2003 Volume 377(3–4) pp:462-468
Publication Date(Web):15 August 2003
DOI:10.1016/S0009-2614(03)01199-0
Abstract
The geometries and the vibrational frequencies for several SiSO+ and GeSO+ species at doublet state have been predicted at density functional theory level with a 6-311+G* basis set. The detailed bonding character is discussed, and the state–state energy separations of various stable states relative to the ground state are calculated. The ground states are linear Si–OS+ and cyclic GeSO+ for two systems, respectively. Result analysis indicates that the cyclic state should be classified as the thiosuperoxide, the bent structure and the linear M–OS+ structure have some thiosuperoxide characters, but the linear S–M–O+ may be classified as thio-oxide.
Chinese Journal of Chemistry 2001 Volume 19(Issue 7) pp:637-640
Publication Date(Web):26 AUG 2010
DOI:10.1002/cjoc.20010190702
The geometries, the harmonic vibrational frequencies and the bonding properties have been predicted for cyclic AlS2 and GaS2 species at the density functional theory (DFT), MPn (n = 2, 3, 4), QCISD(T) and CCSD(T) levels with 6-311 + G (2df) basis set. The novel bonding character was discussed.
Biophysical Journal (7 April 2010) Volume 98(Issue 7) pp:
Publication Date(Web):7 April 2010
DOI:10.1016/j.bpj.2009.12.4274
The sliding and hopping models encapsulate the essential protein-DNA binding process for binary complex formation and dissociation. However, the effects of a cofactor protein on the protein-DNA binding process that leads to the formation of a ternary complex remain largely unknown. Here we investigate the effect of the cofactor Sox2 on the binding and unbinding of Oct1 with the Hoxb1 control element. We simulate the association of Oct1 with Sox2-Hoxb1 using molecular dynamics simulations, and the dissociation of Oct1 from Sox2-Hoxb1 using steered molecular dynamics simulations, in analogy to a hopping event of Oct1. We compare the kinetic and thermodynamic properties of three model complexes (the wild-type and two mutants) in which the Oct1-DNA base-specific interactions or the Sox2-Oct1 protein-protein interactions are largely abolished. We find that Oct1-DNA base-specific interactions contribute significantly to the total interaction energy of the ternary complex, and that nonspecific Oct1-DNA interactions are sufficient for driving the formation of the protein-DNA interface. The Sox2-Oct1 protein-protein binding interface is largely hydrophobic, with remarkable shape complementarity. This interface promotes the formation of the ternary complex and slows the dissociation of Oct1 from its DNA-binding site. We propose a simple two-step reaction model of protein-DNA binding, called the tethered-hopping model, that explains the importance of the cofactor Sox2 and may apply to similar ternary protein-DNA complexes.
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 34) pp:NaN23821-23821
Publication Date(Web):2016/08/03
DOI:10.1039/C6CP04224D
Diffuse functions have been proved to be especially crucial for the accurate characterization of excess electrons which are usually bound weakly in intermolecular zones far away from the nuclei. To examine the effects of diffuse functions on the nature of the cavity-shaped excess electrons in water cluster surroundings, both the HOMO and LUMO distributions, vertical detachment energies (VDEs) and visible absorption spectra of two selected (H2O)24− isomers are investigated in the present work. Two main types of diffuse functions are considered in calculations including the Pople-style atom-centered diffuse functions and the ghost-atom-based floating diffuse functions. It is found that augmentation of atom-centered diffuse functions contributes to a better description of the HOMO (corresponding to the VDE convergence), in agreement with previous studies, but also leads to unreasonable diffuse characters of the LUMO with significant red-shifts in the visible spectra, which is against the conventional point of view that the more the diffuse functions, the better the results. The issue of designing extra floating functions for excess electrons has also been systematically discussed, which indicates that the floating diffuse functions are necessary not only for reducing the computational cost but also for improving both the HOMO and LUMO accuracy. Thus, the basis sets with a combination of partial atom-centered diffuse functions and floating diffuse functions are recommended for a reliable description of the weakly bound electrons. This work presents an efficient way for characterizing the electronic properties of weakly bound electrons accurately by balancing the addition of atom-centered diffuse functions and floating diffuse functions and also by balancing the computational cost and accuracy of the calculated results, and thus is very useful in the relevant calculations of various solvated electron systems and weakly bound anionic systems.
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 21) pp:NaN13818-13818
Publication Date(Web):2017/04/27
DOI:10.1039/C7CP01847A
In this work, we present an ab initio molecular dynamics simulation study on the interaction of an excess electron (EE) with histidine in its aqueous solution. Two different configurations of histidine (imidazole group protonated or not) are considered to reflect its different existing forms in neutral or slightly acidic surroundings. The simulation results indicate that localizations of EEs in different aqueous histidine solutions are quite different and are strongly affected by protonation of the side chain imidazole group and are thus pH-controlled. In neutral aqueous histidine solution, an EE localizes onto the carboxyl anionic group of the amino acid backbone after a relatively lengthy diffuse state, performing just like in an aliphatic amino acid solution. But in weakly acidic solution in which the side chain imidazole group is protonated, an EE undergoes a short lifetime diffuse state and finally localizes on the protonated imidazole group. We carefully examine these two different localization dynamics processes and analyze the competition between different dominating groups in their corresponding electron localization mechanisms. To explain the difference, we investigate the frontier molecular orbitals of these two systems and find that their energy levels and compositions are important to determine these differences. These findings can provide helpful information to understand the interaction mechanisms of low energy EEs with amino acids and even oligopeptides, especially with aromatic rings.
Co-reporter:Yiwei Feng, Fengying Zhang, Xinyu Song and Yuxiang Bu
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 8) pp:NaN5943-5943
Publication Date(Web):2017/01/30
DOI:10.1039/C6CP08201G
While the conductance behavior of carbon-based couplers has been successfully investigated, insight into the magnetic properties of such carbon-based molecule coupled diradical systems is still scarce, and especially the structural effect of such couplers on the magnetic properties is poorly understood. The present work reports three different interference effects on the magnetic properties of carbon-based molecule coupled nitroxide diradicals: twisting, sideways group, and position effects. DFT calculations reveal that (i) torsion does not change their broken-symmetry singlet ground state and antiferromagnetic coupling, but decreases their magnetism; (ii) different linkages of two radical moieties result in different ground states and thus different magnetisms, depending on a combination of meta-sites and para-sites; (iii) the antiferromagnetic coupling with a broken-symmetry singlet ground state is not changed by adding sideways groups, but the coupling magnitude can be tuned by modifying the side-bridge. Discussions on geometries, magnetic properties, SOMO–SOMO splittings, and spin density distributions are made to clarify relevant magnetic behaviors. Clearly, the findings concerning the regulation of the diradicalized material molecules through modifying the carbon-based bridges provide a comprehensive understanding of the magnetism of such carbon-based diradicals and new prospects for the design of building blocks of magnetic functional molecular materials.
Co-reporter:Zhiping Wang, Liang Zhang, Robert I. Cukier and Yuxiang Bu
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 8) pp:NaN1861-1861
Publication Date(Web):2010/01/07
DOI:10.1039/B921104G
The structural and electronic properties of an excess electron (EE) in the ionic liquid (IL) 1-methylpyridinium chloride were explored using ab initio molecular dynamics simulations and quantum chemical calculations to give an overall understanding of the solvation and transport behavior of an EE in this IL. The results show that the EE resides in cation π*-type orbitals and that the electronic states can be characterized by the alternating appearance of localized and delocalized states during the time evolution. The characters of the EE electronic states are determined by the number of cations contributing to the LUMO of the IL. In a localized state one or two cations contribute to the LUMO of the bulk ionic liquid, while in the delocalized state the IL LUMO is composed of π*-type orbitals spanning nearly all the cations in the cell. The arrangement and fluctuation-induced changes of the orbital components in the empty band produce an alternation of different states and leads to the migration of the excess electron. These findings can be attributed to the special features of the electronic structures and geometries of the IL, and they can be used to explain similarities and differences between pyridinium-based and imidazolium-based ILs in mediating electron migration.
Co-reporter:Jun Wang, Huifang Li, Liang Zhang and Yuxiang Bu
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 40) pp:NaN13106-13106
Publication Date(Web):2010/09/08
DOI:10.1039/B927202J
We present here a theoretical investigation of the electronic and energetic properties of Na+GC, a DNA motif bound to a sodium ion (Na+) at the N7 and O6 sites of guanine (G), and its hole-trapped derivative [Na+GC]+ using density functional theory calculations. Normally, Na+GC has positive dissociation energies along various dissociation channels. However, hole-trapping of the Na+GC motif can lead to an unusual energetic phenomenon. Hole-trapping can reduce not only the dissociation barrier by destabilizing the Na+GC motif to a metastable state, but also the dissociation energy of the Na+⋯N7/O6 bond with an unexpected change from a positive to a negative value (61.51 versus −16.18 kcal mol−1). This unexpected negative dissociation energy phenomenon implies that this motif can store energy (∼16 kcal mol−1) in the Na+⋯N7/O6 bond zone due to hole-trapping. The topological properties of electron densities and the Laplacian values at the bond critical points indicate that this energetic phenomenon mainly originates from additional electrostatic repulsions between two moieties linked via a high-energy bond (Na+⋯N7/O6). Proton transfer from G induced by hole-trapping can expand the negative dissociation energy zone to both Na+⋯N7/O6 and Watson–Crick (WC) H-bond zones. Similar phenomena can be observed for the Na+ binding at the minor groove. Solvation of the hole-trapped Na+GC motif can change the negative dissociation energies by varying degrees, depending on the solvent-binding sites and the polarity of the solvents.
Co-reporter:Ping Li, Zhitao Shen, Weihua Wang, Zhiying Ma, Siwei Bi, Haitao Sun and Yuxiang Bu
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 20) pp:NaN5267-5267
Publication Date(Web):2010/04/01
DOI:10.1039/B924058F
To gain a better understanding of the antioxidation behaviors of vitamin C, the reactions between vitamin C (monoanionic form, AAH−) and two radicals, ˙H and ˙OH, have been investigated employing the B3LYP and BHandHLYP methods in combination with the atoms in molecules (AIM) theory and energy decomposition analyses (EDA). Both the radical additions to the five-membered ring of AAH− and H-abstraction reactions are explored. The reaction profiles of various reactions have been obtained. The most favorable active site to be attacked by radical addition has been confirmed to be the C2 site of AAH−, which is different from that of the C3 site in the neutral vitamin C. The ˙OH addition reactions are essentially diffusion-controlled processes, which is in contrast to the previous reports. A new source for the formation of the principal anion free radical (AFR) of AAH− has been observed in the ˙OH attack process, i.e., AFR can be formed mainly from the H13 abstraction reaction involving two types of concerted proton–electron transfer (CPET) mechanisms. Moreover, the binding characters and formation mechanisms of the stable reaction complex formed during the formation of AFR have been systematically investigated.
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 13) pp:NaN5914-5914
Publication Date(Web):2011/02/18
DOI:10.1039/C0CP02297G
Motivated by a promising expansion of the genetic alphabet and a successful design of conductive DNA bases justified from the hetero-ring-expanded purine base (G and A) analogs, we extend our hetero-ring expansion scheme to the pyrimidine bases (C and T) to examine the ring-expansion effects on various properties of these single-ring bases with a comparison with those in the double-ring purine case. Four kinds of the hetero-rings are considered to expand C and T, forming the C and T analogs (nC and nT), respectively. The relevant structures and properties were investigated by means of quantum calculations and molecular dynamics simulations. The results reveal that all the modified bases can form base pairs specifically with their natural counterparts and assemble duplex helices which have comparable stability to native ones. The HOMO–LUMO gaps of G–nC and A–nT are smaller than those of the natural pairs, and the assembled duplex helices ((G–nC)12 and (A–nT)12) are diameter-enlarged but with smaller rise and twist, both of which favor DNA-conduction, as confirmed by ionization potentials and spin density distributions. In addition, the hetero-ring expansion can lower the activation barriers and reduce the reaction heats of the inter-base double proton transfers. In particular, as evidenced by NMR parameters and the excited states, the hetero-ring expansion leads to an enhancement of the transverse electronic communication between two pairing bases, clearly facilitating the conduction along the helices. Furthermore, the hetero-ring expansion effect on the pyrimidine bases is larger than that on the purine bases. In summary, this work presents clear theoretical evidence for the possibility of hetero-ring expanded pyrimidine bases as promising candidates for the motifs of the genetic alphabet and DNA nanowires.
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 42) pp:NaN18463-18463
Publication Date(Web):2013/09/02
DOI:10.1039/C3CP52745J
We present a computational study of the double-electron oxidized guanine–cytosine base pair as well as its deprotonated derivatives, focusing on their structural and electronic properties. Some novel electromagnetic characteristics are found. A hydrazine-like (N–N) cross-linked structure between the G and C radical moieties is the lowest-energy one for the [GC]2+ complexes. Double-electron oxidation can considerably destabilize the GC unit and leads to a barrier-hindered dissociation channel with negative dissociation energy. This channel is governed by a balance between electrostatic repulsion and attractive hydrogen-bonding interaction co-existing between G˙+ and C˙+. The proton/electron transfer reactions in the double-electron oxidized Watson–Crick base pair occur through a proton transfer induced charge migration mechanism. For the deprotonated [GC]2+ derivative, the [G(–H+)C]+ series prefers to accompany by transfer of an electron from the G to C moiety when the G+ is deprotonated, and its highest-doubly occupied molecular orbital mainly localizes over the C moiety with a π-bonding character. For the diradical G˙+C(–H)˙ series in which the C moiety is deprotonated, the two unpaired electrons reside one on each moiety in the π system. The diradical base pairs possess open-shell broken symmetry singlet states, and their magnetic coupling interactions are controlled by both intra- and inter-molecular interactions. The double-electron oxidized Watson–Crick base pair shows strong antiferromagnetic coupling, whereas the magnetic interactions of other diradical derivatives are relatively weak. This study highlights the crucial role of H-bonding in determining the magnetic interactions.
Co-reporter:Xiuxiu Wu, Liang Gao, Jinxiang Liu, Hongfang Yang, Shoushan Wang and Yuxiang Bu
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 40) pp:NaN26863-26863
Publication Date(Web):2015/09/15
DOI:10.1039/C5CP03720D
Studies on the structure, states, and reactivity of excess electrons (EEs) in biological media are of great significance. Although there is information about EE interaction with desolvated biological molecules, solution effects are hardly explored. In this work, we present an ab initio molecular dynamics simulation study on the interaction and reactivity of an EE with glycine in solution. Our simulations reveal two striking results. Firstly, a pre-solvated EE partially localizes on the negatively charged –COO− group of the zwitterionic glycine and the remaining part delocalizes over solvent water molecules, forming an anion-centered quasi-localized structure, due to relative alignment of the lowest unoccupied molecular orbital energy levels of potential sites for EE residence in the aqueous solution. Secondly, after a period of anion-centered localization of an EE, the zwitterionic glycine is induced to spontaneously fragment through the cleavage of the N–Cα bond, losing ammonia (deamination), and leaving a ˙CH2–COO− anion radical, in good agreement with experimental observations. Introduction of the same groups (–COO− or –NH3+) in the side chain (taking lysine and aspartic acid as examples) can affect EE localization, with the fragmentation of the backbone part of these amino acids dependent on the properties of the side chain groups. These findings provide insights into EE interaction mechanisms with the backbone parts of amino acids and low energy EE induced fragmentation of amino acids and even peptides and proteins.
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 30) pp:NaN19805-19805
Publication Date(Web):2015/06/24
DOI:10.1039/C5CP02693H
Radiation-generated secondary electrons can induce resonance processes in a target molecule and fragment it via different pathways. Although the associating electronic resonant states at equilibrium geometry have been well studied for many target molecules in the gas phase, vibrational resonance contributions and the solvent effect are still poorly understood for relevant processes in solution. Taking a radiosensitive drug, 5-bromopyrimidine (5-BrPy), as an example, we here present a combined ab initio molecular dynamics simulation and time-dependent wave packet study with an emphasis on vibrational resonance and solvation effects on excess electron interaction with 5-BrPy in solution. The gaseous results reveal two primary channels for the electron induced C–Br bond cleavage: the highest vibrational resonance on vertical potential energy curve via a tunneling mechanism , and auto-dissociation along repulsive relaxed potential energy curve , which account for the two peaks at 0.2 and 0 eV observed in Modelli's experiment. However, a strong solvation effect modifies the mechanism and dynamics of the dissociation of the electron⋯5-BrPy system. On one hand, the spontaneous dissociation becomes unfavorable due to a barrier on the relaxed free energy surface created by the coupling between the π* and σ* states. Seven vibrational resonances (v = 0–6) are identified for the solution process and only the high-level v = 5, 6 with non-negligible quantum tunneling coefficient can cause the dissociation . On the other hand, protonation is also observed at the N sites of the hydrated 5-BrPy anion , and this inhibits the dissociation along the C–Br bond, suggesting a competing pathway against C–Br bond cleavage. Clearly, this work provides a combination strategy using an ab initio molecular dynamics technique and time-dependent wave packet method to explore the effects of vibrational resonances and solvation on the interaction of radio-generated excess electrons with target biological molecules in complicated solution surroundings.
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 28) pp:NaN18879-18879
Publication Date(Web):2016/06/14
DOI:10.1039/C6CP03552C
In this work, we conduct ab initio molecular dynamics simulations on the localization dynamics of an excess electron (EE) in acetamide/Ca2+ aqueous solutions with three different interaction modes of Ca2+ with acetamide: tight contact, solvent-shared state, and separated interaction. The simulated results reveal that an EE could exhibit two different localization behaviors in these acetamide/Ca2+ aqueous solutions depending on different amide⋯Ca2+ interactions featuring different contact distances. For the tight contact and solvent-shared state of amide⋯Ca2+ solutions, vertically injected diffuse EEs follow different mechanisms with different dynamics, forming a cavity-shaped hydrated electron or a hydrated amide anion, respectively. Meanwhile, for the separated state, only one localization pattern of a vertically injected diffuse EE towards the formation of hydrated amide anion is observed. The hindrance of hydrated Ca2+ and the attraction of the hydrated amide group originating from its polarity and low energy π* orbital are the main driving forces. Additionally, different EE localization modes have different effects on the interaction between the amide group and Ca2+ in turn. This work provides an important basis for further understanding the mechanisms and dynamics of localizations/transfers of radiation-produced EEs and associated EE-induced lesions and damage to biological species in real biological environments or other aqueous solutions.
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 4) pp:NaN2923-2923
Publication Date(Web):2015/12/21
DOI:10.1039/C5CP06133D
Novel DNA base pair derivatives (A2CunU, A2CunC, G3CunU, and G3CunC) are designed by aromatic expansion of pyrimidine bases with four kinds of hetero-rings (denoted by nC and nU, n = 1, 2, 3, and 4) and metal-decoration through Cu replacement of hydrogens in the Watson–Crick hydrogen bond region. Their structures and properties are calculated for examining the cooperative effects of the two modification ways. The calculated results reveal that multiple Cu decoration makes up the deficiencies of size-expansion, and exhibits not only increase of structural stability and reduction of ionization potentials, but also ideal shrink of the HOMO–LUMO gaps, notable enhancement of interbase coupling as well as remarkable redshifts of π → π* transitions for all M-x modified base pairs. The decrease extents of the gaps and ionization potentials follow the same order G3CunU > G3CunC > A2CunU > A2CunC, and in each series (denoted by different n), the gaps, ionization potentials and first π → π* transition energies have an order of 4 < 1 < 2 < 3. The Cu d orbitals function as bridges for π electron delocalization on the conjugated aromatic rings of two bases, leading to an enhancement of transverse electronic communication, as verified by spin density delocalization, orbital composition changes, redshift of the π → π* transition and also advocated by the electron-sharing indexes such as delocalization index, Mayer bond orders and multicenter bonding. Electron localization function ELF-π isosurfaces above the molecular plane further suggested that effective longitudinal conduction is closely relevant with the bicyclic domain involving good electron delocalization and strong π–π stacking between layers. This work presents theoretical evidence for the cooperative effects of metal decoration and ring-expansion modifications on the electronic properties of the modified base pairs and also proves that the base pairs designed here could be competent building blocks for the DNA-based nanowires with improved electron activity and excellent conductivity.
Co-reporter:Boran Han, Xiaohua Chen, Jing Zhao and Yuxiang Bu
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 45) pp:NaN15859-15859
Publication Date(Web):2012/10/01
DOI:10.1039/C2CP41566F
This work presents a density functional theory calculational study for clarifying that peptide loops (–[peptide]n–) including the N-terminal and the C-terminal oligopeptides and the α-helix N-terminal can serve as an intriguing kind of relay elements, as an addition to the known relay stations served by aromatic amino acids for electron hopping migration. For these protein motifs, an excess electron generally prefers to reside at the –NH3+ group in a Rydberg state for the N-terminal peptides, or at the –COOH group in a dipole-bound state for the C-terminal peptides, and at the N-terminal in a dipole-bound π*-orbital state for the peptide loops and α-helices. The electron binding ability can be effectively enhanced by elongation for the α-helix N-terminal, and by bending, twisting, and even β-turning for the peptide chains. The relay property is determined by the local dipole instead of the total dipole of the peptide chains. Although no direct experiment supports this hypothesis, a series of recent studies regarding charge hopping migration associated with the peptide chains and helices could be viewed as strong evidence. But, further studies are still needed by considering the effects from relative redox potential between the donor and acceptor sites, protein environment, and structure water molecules.
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 4) pp:NaN2825-2825
Publication Date(Web):2016/12/16
DOI:10.1039/C6CP07628A
In this work, the effect of diffuse function types (atom-centered diffuse functions versus floating functions and s-type versus p-type diffuse functions) on the structures and properties of three representative water cluster anions featuring a surface-bound excess electron is studied and we find that an effective combination of such two kinds of diffuse functions can not only reduce the computational cost but also, most importantly, considerably improve the accuracy of results and even avoid incorrect predictions of spectra and the EE shape. Our results indicate that (a) simple augmentation of atom-centered diffuse functions is beneficial for the vertical detachment energy convergence, but it leads to very poor descriptions for the singly occupied molecular orbital (SOMO) and lowest unoccupied molecular orbital (LUMO) distributions of the water cluster anions featuring a surface-bound excess electron and thus a significant ultraviolet spectrum redshift; (b) the ghost-atom-based floating diffuse functions can not only contribute to accurate electronic calculations of the ground state but also avoid poor and even incorrect descriptions of the SOMO and the LUMO induced by excessive augmentation of atom-centered diffuse functions; (c) the floating functions can be realized by ghost atoms and their positions could be determined through an optimization routine along the dipole moment vector direction. In addition, both the s- and p-type floating functions are necessary to supplement in the basis set which are responsible for the ground (s-type character) and excited (p-type character) states of the surface-bound excess electron, respectively. The exponents of the diffuse functions should also be determined to make the diffuse functions cover the main region of the excess electron distribution. Note that excessive augmentation of such diffuse functions is redundant and even can lead to unreasonable LUMO characteristics.



![4-amino-1,3-dihydro-2H-Pyrrolo[2,3-d]pyrimidin-2-one](http://img.cochemist.com/ccimg/100100/100047-45-8.png)
![4-amino-1,3-dihydro-2H-Pyrrolo[2,3-d]pyrimidin-2-one](http://img.cochemist.com/ccimg/100100/100047-45-8_b.png)
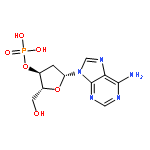
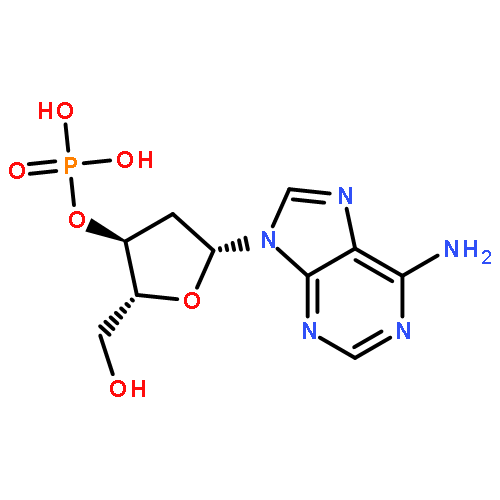






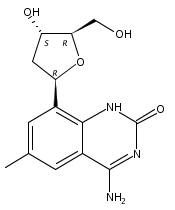





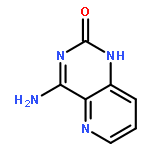








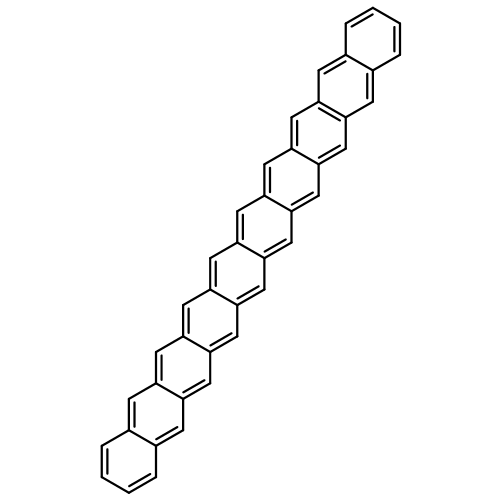


![Glycine, N-[N-(N-glycyl-L-tyrosyl)glycyl]-](http://img.cochemist.com/ccimg/20300/20234-74-6.png)
![Glycine, N-[N-(N-glycyl-L-tyrosyl)glycyl]-](http://img.cochemist.com/ccimg/20300/20234-74-6_b.png)






![6-Methylpyrido[3,2-d]pyrimidine-2,4(1H,3H)-dione](http://img.cochemist.com/ccimg/2500/2499-96-9.png)
![6-Methylpyrido[3,2-d]pyrimidine-2,4(1H,3H)-dione](http://img.cochemist.com/ccimg/2500/2499-96-9_b.png)