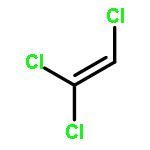
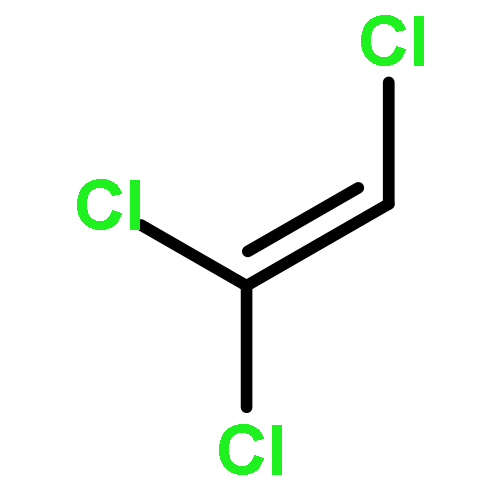
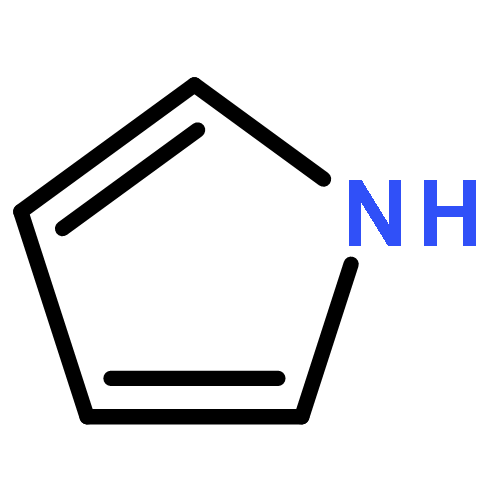
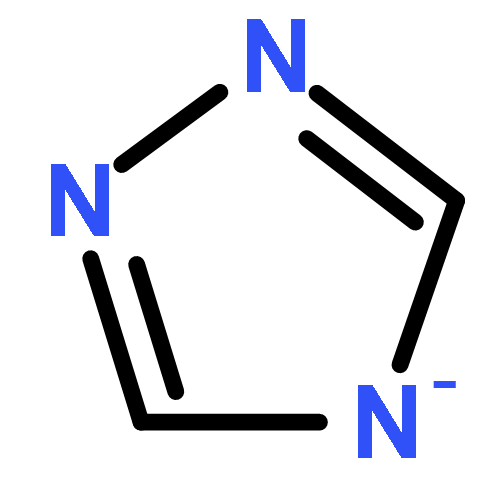
Co-reporter:Ihsan Erden;Christian Gärtner;Jingxiang Ma;Gabriel Cabrera;Kate Markham;Saeed Azimi
European Journal of Organic Chemistry 2017 Volume 2017(Issue 34) pp:5147-5153
Publication Date(Web):2017/09/15
DOI:10.1002/ejoc.201700915
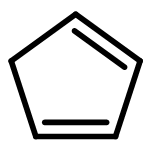
Aldonitrones derived from spiro[2.4]hepta-4,6-diene-1-carbaldehyde and its benzo analog undergo a tandem uncatalyzed intramolecular cyclopropane–nitrone cyclization-5,6-dihydro-1,2-oxazine cycloreversion to give cyclopentadienones. Similarly, the NH-nitrone generated in situ from spiro[cyclopropane-1,1′-indene]carbaldehyde oxime leads to benzocyclopentadienone (1H-inden-1-one) by the same mechanism. DFT calculations are in favor of a concerted yet highly asynchronous pathway for the cyclizations. Control experiments with the dihydro and tetrahydro derivatives show that the spirocyclopentadiene unit is essential for the success of the reaction, invoking spiroconjugative effects for increased cyclopropane reactivity.
Co-reporter:Ihsan Erden;Gabriel Cabrera;Necdet Coskun;Marco Tapken
European Journal of Organic Chemistry 2017 Volume 2017(Issue 20) pp:2925-2931
Publication Date(Web):2017/05/26
DOI:10.1002/ejoc.201700442
The title compound exhibits a number of modes of reactivity toward nucleophiles/bases owing to the presence of several electrophilic and potentially nucleophilic sites in the molecule. We explored the reactions of 6-(chloromethyl)-6-methylfulvene with oxygen and nitrogen nucleophiles and bases as well as a carbon-based nucleophile (an enamine) and realized all possible reactivity modes predicted on the basis of electrophilic and nucleophilic positions in this compound.
Co-reporter:Mariah L. Parker, Scott Gronert
International Journal of Mass Spectrometry 2017 Volume 418(Volume 418) pp:
Publication Date(Web):1 July 2017
DOI:10.1016/j.ijms.2016.11.018
•ETD is used to complete selective gas-phase reductions of metal complexes.•The reduction products are probed in subsequent ion/molecule reactions.•The reactivity parallels that of known condensed-phase reactive intermediates.Sequential ion/ion and ion/molecule reactions are demonstrated in a linear ion trap mass spectrometer and are used to probe the reactivity of metal complexes in unusual ionization states. Taking advantage of the instrument’s electron transfer dissociation (ETD) capabilities, the mono- and bis-phenanthroline complexes of Fe(I), Co(I), Ni(I), Cu(I), and Zn(I) were formed by reduction of the corresponding M(II) species in an ion/ion reaction with the fluoranthene radical anion. The chemistry of the M(I) species was probed in ion/molecule reactions with allyl iodide. The bis-phenanthroline complexes generally give slow reactions and the metals were oxidized to M(II) iodide complexes with presumably the release of allyl radicals. The mono-phenanthroline complexes are much more reactive and give M(II) iodide complexes, M(II) allyl complexes, and adducts. The overall reactivity is in accord with density functional theory calculations and mirrors that of proposed intermediates in condensed-phase catalytic cycles.Download high-res image (129KB)Download full-size image
Co-reporter:Ihsan Erden, John Basada, Daniela Poli, Gabriel Cabrera, Fupei Xu, Scott Gronert
Tetrahedron Letters 2016 Volume 57(Issue 20) pp:2190-2193
Publication Date(Web):18 May 2016
DOI:10.1016/j.tetlet.2016.04.024
•Fulvene endoperoxides ordinarily decompose via allene oxide intermediates.•A hydroxyl substituent has a significant effect on the decomposition mechanism.•In some cases the hydroxyl group causes intramolecular 1,7-H shifts.•Allene oxide/oxy-allyl species have been implicated in some of the isomerizations.•Molecular modeling studies support the proposed pathways.The thermal decomposition of fulvene endoperoxides ordinarily proceeds via an allene oxide intermediate affording oxepin-2(3H)-one derivatives. We have now uncovered new, unusual pathways in these decompositions where the presence of a hydroxyl group on the alkyl or aryl attached to the fulvene exocyclic double bond has a profound effect on the fate of the reactive intermediates derived from the unstable endoperoxides. Computational work supports the proposed mechanistic pathways.
Co-reporter:Joseph Clarke, Patrick W. Fowler, Scott Gronert, and James R. Keeffe
The Journal of Organic Chemistry 2016 Volume 81(Issue 19) pp:8777-8788
Publication Date(Web):September 6, 2016
DOI:10.1021/acs.joc.6b01261
Suprafacial sigmatropic shift reactions of 5-substituted cyclopentadienes, 3-substituted cyclopropenes, and 7-substituted cycloheptatrienes have been studied computationally at the MP2/6-31+G* level for structures and energetics and with the ipsocentric method at the CHF/6-31G** level to calculate current–density maps. The hydrogen shifts in cyclopentadienes have a diatropic ring current indicating aromatic, cyclopentadienide anion character. This result stands in contrast to the fluorine shift in 5-fluorocyclopentadiene which requires much more energy and has a paratropic ring current in the TS pointing to antiaromatic, cyclopentadienyl cation character. [1,3] hydrogen shifts in cyclopropenes are very difficult, passing through transition states that have an extended C–C bond. For 3-fluorocyclopropene, the [1,3] fluorine shift is much easier than the hydrogen shift. For 7-fluorocycloheptatriene, the [1,7] hydrogen shift is predicted but requires very high energy and has a paratropic ring current and antiaromatic character. The [1,7] suprafacial fluorine shift is relatively easy, having a TS with cycloheptatrienyl cation character. Patterns of currents, and the reversal for H and F migration, are rationalized by orbital analysis based on the ipsocentric method. Calculated charges and structural features for reactants and transition states support these conclusions.
Co-reporter:David Derkits;Alex Wiseman
Journal of The American Society for Mass Spectrometry 2016 Volume 27( Issue 2) pp:339-343
Publication Date(Web):2016 February
DOI:10.1007/s13361-015-1284-x
A new, variable-temperature mass spectrometer system is described. By applying polyimide heating tape to the end-cap electrodes of a Bruker (Bremen, Germany) Esquire ion trap, it is possible to vary the effective temperature of the system between 40 and 100°C. The modification does not impact the operation of the ion trap and the heater can be used for extended periods without degradation of the system. The accuracy of the ion trap temperatures was assessed by examining two gas-phase equilibrium processes with known thermochemistry. In each case, the variable-temperature ion trap provided data that were in good accord with literature data, indicating the effective temperature in the ion trap environment was being successfully modulated by the changes in the set-point temperatures on the end-cap electrodes. The new design offers a convenient and effective way to convert commercial ion trap mass spectrometers into variable-temperature instruments.
Co-reporter:Christopher A. Swift and Scott Gronert
Organometallics 2016 Volume 35(Issue 22) pp:3844-3851
Publication Date(Web):November 8, 2016
DOI:10.1021/acs.organomet.6b00719
The gold(I)-induced rearrangements of a variety of propargyl derivatives (ethers, acetals, acetates, and carbonates) were explored in the gas phase with experiments in an ion-trap mass spectrometer as well as with computations at the M06/QZVP level. In accord with condensed-phase studies, it appears that propargyl ethers and acetals prefer 1,3-migrations to give allenes with the release of aldehydes. With propargyl acetates, we show that the preferred path is also a 1,3-migration of the acetate to give an allene species, but that a 1,2-migration to give a gold(I) carbene species is competitive. However, with the kinetic window of our gas-phase instrumentation, only systems that can be locked into a gold(I) carbene structure give carbenoid chemistry. Finally, we found that propargyl carbonates react with gold(I) species and release CO2 in the gas phase; the likely pathway involves sequential 1,3-migrations, leading to a propargyl ether. Overall, the results highlight the dynamic nature of gold(I)-induced rearrangements and the competition between 1,2-migrations, 1,3-migrations, and the bridged intermediates that link them.
Co-reporter:Leah L. Soukup, Scott Gronert
International Journal of Mass Spectrometry 2015 Volume 378() pp:31-37
Publication Date(Web):15 February 2015
DOI:10.1016/j.ijms.2014.05.020
•Gas-phase examples of unusual SN2 processes on bromine centers.•Unusual leaving group patterns in gas-phase nucleophilic aromatic substitution reactions.•Evidence of non-statistical behavior in reactions of relatively large ions and substrates.The gas-phase reactions of a series of polyfluorobromobenzenes with stabilized carbanions (substituted benzyl and phenyl anions) were studied in an ion trap mass spectrometer. The carbanions were formed via collision-induced decarboxylation of appropriate carboxylate precursors that were generated in an electrospray ionization (ESI) source. The systems show two types of reactivity. Attack on the π-system leads to nucleophilic aromatic substitution reactions (SNAr) with the loss of bromide or fluoride anions. Alternatively, attack on the periphery leads to proton transfer or nucleophilic attack on bromine (SN2@Br), each of which leads to a new phenyl anion. The two types of nucleophiles present different reactivity. The phenyl anion nucleophiles mainly undergo proton transfer reactions, presumably due to their more localized charge as well as destabilizing steric interactions in the SNAr transition states. The benzyl anions offer more diverse reactivity and greater competition among the entire set of possible reaction pathways. Thermodynamic factors have some control over the measured rate constants and branching ratios, but the preference for bromide vs. fluoride loss in the SNAr processes does not show a discernable correlation with structural features in the substrates. In addition, there is evidence suggesting non-statistical behavior in the reactions.
Co-reporter:William H. Saunders Jr. and Scott Gronert
The Journal of Organic Chemistry 2015 Volume 80(Issue 21) pp:10787-10793
Publication Date(Web):October 20, 2015
DOI:10.1021/acs.joc.5b01983
Ab initio methods are used to examine the regio- and stereoselectivities of the elimination reactions of 2-fluorobutane and 2-chlorobutane with a series of nucleophiles (F–, HO–, CH3O–, (CH3)3CO–, NH2–, CH3–, H–, Cl–, HS–, and PH2–). The data suggest that regiochemistry is most closely related to the nature of the transition state on the E2 spectrum with E1cb-like reactions favoring the least-substituted alkene product and E1-like reactions favoring the most-substituted alkene product. There appears to be no correlation between the extent of π-bond formation (as measured by the Cα–Cβ distance) and the preference for forming the more highly substituted alkene. The stereochemistry (E vs Z) is less sensitive to the nucleophile and is relatively constant with the exception of a few systems that appear to have long-range interactions that reduce the bias against the Z product. Comparisons with experimental results in solution show, with a few exceptions, similar reactivity trends in solution and the gas phase.
Co-reporter:Dr. Jamal T. Aldajaei;Dr. James R. Keeffe;Christopher A. Swift;Dr. Scott Gronert
Chemistry - A European Journal 2015 Volume 21( Issue 36) pp:12702-12708
Publication Date(Web):
DOI:10.1002/chem.201501550
Abstract
A novel approach is used to synthesize a stable, ligated copper(I) carbene in the gas phase that is capable of typical metal carbenoid chemistry. However, it is shown that copper(I) carbenes generally undergo rapid unimolecular rearrangements including insertions into copper-ligand bonds and Wolff rearrangements. The results indicate that most copper(I) carbenes are inherently unstable and would not be viable intermediates in condensed-phase applications; an alternative intermediate that is less prone to rearrangements is required. Computational data suggest that ylides formed by the complexation of the carbene with solvent or other weak nucleophiles are viable intermediates in the reactions of copper(I) carbenes.
Co-reporter:Christopher A. Swift ;Dr. Scott Gronert
Angewandte Chemie International Edition 2015 Volume 54( Issue 22) pp:6475-6478
Publication Date(Web):
DOI:10.1002/anie.201500863
Abstract
It is demonstrated that a cationic iridium(III) dichloride phenanthroline complex is capable of CH activation and H/D exchange. It can cleave benzylic and unactivated secondary CH bonds, but exhibits unique selectivity when compared to similar systems that have been studied in the condensed phase. Gas-phase rate constants and kinetic isotope effects are reported for a variety of substrates and the analysis is supported by DFT calculations at the M06/QZVP level.
Co-reporter:Christopher A. Swift ;Dr. Scott Gronert
Angewandte Chemie 2015 Volume 127( Issue 22) pp:6575-6578
Publication Date(Web):
DOI:10.1002/ange.201500863
Abstract
It is demonstrated that a cationic iridium(III) dichloride phenanthroline complex is capable of CH activation and H/D exchange. It can cleave benzylic and unactivated secondary CH bonds, but exhibits unique selectivity when compared to similar systems that have been studied in the condensed phase. Gas-phase rate constants and kinetic isotope effects are reported for a variety of substrates and the analysis is supported by DFT calculations at the M06/QZVP level.
Co-reporter:Necdet Coskun, Meliha Çetin, Scott Gronert, Jingxiang Ma, Ihsan Erden
Tetrahedron 2015 Volume 71(Issue 18) pp:2636-2642
Publication Date(Web):6 May 2015
DOI:10.1016/j.tet.2015.03.042
A systematic study of the reactions of cyclopentadiene with α,β-unsaturated carbonyl compounds in the presence of catalytic pyrrolidine-H2O revealed that the reactions can either proceed with a Michael attack at the β-carbon of enone, or 1,2-addition to the carbonyl, leading either to 4-cyclopentadienyl-2-butanones or 6-vinylfulvenes. The former can be isolated and/or converted to the corresponding 1,2-dihydropentalenes with base (or in one-pot at longer reaction times). Substitution pattern on the enones on the competing pathways have been studied and consistent mechanisms are proposed.
Co-reporter:Jeannette T. Bowler, Freeman M. Wong, Scott Gronert, James R. Keeffe and Weiming Wu
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 32) pp:6175-6180
Publication Date(Web):25 Jun 2014
DOI:10.1039/C4OB00946K
The “element effect” in nucleophilic aromatic substitution reactions (SNAr) is characterized by the leaving group order, L = F > NO2 > Cl ≈ Br > I, in activated aryl substrates. A different leaving group order is observed in the substitution reactions of ring-substituted N-methylpyridinium compounds with piperidine in methanol: 2-CN ≥ 4-CN > 2-F ∼ 2-Cl ∼ 2-Br ∼ 2-I. The reactions are second-order in [piperidine], the mechanism involving rate determining hydrogen-bond formation between piperidine and the substrate-piperidine addition intermediate followed by deprotonation of this intermediate. Computational results indicate that deprotonation of the H-bonded complex is probably barrier free, and is accompanied by simultaneous loss of the leaving group (E2) for L = Cl, Br, and I, but with subsequent, rapid loss of the leaving group (E1cB-like) for the poorer leaving groups, CN and F. The approximately 50-fold greater reactivity of the 2- and 4-cyano substrates is attributed to the influence of the electron withdrawing cyano group in the deprotonation step. The results provide another example of β-elimination reactions poised near the E2-E1cB mechanistic borderline.
Co-reporter:Scott Gronert, John M. Garver, Charles M. Nichols, Benjamin B. Worker, and Veronica M. Bierbaum
The Journal of Organic Chemistry 2014 Volume 79(Issue 22) pp:11020-11028
Publication Date(Web):October 20, 2014
DOI:10.1021/jo502039h
The gas-phase reactions of carbon- and nitrogen-centered nucleophiles with polyfluorobromobenzenes were examined in a selected-ion flow tube (SIFT) and modeled computationally at the MP2/6-31+G(d,p)//MP2/6-31+G(d) level. In the gas-phase experiments, rate constants and branching ratios were determined. The carbon nucleophiles produce expected nucleophilic aromatic substitution (SNAr) and proton transfer products along with unexpected products that result from SN2 reactions at the bromine center (polyfluorophenide leaving group). With nitrogen nucleophiles, the SN2 at bromine channel is suppressed. In the SNAr channels, the “element effect” is observed, and fluoride loss competes with bromide loss. The computational modeling indicates that all the substitution barriers are well below the entrance channel and that entropy and dynamics effects control the product distributions.
Co-reporter:Christopher A. Swift and Scott Gronert
Organometallics 2014 Volume 33(Issue 24) pp:7135-7140
Publication Date(Web):December 1, 2014
DOI:10.1021/om500926v
A series of ligated gold(I) carbenes (where the ligand is Ph3P, Me2S, or an N-heterocyclic carbene, NHC) were formed in the gas phase by a variety of methods. Gold(I) benzylidenes could be formed using Chen’s method of dissociating an appropriate phosphorus ylide precursor. The resulting carbene undergoes an addition reaction with olefins to give an adduct. The adduct undergoes a second gas-phase reaction with an olefin, where presumably a cyclopropanation product is displaced by the second olefin molecule. Both steps in the process were analyzed with linear free energy relationships (i.e., Hammett plots). Under collision-induced dissociation conditions, the adduct undergoes competing processes: (1) dissociation of the cyclopropanation product to give ligated gold(I) species and (2) metathesis to give a more stable gold(I) carbene. Attempts to form less stable gold(I) carbenes in the gas phase by Chen’s approach or by reactions of diazo species with the ligated gold(I) cations were not successful—processes other than carbene formation are preferred or the desired carbene, after formation, rearranges rapidly to a more stable species. In accord with other recent work, the data suggest that coordination to a ligated gold(I) cation in the gas phase may not offer sufficient stabilization to carbenes to prevent competition from rearrangement processes.
Co-reporter:Ihsan Erden, Scott Gronert, James R. Keeffe, Jingxiang Ma, Nuket Ocal, Christian Gärtner, and Leah L. Soukup
The Journal of Organic Chemistry 2014 Volume 79(Issue 14) pp:6410-6418
Publication Date(Web):June 30, 2014
DOI:10.1021/jo501157s
The activating effects of the benzyl and allyl groups on SN2 reactivity are well-known. 6-Chloromethyl-6-methylfulvene, also a primary, allylic halide, reacts 30 times faster with KI/acetone than does benzyl chloride at room temperature. The latter result, as well as new experimental observations, suggests that the fulvenyl group is a particularly activating allylic group in SN2 reactions. Computational work on identity SN2 reactions, e.g., chloride– displacing chloride– and ammonia displacing ammonia, shows that negatively charged SN2 transition states (tss) are activated by allylic groups according to the Galabov–Allen–Wu electrostatic model but with the fulvenyl group especially effective at helping to delocalize negative charge due to some cyclopentadienide character in the transition state (ts). In contrast, the triafulvenyl group is deactivating. However, the positively charged SN2 transition states of the ammonia reactions are dramatically stabilized by the triafulvenyl group, which directly conjugates with a reaction center having SN1 character in the ts. Experiments and calculations on the acidities of a variety of allylic alcohols and carboxylic acids support the special nature of the fulvenyl group in stabilizing nearby negative charge and highlight the ability of fulvene species to dramatically alter the energetics of processes even in the absence of direct conjugation.
Co-reporter:Allison D. Eanes;Diogo O. Noin
Journal of The American Society for Mass Spectrometry 2014 Volume 25( Issue 1) pp:10-17
Publication Date(Web):2014 January
DOI:10.1007/s13361-013-0758-y
The reactions of a nucleophilic dianion with a series of activated aryl bromides were studied in the gas phase. Nucleophilic aromatic substitution (SNAr) as well as proton transfer reactions were observed. Rate constants and branching ratios were determined for all the reactions and the experimental data are supported by ab initio calculations. Reactions with bis-trifluoromethylbromobenzenes give only SNAr reactions and the rate constants follow the expected pattern, with substituents at the ortho and para positions having the greatest impact. Reactions of polyfluorobromobenzenes give a mix of proton transfer (when possible) and SNAr, with both bromide and fluoride acting as leaving groups. The latter is much less thermodynamically favorable but is the dominant pathway in each case. The selectivity of the reactions indicate that the products are determined early on the potential energy surface, before there is significant cleavage of the bond to the leaving group—the reaction is potentially directed by the initial formation of a hydrogen bond with the arene. The computational data also suggest that hydrogen bonding in the product ion–ion complexes can stabilize the system until there is sufficient charge separation to use the internal Coulomb repulsion to drive the reactions to products. Overall, the results highlight (1) the ability of multiply-charged systems to efficiently funnel their Coulomb repulsion into reaction processes that are intrinsically unfavorable, and (2) the high degree of selectivity that can be attained even in systems with multiple, low-barrier pathways.
Co-reporter:Scott Gronert;James R. Keeffe
Journal of Physical Organic Chemistry 2013 Volume 26( Issue 12) pp:1023-1031
Publication Date(Web):
DOI:10.1002/poc.3167
Carbocations and carbenes, as electron-deficient species, require electron donation from the remainder of the molecule to the carbon center by whatever means available. Classical interactions include resonance, polar and polarizability effects, but neighboring group participation of several sorts can also serve as stabilizing factors. Simple carbocations with directly attached electron-withdrawing groups (EWGs), that is, EWG–CH2+ ions, comprise one group by which these interactions may be probed. This article provides computational evidence at the MP2/6-311 + G** level for variable but significant stabilizing interactions between the carbocation center and common EWGs, many via bridging (partial or symmetrical), neighboring group participation, homoconjugation or π interactions. Bridging from atoms possessing nonbonding electron pairs is a common motif. Removal of bridging by application of geometric constraints nullifies bridging stabilization, but does not eliminate the possibility of other stabilizing interactions, for example polar, polarizability and π donation from the EWG. The potential for π donation from strong EWGs has not been widely appreciated in the past. A carbocation stabilization enthalpy, CSE+, is defined as the enthalpy of the isodesmic reaction CH3+ + R–H CH4 + R+. Comparisons are made with singlet carbenes bearing EWGs for which many structural features are similar. Copyright © 2013 John Wiley & Sons, Ltd.
Co-reporter: Scott Gronert
Chemistry - A European Journal 2013 Volume 19( Issue 33) pp:11090-11092
Publication Date(Web):
DOI:10.1002/chem.201002302
Co-reporter:Keyanna M. Conner and Scott Gronert
The Journal of Organic Chemistry 2013 Volume 78(Issue 17) pp:8606-8613
Publication Date(Web):July 29, 2013
DOI:10.1021/jo4013354
The SN2 and E2 reactions of a series of alkyl bromides with varying substitution patterns at the α- and β-carbons have been studied in the gas phase using naphthoate and phenoxide-based nucleophiles. The experimental work is supported by calculations at the MP2/6-31+G(d,p)//MP2/6-31+G(d) level. The results parallel reactivity patterns observed in the condensed phase, but offer new insights into steric factors in SN2 processes. In the gas phase, polarizability is more important, and the highest SN2 reactivity is observed when the β-carbon is 2°. In addition, the data confirm that alkyl substituents at the β-carbon have a greater accelerating effect on E2 reactions than those at the α-carbon. Finally, computed data based on lowest enthalpy pathways provide poor descriptions of the reactions of the larger alkyl bromides and are skewed toward crowded systems that offer stabilizing, nonbonded interactions at the expense of conformational freedom.
Co-reporter:Samuel Nettey ; Christopher A. Swift ; Renan Joviliano ; Diogo O. Noin
Journal of the American Chemical Society 2012 Volume 134(Issue 22) pp:9303-9310
Publication Date(Web):May 14, 2012
DOI:10.1021/ja301557a
For a series of α and β substituted haloethanes and haloethenes, gas-phase experiments and computational modeling have been used to characterize their nucleophilic substitution and elimination reactions. Despite being less thermodynamically favorable, the vinylic eliminations have rate constants and computed barriers that are similar to those of analogous aliphatic eliminations. This is the result of the vinylic systems shifting to more E1cb-like transition states and exploiting the inherent greater acidity of vinylic hydrogens. In general, the α-substituents have a greater impact on the SN2 pathways and stabilize the transition states via field and polarizability effects. Substantial stabilization is also provided to the E2 transition states by the α-substituents, but they have surprisingly little impact on the geometries of the transition states of either pathway. The β-substituents generally lead to a strong bias toward elimination and greatly affect the synchronicity of the elimination (more E1cb-like) as well as its location on the reaction coordinate (early). The experimental and computational data are in good accord, and the full data set provides a comprehensive picture of substituent effects on solvent-free SN2 and E2 processes.
Co-reporter:Jamal T. Aldajaei, Scott Gronert
International Journal of Mass Spectrometry 2012 Volumes 316–318() pp:68-75
Publication Date(Web):15 April 2012
DOI:10.1016/j.ijms.2011.12.012
The Mn(III), Fe(III) and Co(III) complexes of tetraphenylporphyrin were allowed to react with ethyl and t-butyl diazoacetate in an ion trap mass spectrometer. The manganese system produces only adducts, but the iron and cobalt systems give addition with loss of N2 to produce carbene-like species. All the reactions are fast and approach the collision-controlled limit. Fragmentation of the iron and cobalt carbene species follow three major pathways: (a) alkene loss from the ester to give a carboxylic acid (which can subsequently decarboxylate to give CH2 complexed to the metal porphyrin), (b) homolytic cleavage of the ester OR bond with loss of CO2 and an alkyl radical to produce CH complexed to the metal porphyrin, and (c) alcohol loss to give CCO complexed to the metal porphyrin. Computational data from density functional theory (B3LYP) are consistent with the observed reactivity trends and indicate that all the carbene complexes prefer a MN insertion structure where the carbene carbon bonds to the metal and one of the porphyrin nitrogens (metalnitrogen bond is lost). The MN insertion structures are generally more than 25 kcal/mol more stable than the conventional metal carbene structures, MC, at the B3LYP level and should dominate their reactivity.Graphical abstractHighlights► Gas-phase reactions of metal porphyrins with diazoacetates to give carbenes. ► Fragmentation gives novel metal species including CH and CCO complexes. ► Computations indicate strong preference for bridging in metal carbenes. ► Bridging will have critical effect on activity of these cationic catalysts.
Co-reporter:William C. Clodfelter, Emileigh M. Wong, Kelly A. Hay, Scott Gronert
International Journal of Mass Spectrometry 2012 Volume 314() pp:63
Publication Date(Web):15 March 2012
DOI:10.1016/j.ijms.2012.02.003
Co-reporter:Zafer Ugur;Chelsea M. Coffey
Analytical and Bioanalytical Chemistry 2012 Volume 404( Issue 5) pp:1399-1411
Publication Date(Web):2012 September
DOI:10.1007/s00216-012-6235-9
In this work, we establish a methodology for comparing the efficiencies of different hydrazide labels for detecting protein carbonyls. We have chosen acrolein-modified human serum albumin as a model. This system provides a convenient means of reproducibly generating carbonylated protein. Five hydrazide-based labels were tested. Three carry a biotin affinity tag, and the others are simple fatty acid hydrazides. For the biotin-based labels, the yield of the labeling reaction varies considerably, and the most commonly used label, biotin hydrazide, gives the lowest yield. The total tandem mass spectrometry (MS/MS) spectrum counts of modified peptides are similar for all of the biotin-based tags, indicating that factors beyond the labeling efficiency are important in determining the effectiveness of the label. In addition, there is a large variation in the number of spectra obtained for specific, modified peptides depending on the nature of the labeling group. This variation implies that the relative detectability of a particular modification site is highly dependent on the tagging reagent, and more importantly, titration schemes aimed at identifying the most reactive site based on its threshold concentration will be biased by the choice of tagging reagent. The fatty acid hydrazides are somewhat more effective than the biotin-based hydrazides in generating identifiable MS/MS spectra but offer no opportunity for enrichment. For the biotin-based tags, avidin affinity chromatography was used with the tryptic digests, and each tag led to similar enrichment levels.
Co-reporter:Scott Gronert ; James R. Keeffe ;Rory A. More O’Ferrall
Journal of the American Chemical Society 2011 Volume 133(Issue 10) pp:3381-3389
Publication Date(Web):February 22, 2011
DOI:10.1021/ja1071493
Thermodynamic stabilities of 92 carbenes, singlets and triplets, have been evaluated on the basis of hydrogenation enthalpies calculated at the G3MP2 level. The carbenes include alkyl-, aryl-, and heteroatom-substituted structures as well as cyclic 1,3-diheteroatom carbenes. Over a wide energy range, a good correlation is seen between the singlet−triplet gaps and the hydrogenation enthalpies of the singlets, but there are some clear outliers, which represent cases where the triplet has unusual stability or instability. By use of hydrogenation enthalpies, separate carbene stabilization enthalpy scales (CSEs) have been developed for singlets and triplets, and these highlight structural features that affect the stability of each. The treatment also allows estimates of aromaticity in cyclic carbenes. In this way, imidazol-2-ylidene is estimated to have an aromatic stabilization energy of about 20 kcal/mol.
Co-reporter:William C. Clodfelter, Emileigh H. Wong, Kelly A. Hay, Scott Gronert
International Journal of Mass Spectrometry 2011 Volume 305(Issue 1) pp:40-44
Publication Date(Web):1 August 2011
DOI:10.1016/j.ijms.2011.05.008
Gas-phase equilibrium measurements have been used to determine the relative binding affinities of 18 ligands to Jacobsen's manganese salen catalyst. The group of ligands spans 5.7 kcal/mol and includes seven functional groups (alcohol, ketone, ester, acyclic ether, cyclic ether, epoxide, and amine). The data follow general trends seen in other gas-phase metal cation affinities, but are influenced to a much greater extent by steric effects. For example, a 2° amine is a stronger binder than a 1° amine (as typical for gas-phase cation binding), but a 3° amine is a much weaker binder due to excessive crowding with the bulky salen ligand. The impact of steric effects was also explored with computational modeling at the B3LYP/6-311+G(d,p)/B3LYP/6-31G(d) level. The study demonstrates the utility of using mass spectrometry to probe the ligand binding characteristics of sterically demanding, metal-centered catalysts.Graphical abstractHighlights► Gas-phase binding affinities of 18 ligands to Jacobsen's catalyst. ► Identification of functional group patterns in the binding affinities. ► Identification of strong steric effects in the binding affinities. ► Major differences in binding to atomic cations, proton and lithium.
Co-reporter:Sha Huang, Scott Gronert, Weiming Wu
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 21) pp:6341-6342
Publication Date(Web):1 November 2011
DOI:10.1016/j.bmcl.2011.08.109
The structures of the uracil and thiouracils were examined using NMR spectroscopy and crystal structure data when available. The relationships between the extent of polarization and the C5–C6 bond length as well as the H5–H6 coupling constants were probed. It was found that the bond length and coupling constants correlate well with the proton affinities at the carbonyl or thiocarbonyl groups at C4 but not C2. The possible implication in the tighter binding of thiouracil based nucleotides to orotidine-5′-monophosphate decarboxylase was discussed.
Co-reporter:Scott K. Koehn, Scott Gronert and Jamal T. Aldajaei
Organic Letters 2010 Volume 12(Issue 4) pp:676-679
Publication Date(Web):January 20, 2010
DOI:10.1021/ol902682f
The rates and products from the gas-phase reactions of Co(III) salens with ethyl and t-butyl diazoacetate were examined. Addition with loss of N2 is observed, and substituent effects as well as DFT calculations indicate that addition is rate determining. Calculations suggest that the carbene species involve novel structures with the carbenic carbon bridging between the cobalt and a salen oxygen. Collision-induced dissociation leads to an unusual, bridged metal/ketene species.
Co-reporter:Scott K. Koehn ; Ngoc L. Tran ; Scott Gronert ;Weiming Wu
Journal of the American Chemical Society 2009 Volume 132(Issue 1) pp:390-395
Publication Date(Web):December 11, 2009
DOI:10.1021/ja906473v
The gas phase stability of carbanions centered at various positions on pyridine N-oxide were investigated by computational and experimental methods. In addition, G3MP2 computations were completed on ring-deprotonated pyridine and N-methylpyridinium. With these species, the effect of a nitrogen-centered positive charge on carbanion stability was assessed. Introduction of the nitrogen-oxide group into the benzene ring decreases the ΔHacid by ∼20 kcal/mol, but surprisingly, the effect is nearly independent of the position of the group (ortho, meta, or para). The results indicate that the N-oxide offers a balance of field, resonance, and local effects that cancels out any positional preferences. G3MP2 calculations indicate that a similar lack of positional selectivity is seen in nitrobenzene and benzonitrile. Overall, the data suggest that π-effects are limited in phenyl anions, and as a result, ylide-like, rather than carbene-like, resonance structures are most important in the anions derived from ring deprotonation of arenes and heterocycles of these general types.
Co-reporter:Scott Gronert
Chemistry - A European Journal 2009 Volume 15( Issue 21) pp:5372-5382
Publication Date(Web):
DOI:10.1002/chem.200800282
Co-reporter:Scott Gronert;David C. Simpson
Journal of The American Society for Mass Spectrometry 2009 Volume 20( Issue 11) pp:2116-2123
Publication Date(Web):2009 November
DOI:10.1016/j.jasms.2009.07.006
The proton affinities of the 20 common amino acids have been computed at the G3MP2 level using structures derived from broad conformational searches at a variety of levels including G3MP2. In some cases, the conformational surveys identified more stable species than had been used in previous studies of proton affinities, though the differences in energy are sometimes rather small. The present values are likely the most reliable measure of amino acid proton affinities in the gas phase. An analysis of differences between these values and those obtained experimentally via the kinetic method indicates that the extraction of proton affinities from kinetic method data can potentially lead to large errors linked to the estimation of relative protonation entropies.
Co-reporter:Jeannette T. Bowler, Freeman M. Wong, Scott Gronert, James R. Keeffe and Weiming Wu
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 32) pp:NaN6180-6180
Publication Date(Web):2014/06/25
DOI:10.1039/C4OB00946K
The “element effect” in nucleophilic aromatic substitution reactions (SNAr) is characterized by the leaving group order, L = F > NO2 > Cl ≈ Br > I, in activated aryl substrates. A different leaving group order is observed in the substitution reactions of ring-substituted N-methylpyridinium compounds with piperidine in methanol: 2-CN ≥ 4-CN > 2-F ∼ 2-Cl ∼ 2-Br ∼ 2-I. The reactions are second-order in [piperidine], the mechanism involving rate determining hydrogen-bond formation between piperidine and the substrate-piperidine addition intermediate followed by deprotonation of this intermediate. Computational results indicate that deprotonation of the H-bonded complex is probably barrier free, and is accompanied by simultaneous loss of the leaving group (E2) for L = Cl, Br, and I, but with subsequent, rapid loss of the leaving group (E1cB-like) for the poorer leaving groups, CN and F. The approximately 50-fold greater reactivity of the 2- and 4-cyano substrates is attributed to the influence of the electron withdrawing cyano group in the deprotonation step. The results provide another example of β-elimination reactions poised near the E2-E1cB mechanistic borderline.

![(4s)-4-propan-2-yl-2-[3-[(4s)-4-propan-2-yl-4,5-dihydro-1,3-oxazol-2-yl]pentan-3-yl]-4,5-dihydro-1,3-oxazole](/data/chemimg/520000/160191-65-1.png)
![(4s)-4-propan-2-yl-2-[3-[(4s)-4-propan-2-yl-4,5-dihydro-1,3-oxazol-2-yl]pentan-3-yl]-4,5-dihydro-1,3-oxazole](/data/chemimg/520000/160191-65-1_b.png)
![Gold, chloro[1,3-dihydro-1,3-bis(1-methylethyl)-2H-imidazol-2-ylidene]-](http://img.cochemist.com/ccimg/873100/873070-09-8.png)
![Gold, chloro[1,3-dihydro-1,3-bis(1-methylethyl)-2H-imidazol-2-ylidene]-](http://img.cochemist.com/ccimg/873100/873070-09-8_b.png)



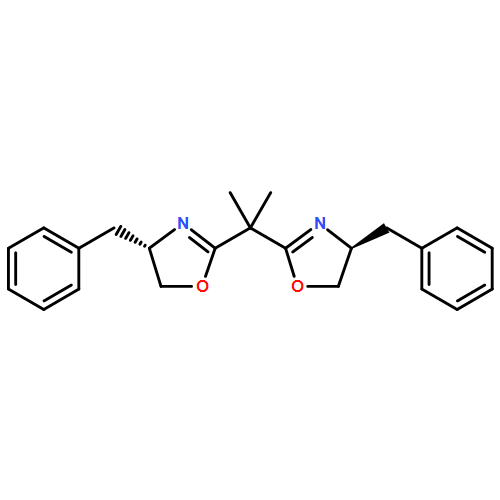
![(+)-2,2'-Isopropylidenebis[(4R)-4-phenyl-2-oxazoline]](/data/chemimg/116500/150529-93-4.png)
![(+)-2,2'-Isopropylidenebis[(4R)-4-phenyl-2-oxazoline]](/data/chemimg/116500/150529-93-4_b.png)


![2,2′-Methylenebis[(4S)-4-tert-butyl-2-oxazoline]](/data/chemimg/106700/132098-54-5.png)
![2,2′-Methylenebis[(4S)-4-tert-butyl-2-oxazoline]](/data/chemimg/106700/132098-54-5_b.png)




![Benzene, [(1R)-1-methoxyethyl]-](http://img.cochemist.com/ccimg/52300/52224-89-2.png)
![Benzene, [(1R)-1-methoxyethyl]-](http://img.cochemist.com/ccimg/52300/52224-89-2_b.png)












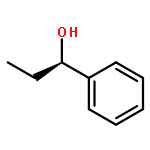
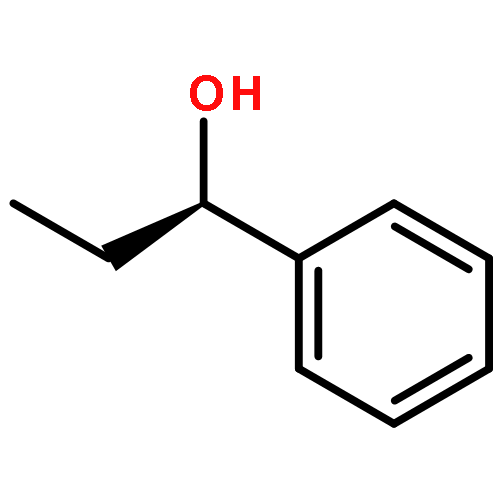


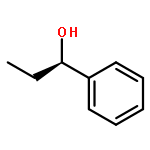
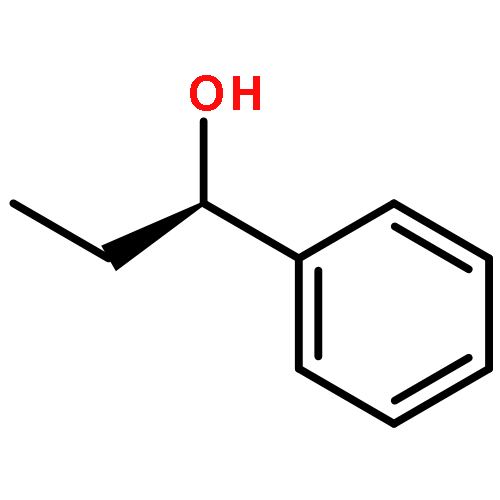
![Benz[c]acridine](http://img.cochemist.com/ccimg/300/225-51-4.png)
![Benz[c]acridine](http://img.cochemist.com/ccimg/300/225-51-4_b.png)
![Benz[a]acridine](http://img.cochemist.com/ccimg/300/225-11-6.png)
![Benz[a]acridine](http://img.cochemist.com/ccimg/300/225-11-6_b.png)
![Dibenz[c,h]acridine](http://img.cochemist.com/ccimg/300/224-53-3.png)
![Dibenz[c,h]acridine](http://img.cochemist.com/ccimg/300/224-53-3_b.png)
![Dibenzo[f,h]quinoline](http://img.cochemist.com/ccimg/300/217-65-2.png)
![Dibenzo[f,h]quinoline](http://img.cochemist.com/ccimg/300/217-65-2_b.png)
![Benzo[f]quinoline](http://img.cochemist.com/ccimg/100/85-02-9.png)
![Benzo[f]quinoline](http://img.cochemist.com/ccimg/100/85-02-9_b.png)