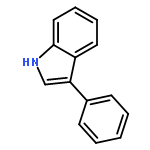
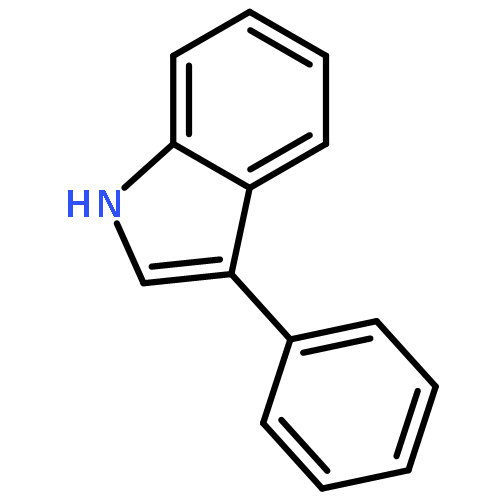
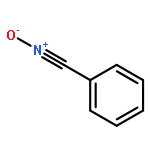
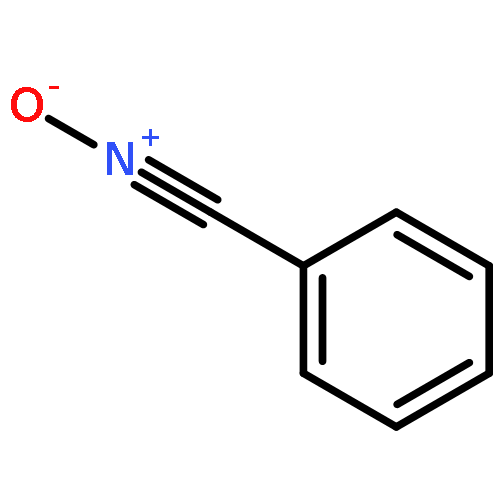
Co-reporter:Abdulselam Adam and Gebhard Haberhauer
Journal of the American Chemical Society July 19, 2017 Volume 139(Issue 28) pp:9708-9708
Publication Date(Web):June 22, 2017
DOI:10.1021/jacs.7b05316
Azobenzene and its derivatives are among the most commonly used switching units in organic chemistry. The switching process consists of two states, in which the trans isomer has a stretched and the cis isomer a compact form. Here, we have designed a system in which all isomeric states of an azobenzene moiety (trans → cis-(M) → cis-(P)) are passed step by step. The first step involves a change in the distance between the benzene units, which is common for azobenzene derivatives. In the second step an inversion of the helicity (M→P) of the cis azobenzene unit takes place. The third step leads back to the stretched trans isomer. This switching cycle is achieved by coupling the azobenzene unit with two chiral clamps and with a further azobenzene switching unit.
Co-reporter:Mathis Kreuzahler, Sven Fabig, Gebhard Haberhauer, and Rolf Gleiter
The Journal of Organic Chemistry December 15, 2017 Volume 82(Issue 24) pp:13572-13572
Publication Date(Web):November 17, 2017
DOI:10.1021/acs.joc.7b02843
In recent years, Au(I)-catalyzed reactions proved to be a valuable tool for the synthesis of substituted cycles by cycloaromatization and cycloisomerization starting from alkynes. Despite the myriad of Au(I)-catalyzed reactions of alkynes, the mono Au(I)-catalyzed pendant to the radical dimerization of nonconjugated alkyne units has not been investigated by quantum chemical calculations. Herein, by means of quantum chemical calculations, we describe the mono Au(I)-catalyzed dimerization of two alkyne units as well as the transannular ring closure reaction of a nonconjugated diyne. We found that depending on the system and the method used either the corresponding cyclopropenylmethyl cation or the butadienyl cation represents the stable intermediate. This circumstance could be explained by different stabilizing effects. Moreover, the calculation reveals a dramatic (>1012-fold) acceleration of the Au(I)-catalyzed reaction compared to that of the noncatalyzed radical variant. Trapping experiments with a substituted 1,6-cyclodecadiyne using benzene as a solvent at room temperature as well as studies with deuterated solvents confirm the calculations. In this context, we also demonstrate that trapping of the cationic intermediate with benzene does not proceed via a Friedel–Crafts-type reaction.
Co-reporter:Abdulselam Adam, Gebhard Haberhauer, and Christoph Wölper
The Journal of Organic Chemistry April 21, 2017 Volume 82(Issue 8) pp:4203-4203
Publication Date(Web):March 30, 2017
DOI:10.1021/acs.joc.7b00185
Cyclic oligomers of azole peptides were isolated from a multitude of marine organisms and were used for a large number of molecular machines. As shown previously, oligomers derived from achiral imidazole amino acids fold into canonical helices. Here we show that a minor change, the introduction of a methyl group in the δ position, results in a significant change in the secondary structure of the corresponding oligomers. Instead of a canonical helix, a noncanonical herringbone helix is formed. In the latter, the slope along the helix changes its sign at least twice per turn. This strategy allows a remarkable change of the secondary structure via a small modification. By means of enantiomerically pure amino acids, we were able to control, for the first time, both the helicity of the helix and the form of the herringbone. The investigation of the underlying herringbone basic element and its folding to a noncanonical helix were conducted by NMR and CD spectroscopy, as well as by X-ray crystallography and quantum chemical calculations.
Co-reporter:Christoph Burkhart
European Journal of Organic Chemistry 2017 Volume 2017(Issue 10) pp:1294-1294
Publication Date(Web):2017/03/10
DOI:10.1002/ejoc.201700273
The cover picture shows the molecular motor in the four different states of its moving cycle, consisting of an azobenzene (blue), a thianthrene unit (yellow), and a chiral clamp. The lightning and the lamp symbolize the two different types of stimuli; on the one hand light of an appropriated wavelength to switch the azobenzene unit from its trans to its cis configuration, and on the other hand electricity of a suitable voltage, which leads to an oxidation and an almost planar structure of the thianthrene group. The wheel in the picture demonstrates the movement of the motor and indicates the pushing movement of the system. Details are discussed in the Full Paper by C. Burkhart and G. Haberhauer et al. on page 1308 ff (DOI: 10.1002/ejoc.201601371).
Co-reporter:Christoph Burkhart
European Journal of Organic Chemistry 2017 Volume 2017(Issue 10) pp:1308-1317
Publication Date(Web):2017/03/10
DOI:10.1002/ejoc.201601371
The synthesis and investigation of molecular artificial motors have been subject to massive developments in recent years. One challenge is to design machines driven by “clean” stimuli. Types of energies are considered as “clean” stimuli if their use leads to (almost) no side-products, no byproducts, no dilution, or any other disruptive effects. We present herein a pushing motor that is driven by light and electricity, both being considered as “clean” stimuli. The switching units of the motor are an azobenzene and a thianthrene. The former is triggered by light, whereas the latter changes its structure as a result of a redox process achieved by cyclic voltammetry. The four states of the molecular motor were identified by quantum chemical methods and UV/Vis spectroscopy. The switching cycles can be performed several times without significant changes in the signals.
Co-reporter:Rolf Gleiter, Gebhard Haberhauer
Coordination Chemistry Reviews 2017 Volume 344(Volume 344) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.ccr.2017.03.003
•Bonding models for trithiapentalene and related species.•Rules to rationalize the structures of polycyclic systems with NSN bridges.•Combination of π and σ aromaticity to a three dimensional σ/π-aromaticity.•Double pancake versus long chalcogen-chalcogen bonds in dimeric C,N,S-heterocycles.•Qualitative concept for predicting the length of long chalcogen-chalcogen bonds.The focus of this review is the nature of a third covalent bond between two divalent chalcogen centers. This bond is longer than a single bond and less than the van der Waals distance between two chalcogen atoms. Such a bond is only possible when electron density is withdrawn from the divalent chalcogens. For two sulfur centers this means S⋯S bond lengths between 2.3 Å and 3.1 Å. Our discussion is based on model systems, such as various trithiapentalene derivatives, 1,5-dithia-2,4,6,8-tetrazocine and related SN cage systems, the dimers of a 1,3-dithia-2,4,6-triazine radical, and systems adopting 2-center-3-electron and 4-center-6-electron bonds, respectively, between two or four sulfur centers. The leitmotiv of this review is that the loss of electron density at the divalent sulfides can lead to trivalent sulfur centers. This behavior is first rationalized qualitatively by simple MO models. The properties of the models are reproduced by high level quantum chemical methods.Download high-res image (96KB)Download full-size image
Co-reporter:Dr. Gebhard Haberhauer;Dr. Rolf Gleiter;Christoph Burkhart
Chemistry - A European Journal 2016 Volume 22( Issue 3) pp:971-978
Publication Date(Web):
DOI:10.1002/chem.201503927
Abstract
Fluorophores were successfully used in several areas of chemistry and biochemistry. For many purposes, however, it is necessary that the fluorescence compound features a high fluorescence quantum yield as well as a large Stokes shift. The latter is, for example, achieved by the use of a twisted intramolecular charge-transfer (TICT) compound, which shows a twisted geometry in the excited state. However, the higher the twisting is, the lower becomes in general the fluorescence quantum yield as the resulting emission from the twisted state is forbidden. In order to escape this dilemma, we propose the model of planarized intramolecular charge-transfer (PLICT) states. These compounds are completely twisted in the ground states and planar in the excited states. By means of quantum chemical calculations (time-dependent (TD)-B3LYP and CC2) and experimental studies, we could demonstrate that 1-aminoindole and its derivatives form photoinduced PLICT states. They show both very large Stokes shifts ( =9000–13 500 cm−1, i.e., λ=100–150 nm) and high fluorescence quantum yields. These characteristics and their easy availability starting from the corresponding indoles, make them very attractive for the use as optical switches in various fields of chemistry as well as biological probes.
Co-reporter:Dr. Gebhard Haberhauer;Dr. Rolf Gleiter
Chemistry - A European Journal 2016 Volume 22( Issue 25) pp:8646-8653
Publication Date(Web):
DOI:10.1002/chem.201601121
Abstract
The double “pancake” bonding in the dimers of the six-membered heterocycles 1,3-dithia-2,4,6-triazine (4) and 1,3-dithia-2,4-diazine (16) were investigated by means of high-level quantum chemical calculations (B3LYP and CCSD(T)). It was found that the S–S dimers, 20 a and 27, are not the most stable isomers, but the dimers showing short S−N (21 a) and S−C (25, 28) bonds. An investigation of the 5-phenyl-1,3-dithia-2,4,6-triazine (4 b) yields that the syn dimer with two S−S bonds (2.57 Å) is the most stable one. In this dimer, the phenyl groups are placed on top of each other. The additional dispersion energy of the phenyl rings causes a stabilization of the syn-S–S (C2v-like) isomer. As a result, two weak albeit relevant single S−S bonds (2.57 Å) are predicted. These findings contradict the recently published concept of double “pancake” bonding in the dimer 4 b2.
Co-reporter:Sven Fabig; Gebhard Haberhauer;Rolf Gleiter
Journal of the American Chemical Society 2015 Volume 137(Issue 5) pp:1833-1843
Publication Date(Web):January 16, 2015
DOI:10.1021/ja510699b
By means of high level quantum chemical calculations (B2PLYPD and CCSD(T)), the dimerization of alkynes substituted with different groups such as F, Cl, OH, SH, NH2, and CN to the corresponding diradicals and dicarbenes was investigated. We found that in case of monosubstituted alkynes the formation of a bond at the nonsubstituted carbon centers is favored in general. Furthermore, substituents attached to the reacting centers reduce the activation energies and the reaction energies with increasing electronegativity of the substituent (F > OH > NH2, Cl > SH, H, CN). This effect was explained by a stabilizing hyperconjugative interaction between the σ* orbitals of the carbon-substituent bond and the occupied antibonding linear combination of the radical centers. The formation of dicarbenes is only found if strong π donors like NH2 and OH as substituents are attached to the carbene centers. The extension of the model calculations to substituted phenylacetylenes (Ph–C≡C–Y) predicts a similar reactivity of the phenylacetylenes: F > OCH3 > Cl > H. Trapping experiments of the proposed cyclobutadiene intermediates using maleic anhydride as dienophile as well as kinetic studies confirm the calculations. In the case of phenylmethoxyacetylene (Ph–C≡C–OCH3) the good yield of the corresponding cycloaddition product makes this cyclization reaction attractive for a synthetic route to cyclohexadiene derivatives from alkynes.
Co-reporter:Gebhard Haberhauer, Rolf Gleiter, and Sven Fabig
Organic Letters 2015 Volume 17(Issue 6) pp:1425-1428
Publication Date(Web):March 10, 2015
DOI:10.1021/acs.orglett.5b00296
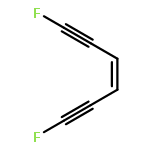
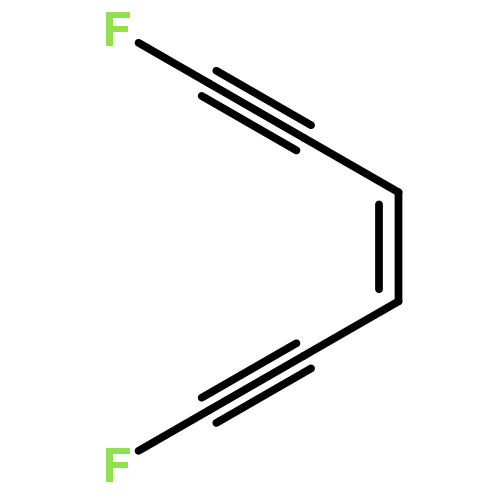
High-level quantum chemical calculations reveal that the dimerization of enediynes to 1,3-butadiene-1,4-diyl diradicals is energetically more favored than the corresponding Bergman cyclization of enediynes. Moreover, the activation barrier of both reactions can be drastically reduced by the introduction of electron-withdrawing substituents like fluoro groups at the reacting carbon centers of the triple bonds.
Co-reporter:Abdulselam Adam ;Dr. Gebhard Haberhauer
Chemistry - A European Journal 2015 Volume 21( Issue 11) pp:4333-4339
Publication Date(Web):
DOI:10.1002/chem.201406277
Abstract
Oligomers of azole peptides have been isolated from a multitude of marine organisms. Up to now, these azole-containing dipeptide-analogue oligomers have only been found as cyclic n-mers (mostly tri- and tetramers) in nature. Herein, we show that imidazole-containing pseudopeptides form helixlike secondary structures in different solvents. The screw sense of the helix can be determined by attaching a single chiral imidazole unit to the N terminus of the oligomer. Investigation by means of circular dichroism (CD) spectroscopy showed that the folding process of the helix depends on the water content of the solvent in a parabolic way. In a pure organic medium, the helix is stabilized by hydrogen bonds between the hydrogen atoms of the amide groups and the nitrogen atoms of the azole ring. In aqueous solution, the formation of the helix is driven by dispersion interactions. The formation of the helix is more pronounced in aqueous solution than in organic solvents.
Co-reporter:Gebhard Haberhauer, Rolf Gleiter, and Sascha Woitschetzki
The Journal of Organic Chemistry 2015 Volume 80(Issue 24) pp:12321-12332
Publication Date(Web):November 11, 2015
DOI:10.1021/acs.joc.5b02230
By means of high level quantum chemical calculations (B2PLYPD and CCSD(T)), the mechanisms of the reaction of nitrile oxides with alkenes and alkynes were investigated. We were able to show that in the case of alkenes, regardless of the chosen substituents, the concerted mechanism is always energetically favored as compared to a two-step process, which runs through an anti-diradical species. In the case of alkynes, the concerted mechanism is favored only for the reaction of alkyl-substituted acetylenes. For aryl-substituted acetylenes, the activation barrier toward the anti-diradical is equal to or lower than the activation barrier of the concerted reaction. This reversal of the reaction paths is not only limited to nitrile oxides as dipolarophiles. Conditions favoring the anti-diradical path are the presence of a triple bond in both the 1,3-dipole and the dipolarophile and additionally an aryl substituent attached to the alkyne. The featured energy relationships between the reaction paths are able to explain the experimentally observed byproducts of the reaction of nitrile oxides with arylacetylenes. The discovered differences for the preferred reaction path of 1,3-dipolar cycloadditions to acetylenes should be of considerable interest to a broader field of chemists.
Co-reporter:Gebhard Haberhauer, Rolf Gleiter, and Sven Fabig
The Journal of Organic Chemistry 2015 Volume 80(Issue 10) pp:5077-5083
Publication Date(Web):April 22, 2015
DOI:10.1021/acs.joc.5b00461
By means of high-level quantum chemical calculations (B2PLYPD and CCSD(T)), the dimerization of 1,3-diacetylenes was studied and compared to the dimerization of acetylene. We found that substituted 1,3-diacetylenes are more reactive than the corresponding substituted acetylenes having an isolated triple bond. The most reactive centers for a dimerization are always the terminal carbon atoms. The introduction of a test reaction allows the calculation of the relative reactivity of individual carbon centers in phenylacetylene, phenylbutadiyne, and phenylhexatriyne. A comparison shows that the reactivity of the terminal carbon atoms increases with increasing numbers of alkyne units, whereas the reactivity of the internal carbon atoms remains very low independent of the number of alkyne units.
Co-reporter:Gebhard Haberhauer, Sascha Woitschetzki, and Christof Füten
The Journal of Organic Chemistry 2015 Volume 80(Issue 16) pp:8065-8072
Publication Date(Web):July 28, 2015
DOI:10.1021/acs.joc.5b01187
Noncovalent interactions play a pivotal role in a variety of biological and chemical processes. The experimental determination and quantum chemical calculations of the forces driving these interactions are of utmost importance. Of special interest are interactions of molecules in small spaces which show phenomena different from conventional behavior in solution. An extension is the encapsulation of guests in smallest spaces: The guests are too large to be included under standard conditions and hence must be forced to intrude into the cavity. Here, we show the design of such a host–guest system which allows to directly compare the measured thermodynamic values to gas-phase quantum chemical calculations. Structural investigation of the complexes reveals that the encapsulation process causes not only an extension of the hollow space of the host but also a shrinking of the included guest by compression.
Co-reporter:Gebhard Haberhauer, Christoph Burkhart, Sascha Woitschetzki, and Christoph Wölper
The Journal of Organic Chemistry 2015 Volume 80(Issue 3) pp:1887-1895
Publication Date(Web):January 13, 2015
DOI:10.1021/acs.joc.5b00026
The imitation of macroscopic movements at the molecular level is a key step in the development of nanomachines. The challenge is the synthesis of molecules that are able to transform external stimuli into a direction-controlled mechanical movement. The more complex such motion sequences are, the more difficult is the construction of the corresponding nanomachine. Here, we present a system that demonstrates a unidirectional, four-state switching cycle that bears similar characteristics to the arm movements of a human breaststroke swimmer. Like the latter, the molecules have a torso and two arms. The arms consist of bipyridine units and can be folded and stretched by addition and removal of copper ions. The unidirectional rotation of the arms is achieved by light-induced switching of an azo unit.
Co-reporter:Dr. Rolf Gleiter;Dr. Gebhard Haberhauer;Sascha Woitschetzki
Chemistry - A European Journal 2014 Volume 20( Issue 42) pp:13801-13810
Publication Date(Web):
DOI:10.1002/chem.201402481
Abstract
A simple way of rationalizing the structures of cyclic, bicyclic, and tricyclic sulfur–nitrogen species and their congeners is presented. Starting from a planar tetrasulfur tetranitride with 12π electrons, we formally derived on paper a number of heterocyclic eight-membered 10π electron species by reacting the 3p orbitals of two opposite sulfur centers with one radical each, or by replacing these centers by other atoms with five (P) or four (Si, C) valence electrons. This led to planar aromatic 10π electron systems, nonplanar bicyclic structures with a transannular SS bond, and tricyclic structures by bridging the planar rings with an acceptor or donor unit. The final structures depend on the number of π electrons in the bridges. Intermediate biradicals are stabilized by Jahn–Teller distortion, giving transannular SS bonds between the NSN units. This procedure may be summarized by two rules, which provide a rationale for the structures of a large number of sulfur–nitrogen-based molecules. The long bonds between the NSN units show a p character of >95 %. The qualitative results have been compared with known molecular structures and the results of B3LYP/cc-pVTZ calculations as well as CASSCF and CASVB calculations. B3LYP/cc-pVTZ calculations have also provided the UV/Vis spectra and the NICS values of the planar 10π systems.
Co-reporter:Christine Kallweit;Dr. Gebhard Haberhauer;Sascha Woitschetzki
Chemistry - A European Journal 2014 Volume 20( Issue 21) pp:6358-6365
Publication Date(Web):
DOI:10.1002/chem.201304166
Abstract
A chirality switch in which the intrinsic chirality of a 4,4′-bipyridine is combined with a metal-ion-induced switching principle is described. In the uncomplexed state the 4,4′-bipyridine unit, which is linked to an S,S,S,S-configured cyclic imidazole peptide, is P-configured. The addition of zinc ions leads to a rotation around the CC bond axis of the 4,4′-bipyridine and the M isomer of the metal complex is formed. By addition of a stronger complexing agent the metal ions are removed and the switch returns to its initial position. The combination of the chirality switch with a second switching unit allows the construction of a molecular pushing motor, which is driven chemically and by light.
Co-reporter:Rolf Gleiter and Gebhard Haberhauer
The Journal of Organic Chemistry 2014 Volume 79(Issue 16) pp:7543-7552
Publication Date(Web):July 10, 2014
DOI:10.1021/jo501277h
Quantum chemical calculations were carried out by applying density functional theory to study the two center-three electron (2c-3e) bonds between the sulfur centers of cyclic dithioethers. Calculated were the S–S distance, the stabilization energy, and the energy of the σ → σ* transition. The extension of the calculations to two (2c-3e) bonds in one molecule shows that a rearrangement to one σ bond and two lone pairs on sulfur is usually more favorable. Exceptions are [H2S2+]2, the dimer of the 1,2-dithia-3,5-diazolyl radical (27a), the dimer of the 1,2,4-trithia-3,5-diazolyl radical cation (26a2+), and its Selena congeners and derivatives. In the case of [H2S2+]2, the (4c-6e) bond between the chalcogen centers is a good description of this dimer. To describe the binding situation in the dimer 26a2+ and 27a, the concept of a “simple” (4c-6e) bond was extended. Our calculations reveal a strong σ-aromaticity within the plane of the four sulfur centers in addition to a strong π-conjugation within the five-membered rings. The whole phenomenon can best be described as a three-dimensional σ/π-aromaticity within the 14π dimers.
Co-reporter:Gebhard Haberhauer ;Rolf Gleiter
Journal of the American Chemical Society 2013 Volume 135(Issue 21) pp:8022-8030
Publication Date(Web):April 28, 2013
DOI:10.1021/ja4020937
By means of high level quantum chemical calculations, the influence of electron-donating heteroatomic groups (O, NH) was investigated on the 1,6-transannular ring closure of 1,6-cyclodecadiyne (8a). In the case of 8a, the bicyclo[4.4.0]deca-1,6-dien-2,7-diyl biradical 12 is generated. It was found that oxygen centers or NH groups next to the triple bond reduce the activation energy of the ring closure considerably. For the intermediate, a 2-buten-1,4-dicarbene derivative is predicted. The extension of the model calculations to two hydroxyl- or aminoacetylenes predicts the formation of the corresponding 1,3-butadien-1,4-diyl intermediates or the 2-buten-1,4-dicarbene derivatives, a member of the nucleophilic carbene family. Moreover, the calculations predict that two separated dimethoxyacetylenes are more than 7 kcal/mol less stable than the corresponding biradical and dicarbene, respectively. Possible reactions of the dicarbenes with transition metal compounds are discussed.
Co-reporter:Gebhard Haberhauer;Christina Tepper;Christoph Wölper ;Dieter Bläser
European Journal of Organic Chemistry 2013 Volume 2013( Issue 12) pp:2325-2333
Publication Date(Web):
DOI:10.1002/ejoc.201300087
Abstract
Biphenyl derivatives with small substituents in the ortho and ortho′ positions are called tropos. Due to the low rotation barrier around the C–C bond connecting the two phenyl units, the isolation of only one conformer is not possible; thus they are conformationally unstable. Using DFT calculations, we were able to show that using a suitable peptidic bridging unit, biphenyl systems can become conformationally stable. This stabilization should be independent of the type of substituent in the ortho and ortho′ positions. Some of the proposed biphenyl derivatives were successfully synthesized and studied in solution and solid state. The recorded VT-NMR, 2D-NMR and CD spectra show that all biphenyl derivatives exhibit the P conformation. The preference for the P conformation is confirmed by the structure of a biphenyl derivative in solid state.
Co-reporter:Dr. Gebhard Haberhauer;M.Sc. Christine Kallweit;Dr. Christoph Wölper ;Dieter Bläser
Angewandte Chemie International Edition 2013 Volume 52( Issue 30) pp:7879-7882
Publication Date(Web):
DOI:10.1002/anie.201301516
Co-reporter:Dr. Gebhard Haberhauer;M.Sc. Christine Kallweit;Dr. Christoph Wölper ;Dieter Bläser
Angewandte Chemie 2013 Volume 125( Issue 30) pp:8033-8036
Publication Date(Web):
DOI:10.1002/ange.201301516
Co-reporter:Dr. Gebhard Haberhauer
Angewandte Chemie 2011 Volume 123( Issue 28) pp:6539-6543
Publication Date(Web):
DOI:10.1002/ange.201101501
Co-reporter:Dr. Gebhard Haberhauer
Angewandte Chemie International Edition 2011 Volume 50( Issue 28) pp:6415-6418
Publication Date(Web):
DOI:10.1002/anie.201101501
Co-reporter:Christina Tepper ;Dr. Gebhard Haberhauer
Chemistry - A European Journal 2011 Volume 17( Issue 29) pp:8060-8065
Publication Date(Web):
DOI:10.1002/chem.201003682
Abstract
A remarkable challenge for the design of molecular machines is the realization of a synchronized and unidirectional movement caused by an external stimulus. Such a movement can be achieved by a unidirectionally controlled change of the conformation or the configuration. Biphenol derivatives are one possibility to realize a redox-driven unidirectional molecular switch. For this reason, a 4,4′-biphenol derivative was fixed to a chiral cyclopeptidic scaffold and stimulated by chemical oxidants and reduction agents. The conformation of the switch was determined by DFT calculations by using B3LYP and the 6-31G* basis set. The switching process was observed by UV and circular dichroism (CD) spectroscopic measurements. Several oxidation agents and various conditions were tested, among which (diacetoxy)iodobenzene (DAIB) in methanol proved to be the best. In this way it was possible to synthesize a redox-stimulated molecular switch with a movement that is part of a rotation around a biaryl binding axis.
Co-reporter:Dr. Gebhard Haberhauer;Dipl.-Chem. Silvia Ernst ;Dipl.-Chem. Constanze Wilch
Chemistry - A European Journal 2011 Volume 17( Issue 31) pp:8643-8647
Publication Date(Web):
DOI:10.1002/chem.201002073
Abstract
Configurationally stable triaryl phosphane oxides are important for reactions with transfer of chiral information. Apart from introducing bulky substituents to suppress fast inversion of helicity at room temperature, the use of a second chiral element which induces chirality in the triaryl phosphane oxide, so that it adopts only one configuration, is suitable. With regard to chirality transfer, C2-symmetric imidazole cyclopeptides were tested for obtaining a configurationally stable phosphane oxide. Density functional calculations showed almost equal energies of the three possible triaryl phosphane oxides (MMM)-1, (PPP)-1, and (MP)-1. Surprisingly, after synthesis only the MMM conformer is present in solution, and its configurational stability was proved by variable-temperature and 2D NMR experiments as well as CD measurements. In view of the results of the DFT calculations, formation of stable (MMM)-1 cannot be explained thermodynamically but by kinetic reaction control. This concept of freezing the conformation of a triaryl phosphane oxide can in future be used to easily prepare configurationally stable stereoisomeric propellerlike compounds.
Co-reporter:Gebhard Haberhauer Dr. ;Christine Kallweit
Angewandte Chemie 2010 Volume 122( Issue 13) pp:2468-2471
Publication Date(Web):
DOI:10.1002/ange.200906731
Co-reporter:Dr. Gebhard Haberhauer
Angewandte Chemie International Edition 2010 Volume 49( Issue 48) pp:9286-9289
Publication Date(Web):
DOI:10.1002/anie.201004460
Co-reporter:Gebhard Haberhauer Dr. ;Christine Kallweit
Angewandte Chemie International Edition 2010 Volume 49( Issue 13) pp:2418-2421
Publication Date(Web):
DOI:10.1002/anie.200906731
Co-reporter:Dr. Gebhard Haberhauer
Angewandte Chemie 2010 Volume 122( Issue 48) pp:9474-9477
Publication Date(Web):
DOI:10.1002/ange.201004460
Co-reporter:Markus Schnopp
European Journal of Organic Chemistry 2009 Volume 2009( Issue 26) pp:4458-4467
Publication Date(Web):
DOI:10.1002/ejoc.200900510
Abstract
A straightforward synthesis of C3-symmetric, imidazole-containing, macrocyclic peptides with different binding arms is presented. The chirality of the backbone and the selection of adequate receptor arms make these systems highly selective receptors for α-chiral primary organoammonium ions. Furthermore, the receptors have the ability to discriminate between enantiomeric guests with selectivity ratios of up to 87:13. The binding constants and the selectivity ratios were estimated by standard 1H NMR titration techniques in CDCl3. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2009)
Co-reporter:Markus Schnopp;Silvia Ernst
European Journal of Organic Chemistry 2009 Volume 2009( Issue 2) pp:213-222
Publication Date(Web):
DOI:10.1002/ejoc.200800811
Abstract
A straightforward synthesis of C2-symmetric azole-containing macrocyclic peptides is presented. This type of macrocycle possesses four amide groups directed into the interior of the scaffold that act as hydrogen-bond donors and two nitrogen atoms from the azole unit that act as hydrogen-bond acceptors. This arrangement makes them sensitive receptors for Y-shaped anions like AcO– and H2PO4– with a selectivity for dihydrogen phosphate versus acetate, as was shown with 1H NMR titration techniques in [D6]DMSO/5 % CDCl3.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2009)
Co-reporter:Eva Ziegler
European Journal of Organic Chemistry 2009 Volume 2009( Issue 20) pp:3432-3438
Publication Date(Web):
DOI:10.1002/ejoc.200900250
Abstract
A straightforward synthesis of a C3-symmetric imidazole-containing macrocyclic peptide with three hydroxyquinoline side arms is presented. Complex formation with various metal ions (Al3+, Ga3+, Fe3+, La3+ and Y3+) was investigated by spectrophotometric methods. CD spectroscopy revealed a highly diastereoselective binding of these ions at room temperature. In the case of Al3+, Ga3+ and La3+ complexes, Job plot analyses gave evidence of pure 1:1 stoichiometry. Ab initio calculations for the Al3+ and Ga3+ complexes showed that the Λ isomers are considerably stabilized relative to the Δ isomers. Furthermore, the calculated CD spectra for the Al3+ complexes confirm the formation of the Λ isomers in solution. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2009)
Co-reporter:Silvia Ernst
Chemistry - A European Journal 2009 Volume 15( Issue 48) pp:13406-13416
Publication Date(Web):
DOI:10.1002/chem.200901940
Abstract
A basic requirement for each molecular system that is supposed to perform work is a synchronized and unidirectional movement. Unidirectionality can be achieved by a change of configuration or conformation that is controllable by external stimulation. Molecular hinges based on a bipyridine unit work unidirectionally and are able to reach an amplitude of motion that amounts to about 180°. To analyze if it is possible to adjust the height of the unidirectional amplitude of motion, three planar chiral molecular hinge systems with a 2,2′-bipyridine unit as functional element were designed and stimulated with various divalent metal ions in different solvents. The configurations of the hinges were determined by DFT calculations using B3LYP and the 6-31G* basis set and experimentally verified by 2D NMR NOESY spectra. Circular dichroism (CD) and UV spectroscopy were used to study the properties of the hinges by the addition of metal ions (primarily Zn2+ and Hg2+) in dichloromethane and methanol. The choice of metal ions and solvents determines whether or not and how far the hinges are closed. Furthermore, a drastic change in the height of the amplitude of motion can be reached by modifying the position of the bipyridine unit in the hinge. Amplitude values from 45 up to 190° were obtained from quantum mechanical calculations. This control of the amplitude of motion can in the future be used for more complex switching processes of molecular machines.
Co-reporter:Áron Pintér, Gebhard Haberhauer
Tetrahedron 2009 65(11) pp: 2217-2225
Publication Date(Web):
DOI:10.1016/j.tet.2009.01.047
Co-reporter:Áron Pintér
European Journal of Organic Chemistry 2008 Volume 2008( Issue 14) pp:2375-2387
Publication Date(Web):
DOI:10.1002/ejoc.200701153
Abstract
A straightforward synthesis of C3-symmetric oxazole-containing macrocyclic peptide scaffolds is presented. This type of macrocycles bears three functional groups on the oxazole rings, which allows fixing of various receptor arms on them in an easy manner. The chiral backbone of the macrocycle proved to be a powerful tool for chirality induction, thus predetermining the configuration of helically coordinated metal centres. The diastereoselective formation of CoII, NiII, CuII and ZnII complexes with tripodal bipyridyl ligand 4 was proved by UV- and CD-absorption spectrophotometric titration experiments.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2008)
Co-reporter:Áron Pintér Dr. Dr.
Chemistry - A European Journal 2008 Volume 14( Issue 35) pp:11061-11068
Publication Date(Web):
DOI:10.1002/chem.200801552
Abstract
A chiral C3-symmetric enterobactin analogue (1) has been synthesized by attachment of three 2,3-dihydroxybenzoyl units to a chiral oxazole-containing macrocyclic peptide scaffold. Complex formation kinetics and stoichiometry with various metal ions were investigated by spectrophotometric methods. In the cases of AlIII, InIII and FeIII complexes, UV absorption and CD kinetics showed nonlinearity, which results from slow conformational changes of the octahedral complexes. Virtual binding constants were determined from UV absorption data and showed selective binding of GaIII in preference to FeIII, by two orders of magnitude. CD spectroscopy revealed highly diastereoselective binding of AlIII, GaIII, InIII, FeIII and GeIV ions at room temperature, corresponding to the helical chirality opposite to that of the analogous enterobactin complexes. Ab initio calculations confirmed the energetic stabilization of the Λ isomers relative to the Δ isomers.
Co-reporter:Gebhard Haberhauer Dr.
Angewandte Chemie 2008 Volume 120( Issue 19) pp:3691-3694
Publication Date(Web):
DOI:10.1002/ange.200800062
Co-reporter:Gebhard Haberhauer Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 19) pp:3635-3638
Publication Date(Web):
DOI:10.1002/anie.200800062
Co-reporter:Áron Pintér, Gebhard Haberhauer, Isabella Hyla-Kryspin and Stefan Grimme
Chemical Communications 2007 (Issue 36) pp:3711-3713
Publication Date(Web):13 Aug 2007
DOI:10.1039/B709655K
Configurationally stable, propeller-like triarylphosphine and triarylphosphine oxide can be synthesized; a chiral scaffold based on Lissoclinum-cyclopeptides linked via three peptide bonds with a triphenylphosphine and triphenylphosphine oxide moiety, respectively, prevents effectively epimerization at the chiral phosphorus atom.
Co-reporter:Áron Pintér;Thomas Oeser;Frank Rominger
European Journal of Organic Chemistry 2007 Volume 2007(Issue 11) pp:1779-1792
Publication Date(Web):20 FEB 2007
DOI:10.1002/ejoc.200600942
The syntheses of a C4- and a C2-symmetric scaffold are presented. The first is composed of four imidazole units, while the second contains two imidazole and two oxazole moieties. The synthesis of the key building block for both systems has been fundamentally improved. Starting from these scaffolds, various platforms possessing two or four arms can easily be prepared. The structures of the scaffolds, together with that of a C4-symmetric oxazole scaffold, were investigated in the solid state and in the gas phase. We found that these 24-membered cyclic peptides exist as two different structure types. In one case (type I), the nitrogen atoms of the azole units pointing into the interior of the macrocycle form a square, whereas in the second case (type II), they form a parallelogram. The type of molecular structure does not depend on the symmetry of the system, but on the type of azole used. Cyclic systems composed of four imidazole units are present in the type I structure, whereas systems consisting of four oxazole units exhibit the type II nature. Structure type II is also present in cycles composed of two oxazole units and two imidazole units. The existence of the different structure types can be explained in terms of the different orbital energies of the imidazole and oxazole systems. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2007)
Co-reporter:Gebhard Haberhauer Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 23) pp:
Publication Date(Web):30 APR 2007
DOI:10.1002/anie.200605098
The trick with the chiral clamp: In metacyclophanes such as (P)-2 it is possible to stabilize one chiral conformation by means of a chiral clamp so effectively that at room temperature only this conformation is present. This conformation resembles that of o,o′-bridged binaphthyls (for example (P)-1). New binaphthyl alternatives are thus made accessible.
Co-reporter:Gebhard Haberhauer Dr.
Angewandte Chemie 2007 Volume 119(Issue 23) pp:
Publication Date(Web):30 APR 2007
DOI:10.1002/ange.200605098
Der Trick mit der chiralen Klammer: Mit ihr ist es möglich, bei Metacyclophanen wie (P)-2 eine chirale Konformation so zu stabilisieren, dass sie bei Raumtemperatur ausschließlich eingenommen wird. Da die Konformation der Metacyclophane derjenigen von o,o′-verbrückten Binaphthylen wie (P)-1 gleicht, bieten sie sich als Alternative für diese Verbindungsklasse an.
Co-reporter:Áron Pintér, Gebhard Haberhauer, Isabella Hyla-Kryspin and Stefan Grimme
Chemical Communications 2007(Issue 36) pp:NaN3713-3713
Publication Date(Web):2007/08/13
DOI:10.1039/B709655K
Configurationally stable, propeller-like triarylphosphine and triarylphosphine oxide can be synthesized; a chiral scaffold based on Lissoclinum-cyclopeptides linked via three peptide bonds with a triphenylphosphine and triphenylphosphine oxide moiety, respectively, prevents effectively epimerization at the chiral phosphorus atom.

![Diazene, 1-phenyl-2-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]-, (1E)-](/data/chemimg/3756600/1070337-91-5.png)
![Diazene, 1-phenyl-2-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]-, (1E)-](/data/chemimg/3756600/1070337-91-5_b.png)
![1H-Imidazole-4-carboxylic acid,2-[(1S)-1-[[(1,1-dimethylethoxy)carbonyl]amino]-2-methylpropyl]-5-methyl-1-(phenylmethyl)-](http://img.cochemist.com/ccimg/790700/790663-97-7.png)
![1H-Imidazole-4-carboxylic acid,2-[(1S)-1-[[(1,1-dimethylethoxy)carbonyl]amino]-2-methylpropyl]-5-methyl-1-(phenylmethyl)-](http://img.cochemist.com/ccimg/790700/790663-97-7_b.png)
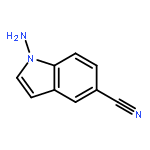
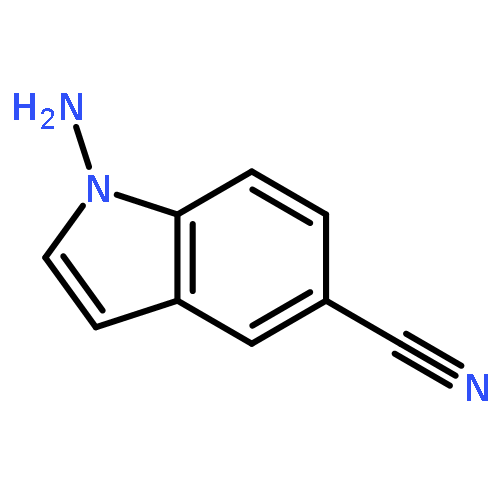







![4-[5-[[2-(AZEPAN-1-YL)-5-CYANO-1-ETHYL-4-METHYL-6-OXOPYRIDIN-3-YL]METHYLIDENE]-4-OXO-2-SULFANYLIDENE-1,3-THIAZOLIDIN-3-YL]BUTANOIC ACID](http://img.cochemist.com/ccimg/7100/7063-95-8.png)
![4-[5-[[2-(AZEPAN-1-YL)-5-CYANO-1-ETHYL-4-METHYL-6-OXOPYRIDIN-3-YL]METHYLIDENE]-4-OXO-2-SULFANYLIDENE-1,3-THIAZOLIDIN-3-YL]BUTANOIC ACID](http://img.cochemist.com/ccimg/7100/7063-95-8_b.png)