Co-reporter:Yumiko Suzuki, Jun-ichi Sawada, Paulina Hibner, Hirosuke Ishii, Kenji Matsuno, Masayuki Sato, Bernhard Witulski, Akira Asai
Dyes and Pigments 2017 Volume 145(Volume 145) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.dyepig.2017.05.050
Anticancer 2-aminoquinazolines were evaluated by their photophysical properties responding to media changes. By binding to the colchicine binding site of β-tubulin, the fluorescence emission of 2-N-morpholino-derivative (1) was blue-shifted with enhanced intensity. These distinguished features were used for the development of a β-tubulin colchicine site competition assay and visualization of the intracellular distribution of 1 by fluorescence microscopy.Download high-res image (260KB)Download full-size image
Co-reporter:Hideshi Yokoyama, Jun-ichi Sawada, Shiori Katoh, Kenji Matsuno, Naohisa Ogo, Yoshinobu Ishikawa, Hiroshi Hashimoto, Satoshi Fujii, and Akira Asai
ACS Chemical Biology 2015 Volume 10(Issue 4) pp:1128
Publication Date(Web):January 26, 2015
DOI:10.1021/cb500939x
Kinesin spindle protein Eg5 is a target for anticancer therapies, and small molecule inhibitors of its ATPase activity have been developed. We herein report for the first time the crystal structure of and biochemical studies on the Eg5 motor domain in complex with a new type of allosteric inhibitor. The biphenyl-type inhibitor PVZB1194 binds to the α4/α6 allosteric pocket 15 Å from the ATP-binding pocket, which differs from conventional allosteric inhibitors that bind to the allosteric L5/α2/α3 pocket of Eg5. Binding of the inhibitor is involved in the neck-linker conformation and also causes conformational changes around the ATP-binding pocket through Tyr104 to affect the interaction of ATP with the pocket. This structure provides useful information for the development of novel types of allosteric drugs as well as a novel insight into the molecular mechanism responsible for regulating the motor activity of kinesins.
Co-reporter:Kenta Kuroiwa, Hirosuke Ishii, Kenji Matsuno, Akira Asai, and Yumiko Suzuki
ACS Medicinal Chemistry Letters 2015 Volume 6(Issue 3) pp:287
Publication Date(Web):January 10, 2015
DOI:10.1021/ml5004684
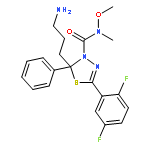
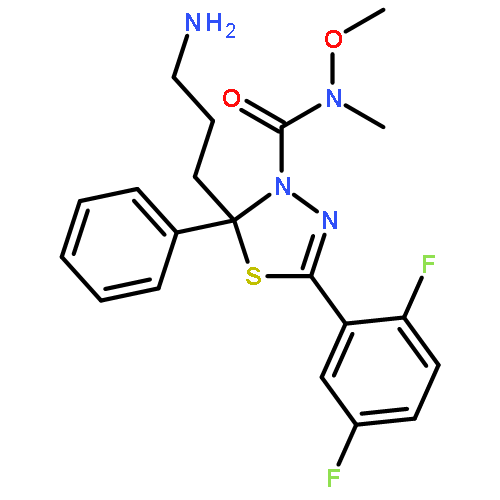
A quinazoline derivative PVHD121 (1a) was shown to have strong antiproliferative activity against various tumor-derived cell lines, including A549 (lung), NCI-H460 (lung), HCT116 (colon), MCF7 (breast), PC3 (prostate), and HeLa (cervical) cells with IC50 values from 0.1 to 0.3 μM. A structure–activity relationship (SAR) study at the 2- and 4-position of the quinazoline core lead to the discovery of more potent anticancer agents (14, 16, 17, 19, 24, and 31). The results of an in vitro tubulin polymerization assay and fluorescent-based colchicine site competition assay with purified tubulin indicated that 1a inhibits tubulin polymerization by binding to the colchicine site.Keywords: anticancer; colchicine binding site; Quinazoline; structure−activity relationship study; tubulin inhibitor
Co-reporter:Naohisa Ogo, Yoshinobu Ishikawa, Jun-ichi Sawada, Kenji Matsuno, Akihiro Hashimoto, and Akira Asai
ACS Medicinal Chemistry Letters 2015 Volume 6(Issue 9) pp:1004
Publication Date(Web):July 22, 2015
DOI:10.1021/acsmedchemlett.5b00221
Kinesin spindle protein (KSP), known as Hs Eg5, a member of the kinesin-5 family, plays an important role in the formation and maintenance of the bipolar spindle. We previously reported S-trityl-l-cysteine derivatives as selective KSP inhibitors. Here, we report further optimizations using docking modeling in the L5 allosteric binding site, which led to the discovery of several high affinity derivatives with two fused phenyl rings in the trityl group giving low nanomolar range KSP ATPase inhibition. The representative derivatives potently inhibited cell growth of HCT116 cells in correlation with KSP inhibitory activities and significantly suppressed tumor growth in the xenograft model in vivo.Keywords: differential scanning fluorimetry; HCT-116 xenograft model; Kinesin spindle protein; l-cysteine derivative; molecular modeling;
Co-reporter:Kenji Matsuno, Hiroshi Yamazaki, Yoshinobu Isaka, Kazushige Takai, Yuka Unno, Naohisa Ogo, Yoshinobu Ishikawa, Satoshi Fujii, Osamu Takikawa and Akira Asai
MedChemComm 2012 vol. 3(Issue 4) pp:475-479
Publication Date(Web):10 Jan 2012
DOI:10.1039/C2MD00278G
IDO is considered a promising therapeutic target for immunological cancer treatment. We found that the antihypertensive agent candesartan cilexetil inhibits IDO. The structural modifications provided a >10-fold more potent inhibitor. Structure–activity relationship and docking studies suggested that candesartan analogues uniquely bind to the entrance of the active site in IDO, and not to the haem region. Analogue synthesis, kinetic analysis and cellular activity are also described herein.
Co-reporter:Kenji Matsuno, Yoshiaki Masuda, Yutaka Uehara, Hiroshi Sato, Ayumu Muroya, Osamu Takahashi, Takane Yokotagawa, Toshio Furuya, Tadashi Okawara, Masami Otsuka, Naohisa Ogo, Tadashi Ashizawa, Chie Oshita, Sachiko Tai, Hidee Ishii, Yasuto Akiyama, and Akira Asai
ACS Medicinal Chemistry Letters 2010 Volume 1(Issue 8) pp:371
Publication Date(Web):July 13, 2010
DOI:10.1021/ml1000273
The signal transducer and activator of transcription 3 (STAT3) is considered to be an attractive therapeutic target for oncology drug development. We identified a N-[2-(1,3,4-oxadiazolyl)]-4-quinolinecarboxamide derivative, STX-0119, as a novel STAT3 dimerization inhibitor by a virtual screen using a customized version of the DOCK4 program with the crystal structure of STAT3. In addition, we used in vitro cell-based assays such as the luciferase reporter gene assay and the fluorescence resonance energy transfer-based STAT3 dimerization assay. STX-0119 selectively abrogated the DNA binding activity of STAT3 and suppressed the expression of STAT3-regulated oncoproteins such as c-myc and survivin in cancer cells. In contrast, a truncated inactive analogue, STX-0872, did not exhibit those activities. Oral administration of STX-0119 effectively abrogated the growth of human lymphoma cells in a SCC-3 subcutaneous xenograft model without visible toxicity. Structure−activity relationships of STX-0119 derivatives were investigated using the docking model of the STAT3-SH2 domain/STX-0119.Keywords (keywords): antitumor; dimerization; inhibitor; protein−protein interaction; STAT3; virtual screening
Co-reporter:Kenji Matsuno, Kazushige Takai, Yoshinobu Isaka, Yuka Unno, Masayuki Sato, Osamu Takikawa, Akira Asai
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 17) pp:5126-5129
Publication Date(Web):1 September 2010
DOI:10.1016/j.bmcl.2010.07.025
S-Benzylisothiourea 3a was discovered by its ability to inhibit indoleamine-2,3-dioxygenase (IDO) in our screening program. Subsequent optimization of the initial hit 3a lead to the identification of sub-μM inhibitors 3r and 10h, both of which suppressed kynurenine production in A431 cells. Synthesis and structure–activity relationship of S-benzylisothiourea analogues as small-molecule inhibitors of IDO are described.Synthesis and structure–activity relationship of S-benzylisothiourea analogues as small-molecule inhibitors of indoleamine-2,3-dioxygenase are described.
Co-reporter:Makiko Shimizu, Hirosuke Ishii, Naohisa Ogo, Kenji Matsuno, Akira Asai
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 5) pp:1578-1580
Publication Date(Web):1 March 2010
DOI:10.1016/j.bmcl.2010.01.088
Biochemical analysis of the cellular target of S-trityl-l-cysteine (STLC) derivatives was performed by using the newly synthesized STLC derivative-immobilized affinity beads (3d). The affinity beads efficiently captured KSP in HCT116 cytoplasmic cell lysate. The results obtained from pull-down and competition experiments using 3d with STLC derivatives provided the first evidence for direct interaction of these derivatives with KSP in cancer cells. Design, synthesis and application of 3d were reported.Synthesis of S-trityl-l-cysteine (STLC) analogue-immobilized affinity beads and biochemical analysis of the cellular target of STLC derivatives were reported.
Co-reporter:Kenji Matsuno, Jun-ichi Sawada, Mina Sugimoto, Naohisa Ogo, Akira Asai
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 4) pp:1058-1061
Publication Date(Web):15 February 2009
DOI:10.1016/j.bmcl.2009.01.018
Synthesis of 4-(4-tert-butylphenyl)pyridine analogues as kinesin spindle protein (KSP) inhibitors, SAR, cytotoxicity and mitotic arrest in HeLa cells are described. Interestingly, PVZB1194 showed potent KSP inhibition only in the presence of microtubules and distinct KSP localization from a known KSP inhibitor S-trytylcysteine analogue in mitosis. The observations would have resulted from a different molecular mechanism of KSP inhibition and suggest a novel biological regulation for KSP in mitosis.Synthesis of 4-(4-tert-butylphenyl)pyridine (1) analogues as KSP inhibitor, SAR, cytotoxicity and mitotic arrest in HeLa cells are described. Distinct KSP distribution by different chemotypes of KSP inhibitor is also reported.
Co-reporter:Naohisa Ogo, Shinya Oishi, Kenji Matsuno, Jun-ichi Sawada, Nobutaka Fujii, Akira Asai
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 14) pp:3921-3924
Publication Date(Web):15 July 2007
DOI:10.1016/j.bmcl.2007.04.101
Inhibition of Eg5 represents a novel approach for the treatment of cancer. Here, we report the synthesis and structure-activity relationship of S-trityl-l-cysteine (STLC) derivatives as Eg5 inhibitors. Some of these derivatives such as 4f demonstrated enhanced inhibitory activity against Eg5 and induced mitotic arrest with characteristic monoastral spindles in HeLa cells.The synthesis and evaluation for Eg5 inhibitory activity of S-substituted-l-cysteine derivatives are reported. Derivative 4f (R1 = H, R2 = OH, R3 = 4-OMe) demonstrated potent and selective inhibitory activity against Eg5 and induced mitotic arrest with characteristic monoastral spindles in HeLa cells.
Co-reporter:Jun-ichi Sawada, Ayumi Osawa, Tomoki Takeuchi, Masato Kaneda, Shinya Oishi, Nobutaka Fujii, Akira Asai, Keiji Tanino, Kosuke Namba
Bioorganic & Medicinal Chemistry Letters (1 December 2016) Volume 26(Issue 23) pp:5765-5769
Publication Date(Web):1 December 2016
DOI:10.1016/j.bmcl.2016.10.047
Co-reporter:Jun-ichi Sawada, Ayumi Osawa, Tomoki Takeuchi, Masato Kaneda, Shinya Oishi, Nobutaka Fujii, Akira Asai, Keiji Tanino, Kosuke Namba
Bioorganic & Medicinal Chemistry Letters (1 December 2016) Volume 26(Issue 23) pp:5765-5769
Publication Date(Web):1 December 2016
DOI:10.1016/j.bmcl.2016.10.047
1,3a,6a-Triazapentalene is a compact fluorescent chromophore. In this study, triazapentalene was used to modify a series of biphenyl-type inhibitors of kinesin spindle protein (KSP) to develop fluorescent probes for the intracellular visualization of this protein. Microscopic studies demonstrated that these novel triazapentalene-labeled compounds exhibited inhibitory activity towards KSP in cultured cells and provided important information concerning the intracellular distribution.

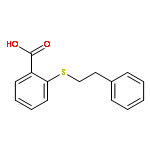
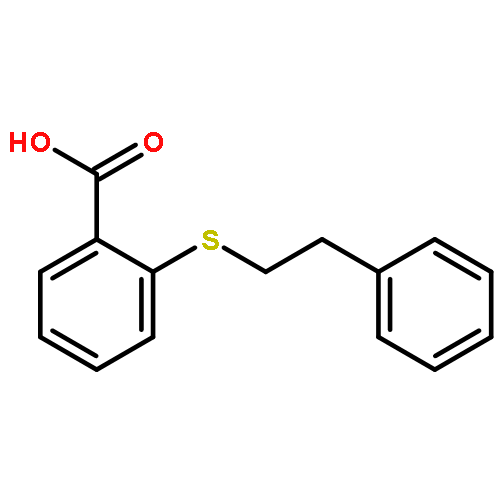
![Benzamide,N-(3-aminopropyl)-N-[(1R)-1-[7-chloro-3,4-dihydro-4-oxo-3-(phenylmethyl)-2-quinazolinyl]-2-methylpropyl]-4-methyl-](http://img.cochemist.com/ccimg/336200/336113-53-2.png)
![Benzamide,N-(3-aminopropyl)-N-[(1R)-1-[7-chloro-3,4-dihydro-4-oxo-3-(phenylmethyl)-2-quinazolinyl]-2-methylpropyl]-4-methyl-](http://img.cochemist.com/ccimg/336200/336113-53-2_b.png)
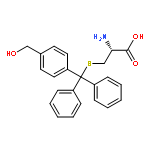
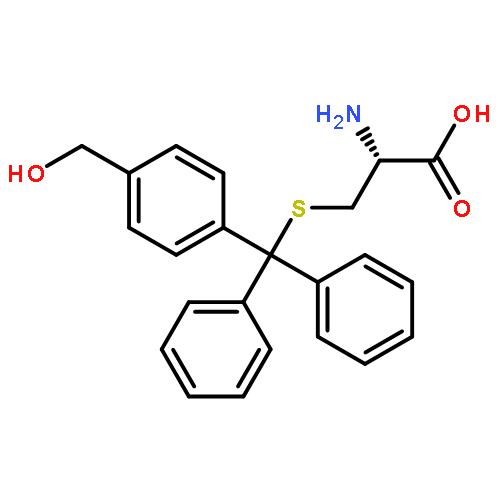
![9H-Xanthen-9-ol, 9-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/112400/112305-07-4.png)
![9H-Xanthen-9-ol, 9-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/112400/112305-07-4_b.png)
![5H-Dibenzo[a,d]cyclohepten-5-ol, 5-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/80700/80623-18-3.png)
![5H-Dibenzo[a,d]cyclohepten-5-ol, 5-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/80700/80623-18-3_b.png)
![5H-Dibenzo[a,d]cyclohepten-5-ol, 10,11-dihydro-5-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/66800/66774-09-2.png)
![5H-Dibenzo[a,d]cyclohepten-5-ol, 10,11-dihydro-5-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/66800/66774-09-2_b.png)
![5H-DIBENZO[A,D]CYCLOHEPTEN-5-OL, 5-(4-CHLOROPHENYL)-10,11-DIHYDRO-](http://img.cochemist.com/ccimg/59700/59631-06-0.png)
![5H-DIBENZO[A,D]CYCLOHEPTEN-5-OL, 5-(4-CHLOROPHENYL)-10,11-DIHYDRO-](http://img.cochemist.com/ccimg/59700/59631-06-0_b.png)
![5H-Dibenzo[a,d]cyclohepten-5-ol, 10,11-dihydro-5-phenyl-](http://img.cochemist.com/ccimg/55100/55090-32-9.png)
![5H-Dibenzo[a,d]cyclohepten-5-ol, 10,11-dihydro-5-phenyl-](http://img.cochemist.com/ccimg/55100/55090-32-9_b.png)