Co-reporter:Stefan J. Dekker, Yongjie Zhang, J. Chris Vos, Nico P. E. Vermeulen, and Jan N. M. Commandeur
Chemical Research in Toxicology 2016 Volume 29(Issue 12) pp:
Publication Date(Web):November 11, 2016
DOI:10.1021/acs.chemrestox.6b00250
Nevirapine (NVP) is a non-nucleoside reverse transcriptase-inhibitor, which is associated with severe idiosyncratic skin rash and hepatotoxicity. These adverse drug reactions are believed to be mediated by the formation of epoxides and/or quinone methide formed by oxidative metabolism by P450s and 12-sulfoxyl-NVP formed by sequential 12-hydroxylation and O-sulfonation. Although different GSH-conjugates and corresponding mercapturic acids have been demonstrated previously in vitro and in vivo, the role of the glutathione S-transferases in the inactivation of the different reactive metabolites has not been studied so far. In the present study the activity of 10 recombinant human glutathione S-transferases (GSTs) in the detoxification of the different reactive metabolites of NVP was studied. The results show that GSTP1–1 is a highly active catalyst of GSH-conjugation of the oxidative metabolites of NVP, even at high GSH-concentration. Experiments with trideuterated NVP suggest involvement of a reactive epoxide rather than quinone methide in the formation of the GSH-conjugate formed after oxidative bioactivation. GSH-conjugation of 12-sulfoxyl-NVP forming NVP-12-GSH was only catalyzed by GSTM1–1, GSTA1–1, and GSTA3–3. Although the exact expression levels of these enzymes in the skin is unknown, the relatively low activity of this catalysis makes it unlikely that GSTs can provide significant protection against this metabolite. However, since NVP-12-GSH is specifically formed via the 12-sulfoxyl-NVP, its corresponding urinary mercapturic acid can be considered as a biomarker for recent internal exposure to this protein-reactive sulfate. However, it has to be taken into account that 12-sulfoxyl-NVP is not completely trapped by GSH and that rates of bioinactivation will differ between patients due to variability in expression of GSTM1, GSTA1, and GSTA3.
Co-reporter:Jan Simon Boerma, Naura S. Elias, Nico P.E. Vermeulen, Jan N.M. Commandeur
Journal of Pharmaceutical and Biomedical Analysis 2015 Volume 103() pp:17-25
Publication Date(Web):25 January 2015
DOI:10.1016/j.jpba.2014.10.025
•A novel procedure for the preparation of drug-modified proteins is presented.•Separation of target protein from bioactivation system by a semi-permeable membrane.•Purified and membrane-bound cytochrome P450s can be used for drug bioactivation.•Analysis of intact protein adducts by mass spectrometry without laborious workup.•The described method is applicable to reactive metabolites of significant half-life.The modification of critical cellular proteins by reactive metabolites (RMs) resulting from P450-dependent drug bioactivation is considered essential to the onset of many idiosyncratic drug reactions. In this study, we report a novel method that can be used to prepare and study drug-protein adducts. Drug bioactivation by P450s was performed in a small container containing a mini-dialysis tube with the model target protein human glutathione-S-transferase P1-1 (hGST P1-1), allowing RMs to translocate from P450 to hGST P1-1 via a semi-permeable membrane (6–8 kDa). GST P1-1 modification was evaluated by LC–MS analysis of intact protein adducts and following digestion of protein with trypsin. As proof of principle, the described methodology was first applied to the direct electrophile monochlorobimane. A highly active P450 BM3 mutant (CYP102A1M11H) was subsequently used for bioactivation of acetaminophen, clozapine, diclofenac (DF) and mefenamic acid (MFA), but hGST P1-1 adducts were only observed for the latter two drugs. CYP2C9 and CYP3A4, which metabolize DF to p-benzoquinone imines, were tested to investigate the applicability of human P450s. Finally, it was evaluated whether bioactivation of MFA by human and rat liver microsomes resulted in modification of hGST P1-1. The results show that our adduct preparation method can also be used in combination with membrane-bound P450 bioactivation systems, as long as formed RMs have sufficient life-time to reach hGST P1-1 inside the dialysis tube.
Co-reporter:Jelle Reinen, Leyla Nematollahi, Alex Fidder, Nico P. E. Vermeulen, Daan Noort, and Jan N. M. Commandeur
Chemical Research in Toxicology 2015 Volume 28(Issue 4) pp:711
Publication Date(Web):February 23, 2015
DOI:10.1021/tx500490v
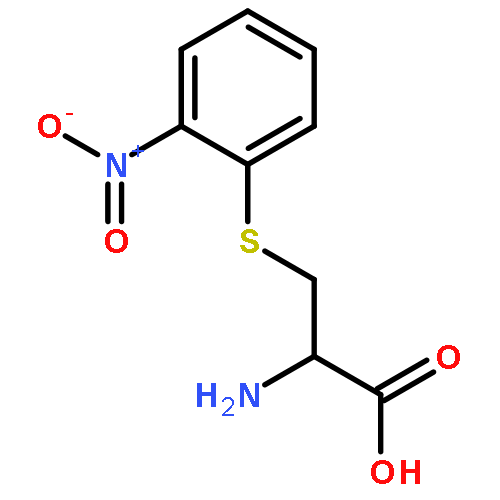
Tri-ortho-cresyl phosphate (ToCP) is a multipurpose organophosphorus compound that is neurotoxic and suspected to be involved in aerotoxic syndrome in humans. It has been reported that not ToCP itself but a metabolite of ToCP, namely, 2-(ortho-cresyl)-4H-1,2,3-benzodioxaphosphoran-2-one (CBDP), may be responsible for this effect as it can irreversibly bind to human butyrylcholinesterase (BuChE) and human acetylcholinesterase (AChE). The bioactivation of ToCP into CBDP involves Cytochrome P450s (P450s). However, the individual human P450s responsible for this bioactivation have not been identified yet. In the present study, we aimed to investigate the metabolism of ToCP by different P450s and to determine the inhibitory effect of the in vitro generated ToCP-metabolites on human BuChE and AChE. Human liver microsomes, rat liver microsomes, and recombinant human P450s were used for that purpose. The recombinant P450s 2B6, 2C18, 2D6, 3A4 and 3A5 showed highest activity of ToCP-bioactivation to BuChE-inhibitory metabolites. Inhibition experiments using pooled human liver microsomes indicated that P450 3A4 and 3A5 were mainly involved in human hepatic bioactivation of ToCP. In addition, these experiments indicated a minor role for P450 1A2. Formation of CBDP by in-house expressed recombinant human P450s 1A2 and 3A4 was proven by both LC-MS and GC-MS analysis. When ToCP was incubated with P450 1A2 and 3A4 in the presence of human BuChE, CBDP-BuChE-adducts were detected by LC-MS/MS which were not present in the corresponding control incubations. These results confirmed the role of human P450s 1A2 and 3A4 in ToCP metabolism and demonstrated that CBDP is the metabolite responsible for the BuChE inactivation. Interindividual differences at the level of P450 1A2 and 3A4 might play an important role in the susceptibility of humans in developing neurotoxic effects, such as aerotoxic syndrome, after exposure to ToCP.
Co-reporter:Harini Venkataraman, Marlies C.A. Verkade-Vreeker, Luigi Capoferri, Daan P. Geerke, Nico P.E. Vermeulen, Jan N.M. Commandeur
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 20) pp:5613-5620
Publication Date(Web):15 October 2014
DOI:10.1016/j.bmc.2014.06.022
Cytochrome P450 BM3 mutants are promising biocatalysts for the production of drug metabolites. In the present study, the ability of cytochrome P450 BM3 mutants to produce oxidative metabolites of structurally related NSAIDs meclofenamic acid, mefenamic acid and tolfenamic acid was investigated. A library of engineered P450 BM3 mutants was screened with meclofenamic acid (1) to identify catalytically active and selective mutants. Three mono-hydroxylated metabolites were identified for 1. The hydroxylated products were confirmed by NMR analysis to be 3′-OH-methyl-meclofenamic acid (1a), 5-OH-meclofenamic acid (1b) and 4′-OH-meclofenamic acid (1c) which are human relevant metabolites. P450 BM3 variants containing V87I and V87F mutation showed high selectivity for benzylic and aromatic hydroxylation of 1 respectively. The applicability of these mutants to selectively hydroxylate structurally similar drugs such as mefenamic acid (2) and tolfenamic acid (3) was also investigated. The tested variants showed high total turnover numbers in the order of 4000–6000 and can be used as biocatalysts for preparative scale synthesis. Both 1 and 2 could undergo benzylic and aromatic hydroxylation by the P450 BM3 mutants, whereas 3 was hydroxylated only on aromatic rings. The P450 BM3 variant M11 V87F hydroxylated the aromatic ring at 4′ position of all three drugs tested with high regioselectivity. Reference metabolites produced by P450 BM3 mutants allowed the characterisation of activity and regioselectivity of metabolism of all three NSAIDs by thirteen recombinant human P450s. In conclusion, engineered P450 BM3 mutants that are capable of benzylic or aromatic hydroxylation of fenamic acid containing NSAIDs, with high selectivity and turnover numbers have been identified. This shows their potential use as a greener alternative for the generation of drug metabolites.
Co-reporter:Harini Venkataraman, Michiel W. den Braver, Nico P. E. Vermeulen, and Jan N. M. Commandeur
Chemical Research in Toxicology 2014 Volume 27(Issue 12) pp:2071
Publication Date(Web):November 5, 2014
DOI:10.1021/tx500288b
Mefenamic acid (MFA) has been associated with rare but severe cases of hepatotoxicity, nephrotoxicity, gastrointestinal toxicity, and hypersensitivity reactions that are believed to result from the formation of reactive metabolites. Although formation of protein-reactive acylating metabolites by phase II metabolism has been well-studied and proposed to be the cause of these toxic side effects, the oxidative bioactivation of MFA has not yet been competely characterized. In the present study, the oxidative bioactivation of MFA was studied using human liver microsomes (HLM) and recombinant human P450 enzymes. In addition to the major metabolite 3′-OH-methyl-MFA, resulting from the benzylic hydroxylation by CYP2C9, 4′-hydroxy-MFA and 5-hydroxy-MFA were identified as metabolites resulting from oxidative metabolism of both aromatic rings of MFA. In the presence of GSH, three GSH conjugates were formed that appeared to result from GSH conjugation of the two quinoneimines formed by further oxidation of 4′-hydroxy-MFA and 5-hydroxy-MFA. The major GSH conjugate was identified as 4′-OH-5′-glutathionyl-MFA and was formed at the highest activity by CYP1A2 and to a lesser extent by CYP2C9 and CYP3A4. Two minor GSH conjugates resulted from secondary oxidation of 5-hydroxy-MFA and were formed at the highest activity by CYP1A2 and to a lesser extent by CYP3A4. Additionally, the ability of seven human glutathione S-transferases (hGSTs) to catalyze the GSH conjugation of the quinoneimines formed by P450s was also investigated. The highest increase of total GSH conjugation was observed with hGSTP1-1, followed by hepatic hGSTs hGSTA2-2 and hGSTM1-1. The results of this study show that, next to phase II metabolites, reactive quinoneimines formed by oxidative bioactivation might also contribute to the idiosyncratic toxicity of MFA.
Co-reporter:Sanja Dragovic, Jan Simon Boerma, Nico P. E. Vermeulen, and Jan N. M. Commandeur
Chemical Research in Toxicology 2013 Volume 26(Issue 11) pp:1632
Publication Date(Web):October 1, 2013
DOI:10.1021/tx400204d
Idiosyncratic adverse drug reactions due to the anti-inflammatory drug diclofenac have been proposed to be caused by the generation of reactive acyl glucuronides and oxidative metabolites. For the oxidative metabolism of diclofenac by cytochromes P450 at least five different reactive intermediates have been proposed previously based on structural identification of their corresponding GSH-conjugates. In the present study, the ability of four human glutathione S-transferases (hGSTs) to catalyze the GSH-conjugation of the different reactive intermediates formed by P450s was investigated. Addition of pooled human liver cytosol and recombinant hGSTA1–1, hGSTM1–1, and hGSTP1–1 to incubations of diclofenac with human liver microsomes or purified CYP102A1M11 L437N as a model system significantly increased total GSH-conjugation. The strongest increase of total GSH-conjugation was observed by adding hGSTP1–1, whereas hGSTM1–1 and hGSTA1–1 showed lower activity. Addition of hGSTT1–1 only showed a minor effect. When considering the effects of hGSTs on GSH-conjugation of the different quinoneimines of diclofenac, it was found that hGSTP1–1 showed the highest activity in GSH-conjugation of the quinoneimine derived from 5-hydroxydiclofenac (5-OH-DF). hGSTM1–1 showed the highest activity in inactivation of the quinoneimine derived from 4′-hydroxydiclofenac (4′-OH-DF). Separate incubations with 5-OH-DF and 4′-OH-DF as substrates confirmed these results. hGSTs also catalyzed GSH-conjugation of the o-iminemethide formed by oxidative decarboxylation of diclofenac as well as the substitution of one of the chlorine atoms of DF by GSH. hGSTP1–1 showed the highest activity for the formation of these minor GSH-conjugates. These results suggest that hGSTs may play an important role in the inactivation of DF quinoneimines and its minor reactive intermediates especially in stress conditions when tissue levels of GSH are decreased.
Co-reporter:Harini Venkataraman;Stephanie B. A. de Beer;Daan P. Geerke;Nico P. E. Vermeulen ;Jan N. M. Commeur
Advanced Synthesis & Catalysis 2012 Volume 354( Issue 11-12) pp:2172-2184
Publication Date(Web):
DOI:10.1002/adsc.201200067
Abstract
The selective hydroxylation of an unactivated CH bond is a crucial step in the synthesis of fine chemicals such as hydroxylated terpenoids. In the present study, the ability of 40 cytochrome P450 BM3 mutants to perform the regio- and stereoselective hydroxylation of α-ionone has been investigated. Based on their activity and selectivity to produce 3-hydroxy-α-ionone from racemic α-ionone, 6 BM3 mutants were selected. Out of these, 3 mutants (M01 A82W, M11 A82W and M11 V87I) showed high selectivity for trans-3-hydroxy-α-ionone formation while 3 other mutants (M11 L437N, M11 L437S and M11 L437T) formed almost equal amounts of both cis-3-hydroxy- and trans-3-hydroxy-α-ionone. Incubation with individual enantiomers showed that M11 L437N, M11 L437S and M11 L437T exhibited opposite stereoselectivity producing (3S,6S)-hydroxy-α-ionone with the (6S)-enantiomer and (3S,6R)-hydroxy-α-ionone with the (6R)-enantiomer. Thus for the first time, BM3 mutants that can selectively produce diastereomers of 3-hydroxy-α-ionone (>90% de), with high turnover numbers and minimal secondary metabolism, have been identified. Docking studies have been performed to rationalize the basis of the experimentally observed selectivity. In conclusion, engineered P450 BM3s are promising biocatalysts for regio- and stereoselective production of hydroxylated α-ionones for industrial applications.
Co-reporter:V. Rea, A. J. Kolkman, E. Vottero, E. J. Stronks, K. A. M. Ampt, M. Honing, N. P. E. Vermeulen, S. S. Wijmenga, and J. N. M. Commandeur
Biochemistry 2012 Volume 51(Issue 3) pp:
Publication Date(Web):December 28, 2011
DOI:10.1021/bi201433h
Cytochrome P450 BM3 from Bacillus megaterium is a monooxygenase with great potential for biotechnological applications. In this paper, we present engineered drug-metabolizing P450 BM3 mutants as a novel tool for regioselective hydroxylation of steroids at position 16β. In particular, we show that by replacing alanine at position 82 with a tryptophan in P450 BM3 mutants M01 and M11, the selectivity toward 16β-hydroxylation for both testosterone and norethisterone was strongly increased. The A82W mutation led to a ≤42-fold increase in Vmax for 16β-hydroxylation of these steroids. Moreover, this mutation improves the coupling efficiency of the enzyme, which might be explained by a more efficient exclusion of water from the active site. The substrate affinity for testosterone increased at least 9-fold in M11 with tryptophan at position 82. A change in the orientation of testosterone in the M11 A82W mutant as compared to the orientation in M11 was observed by T1 paramagnetic relaxation nuclear magnetic resonance. Testosterone is oriented in M11 with both the A- and D-ring protons closest to the heme iron. Substituting alanine at position 82 with tryptophan results in increased A-ring proton–iron distances, consistent with the relative decrease in the level of A-ring hydroxylation at position 2β.
Co-reporter:Jan Simon Boerma, Sanja Dragovic, Nico P. E. Vermeulen, and Jan N. M. Commandeur
Chemical Research in Toxicology 2012 Volume 25(Issue 11) pp:2532
Publication Date(Web):September 21, 2012
DOI:10.1021/tx300334w
Use of the nonsteroidal anti-inflammatory drug diclofenac (DF) is associated with serious idiosyncratic hepatotoxicity. Covalent binding of reactive intermediates of DF to proteins is considered to initiate the process leading to this severe side-effect. The aim of this study was to characterize the nature of covalent protein modifications by reactive metabolites of DF which result from bioactivation by cytochrome P450. DF and its major monohydroxylated metabolites 4′-hydroxydiclofenac (4′-OH-DF) and 5-hydroxydiclofenac (5-OH-DF) were bioactivated using a highly active P450 BM3 mutant (CYP102A1M11H) in the presence of the model target protein human glutathione-S-transferase P1-1 (hGST P1-1). Protein-adducts were subsequently identified by LC-MS/MS analysis of tryptic digests of hGST P1-1. In total, 10 different peptide adducts were observed which result from modifications of Cys-47 and Cys-14 of hGST P1-1. The majority of the protein thiol modifications appeared to be derived from 5-OH-DF, which produced seven different peptide adducts with mass increments of 289.0, 309.0, and 339.0 Da. Remarkably, no peptide adducts were observed upon the bioactivation of 4′-OH-DF. Incubations of P450 BM3 with DF also showed the peptide adducts derived from 5-OH-DF and peptide adducts that are not derived from quinone imine. A peptide adduct with a mass increment of 249.0 Da most likely results from the o-imine methide formed by oxidative decarboxylation of DF. In addition, a peptide adduct was observed with a mass increment of 259.0 Da, which corresponds to the substitution of one of the chlorine atoms of DF by protein thiol. A corresponding GSH-conjugate with a similar mass increment was only observed if incubations of DF with P450 and GSH were supplemented by human GST P1-1. The results of this study not only confirm the importance of 5-OH-DF in covalent protein-binding but also suggest that the nature of protein adduction is not necessarily reflected by chemical conjugation with GSH.
Co-reporter:Harini Venkataraman;Stephanie B. A. de Beer;Laura A. H. van Bergen;Nick van Essen;Dr. Daan P. Geerke; Dr. Nico P. E. Vermeulen ;Dr. Jan N. M. Commeur
ChemBioChem 2012 Volume 13( Issue 4) pp:520-523
Publication Date(Web):
DOI:10.1002/cbic.201100750
Co-reporter:J. S. Boerma, N. P. E. Vermeulen, and J. N. M. Commandeur
Chemical Research in Toxicology 2011 Volume 24(Issue 8) pp:1263
Publication Date(Web):June 3, 2011
DOI:10.1021/tx2001515
Covalent binding of reactive metabolites (RMs) to proteins is considered to be one of the important mechanisms by which drugs can cause tissue damage. To facilitate the study of drug–protein adducts, we developed a potentially generic method for producing high levels of covalently modified proteins. A highly active drug metabolizing P450 BM3 mutant (CYP102A1M11H) is used for drug bioactivation. Because of its His-tag, CYP102A1M11H is easily removed by nickel affinity chromatography, facilitating subsequent characterization of the modified target protein. The applicability of our procedure is demonstrated by the trapping of RMs of acetaminophen (APAP), clozapine (CLOZ), and troglitazone (TGZ) with human glutathione-S-transferase P1-1 (hGST P1-1) as the model target protein. Tryptic digests of hGST P1-1 were subjected to analysis by LC-MS/MS and modified peptides identified by the comparative analysis of tryptic peptides of adducted and nonadducted hGST P1-1. Characteristic MS/MS ions of drug-modified peptides were identified by first searching for expected adduct-masses. Unanticipated drug–peptide adducts were subsequently identified in an unbiased manner by screening for diagnostic MS/MS ions of modified peptides. Reactive intermediates of APAP and CLOZ adducted to cysteine-47 and mass shifts corresponded to the alkylation of N-acetyl-p-benzoquinone imine (NAPQI) and the CLOZ nitrenium ion, respectively. Adduction of TGZ appeared more complex, yielding three different types of adducts to cysteine-47, two adducts to cysteine-14, and a single adduct to cysteine-101. Together, these findings show that P450 BM3 mutants with high capacity to activate drugs into relevant RMs can be employed to produce protein adducts to study the nucleophilic selectivity of highly reactive electrophiles.
Co-reporter:Eduardo Vottero;Vanina Rea
JBIC Journal of Biological Inorganic Chemistry 2011 Volume 16( Issue 6) pp:899-912
Publication Date(Web):2011 August
DOI:10.1007/s00775-011-0789-4
CYP102A1, originating from Bacillus megaterium, is a highly active enzyme which has attracted much attention because of its potential applicability as a biocatalyst for oxidative reactions. Previously we developed drug-metabolizing mutant CYP102A1 M11 by a combination of site-directed and random mutagenesis. CYP102A1 M11 contains eight mutations, when compared with wild-type CYP102A1, and is able to produce human-relevant metabolites of several pharmaceuticals. In this study, active-site residue 87 of drug-metabolizing mutant CYP102A1 M11 was mutated to all possible natural amino acids to investigate its role in substrate selectivity and regioselectivity. With alkoxyresorufins as substrates, large differences in substrate selectivities and coupling efficiencies were found, dependent on the nature of residue 87. For all combinations of alkoxyresorufins and mutants, extremely fast rates of NADPH oxidation were observed (up to 6,000 min−1). However, the coupling efficiencies were extremely low: even for the substrates showing the highest rates of O-dealkylation, coupling efficiencies were lower than 1%. With testosterone as the substrate, all mutants were able to produce three hydroxytestosterone metabolites, although with different activities and with remarkably different product ratios. The results show that the nature of the amino acid at position 87 has a strong effect on activity and regioselectivity in the drug-metabolizing mutant CYP102A1 M11. Because of the wide substrate selectivity of CYP102A1 M11 when compared with wild-type CYP102A1, this panel of mutants will be useful both as biocatalysts for metabolite production and as model proteins for mechanistic studies on the function of P450s in general.
Co-reporter:Michiel W. den Braver, Yongjie Zhang, Harini Venkataraman, Nico P.E. Vermeulen, Jan N.M. Commandeur
Toxicology Letters (25 July 2016) Volume 255() pp:52-62
Publication Date(Web):25 July 2016
DOI:10.1016/j.toxlet.2016.05.015
•DF-1′,4′-QI and DF-2,5-QI are efficiently inactivated by multiple major (hepatic) GST isoforms at low and physiological GSH concentrations.•GSTM1-1 and P1-1 can prevent alkylation of a model nucleophile by DF-1′,4′-QI and DF-2,5-QI in vitro.•Based on the GST-profiles in a panel of 22 individual human livers, a large inter-individual variability in detoxification of DF-1′,4′-QI and DF-2,5-QI is predicted.•A low hepatic activity of GSTs might be considered as risk factor for DF-induced IDILI.Diclofenac is a widely prescribed NSAID that causes severe idiosyncratic drug induced liver injury (IDILI) in a small part of the patient population. Formation of protein-reactive metabolites is considered to play a role in the development of diclofenac-induced IDILI. Therefore, a high hepatic activity of enzymes involved in bioactivation of diclofenac is expected to increase the risk for liver injury. However, the extent of covalent protein binding may also be determined by activity of protective enzymes, such as glutathione S-transferases (GSTs). This is supported by an association study in which a correlation was found between NSAID-induced IDILI and the combined null genotypes of GSTM1 and GSTT1. In the present study, the activity of 10 different recombinant human GSTs in inactivation of protein-reactive quinoneimine (QI) metabolites of diclofenac was tested. Both at low and high GSH concentrations, high activities of GSTA1-1, A2-2, A3-3, M1-1, M3-3 and P1-1 in the inactivation of these QIs were found. By using the expression levels of GSTs in livers of 22 donors, a 6-fold variation in GST-dependent inactivation of reactive diclofenac metabolites was predicted. Moreover, it was shown in vitro that GSTs can strongly increase the efficiency of GSH to protect against the alkylation of the model thiol N-acetylcysteine by reactive diclofenac metabolites. The results of this study demonstrate that variability of GST expression may significantly contribute to the inter-individual differences in susceptibility to diclofenac-induced liver injury. In addition, expression levels of GSTs in in vitro models for hepatotoxicity may be important factors determining sensitivity to diclofenac cytotoxicity.
Co-reporter:Jan Simon Boerma, Nico P.E. Vermeulen, Jan N.M. Commandeur
Chemico-Biological Interactions (25 January 2014) Volume 207() pp:32-40
Publication Date(Web):25 January 2014
DOI:10.1016/j.cbi.2013.11.001
•A novel mechanism of bioactivation of diclofenac is described.•Diclofenac is bioactivated by P450s via one-electron oxidation even after heat-denaturation.•The radical cation of diclofenac is only reactive to GSH in presence of glutathione S-transferases.•The selectivity for reactive cysteines in proteins might be of toxicological relevance.Reactive metabolites have been suggested to play a role in the idiosyncratic hepatotoxicity observed with diclofenac (DF). By structural identification of the GSH conjugates formed after P450-catalyzed bioactivation of DF, it was shown that three types of reactive intermediates were formed: p-benzoquinone imines, o-imine methide and arene-oxide. Recently, detection of 2′-(glutathion-S-yl)-deschloro-diclofenac (DDF-SG), resulting from chlorine substitution, suggested the existence of a fourth type of P450-dependent reactive intermediate whose inactivation by GSH is completely dependent on presence of glutathione S-transferase. In this study, fourteen recombinant cytochrome P450s and three flavin-containing monooxygenases were tested for their ability to produce oxidative DF metabolites and their corresponding GSH conjugates. Concerning the hydroxymetabolites and their GSH conjugates, results were consistent with previous studies. Unexpectedly, all tested recombinant P450s were able to form DDF-SG to almost similar extent. DDF-SG formation was found to be partially independent of NADPH and even occurred by heat-inactivated P450. However, product formation was fully dependent on both GSH and glutathione-S-transferase P1-1. DDF-SG formation was also observed in reactions with horseradish peroxidase in absence of hydrogen peroxide. Because DDF-SG was not formed by free iron, it appears that DF can be bioactivated by iron in hemeproteins. This was confirmed by DDF-SG formation by other hemeproteins such as hemoglobin. As a mechanism, we propose that DF is subject to heme-dependent one-electron oxidation. The resulting nitrogen radical cation, which might activate the chlorines of DF, then undergoes a GST-catalyzed nucleophilic aromatic substitution reaction in which the chlorine atom of the DF moiety is replaced by GSH.
Co-reporter:Michiel W. den Braver, Shalenie P. den Braver-Sewradj, Nico P.E. Vermeulen, Jan N.M. Commandeur
Toxicology Letters (24 June 2016) Volume 253() pp:46-54
Publication Date(Web):24 June 2016
DOI:10.1016/j.toxlet.2016.04.022
•CYP3A4 and CYP2C9 are sequentially involved in the two-step formation of diclofenac-2,5-quinone imine.•Two-step diclofenac-1′,4′-quinone imine formation involves CYP2C9 in both steps.•The involvement of multiple CYPs in the sequential oxidation of DF has important implication for prediction of individual variability in exposure to p-benzoquinone imines, as CYP activity is highly variable.•The results explain previous in vitro data in which DF toxicity was only apparent when multiple CYPs were expressed simultaneously.Idiosyncratic drug-induced lever injury (IDILI) is a rare but severe side effect of diclofenac (DF). Several mechanisms have been proposed as cause of DF-induced toxicity including the formation of protein-reactive diclofenac-1′,4′-quinone imine (DF-1′,4′-QI) and diclofenac-2,5-quinone imine (DF-2,5-QI). Formation of these p-benzoquinone imines result from two-step oxidative metabolism involving aromatic hydroxylation to 4′-hydroxydiclofenac and 5-hydroxydiclofenac followed by dehydrogenation to DF-1′,4′-QI and DF-2,5-QI, respectively. Although the contribution of individual cytochrome P450s (CYPs) in aromatic hydroxylation of DF is well studied, the enzymes involved in the dehydrogenation reactions have been poorly characterized. The results of the present study show that both formation of 4′-hydroxydiclofenac and it subsequent bioactivation to DF-1′,4′-QI is selectively catalyzed by CYP2C9. However, the two-step bioactivation to DF-2,5-QI appears to be catalyzed with highest activity by two different CYPs: 5-hydroxylation of DF is predominantly catalyzed by CYP3A4, whereas its subsequent bioactivation to DF-2,5-QI is catalyzed with 14-fold higher intrinsic clearance by CYP2C9. The fact that both CYPs involved in two-step bioactivation of DF show large interindividual variability may play a role in different susceptibility of patients to DF-induced IDILI. Furthermore, expression levels of these enzymes and protective enzymes might be important factors determining sensitivity of in vitro models for hepatotoxicity.
Co-reporter:Michiel W. den Braver, Nico P.E. Vermeulen, Jan N.M. Commandeur
Journal of Chromatography B (1 March 2017) Volume 1046() pp:185-194
Publication Date(Web):1 March 2017
DOI:10.1016/j.jchromb.2017.02.004

![[1,1'-Biphenyl]-2-ol, 2',3-dibromo-](http://img.cochemist.com/ccimg/868600/868524-25-8.png)
![[1,1'-Biphenyl]-2-ol, 2',3-dibromo-](http://img.cochemist.com/ccimg/868600/868524-25-8_b.png)
![[1,1'-Biphenyl]-2-ol, 4,4'-dibromo-](http://img.cochemist.com/ccimg/868600/868524-21-4.png)
![[1,1'-Biphenyl]-2-ol, 4,4'-dibromo-](http://img.cochemist.com/ccimg/868600/868524-21-4_b.png)


![[1,1'-Biphenyl]-3-ol, 4,4'-dibromo-](http://img.cochemist.com/ccimg/59500/59452-48-1.png)
![[1,1'-Biphenyl]-3-ol, 4,4'-dibromo-](http://img.cochemist.com/ccimg/59500/59452-48-1_b.png)




![[1,1'-Biphenyl]-2-ol, 2',6-dibromo-](http://img.cochemist.com/ccimg/868600/868524-23-6.png)
![[1,1'-Biphenyl]-2-ol, 2',6-dibromo-](http://img.cochemist.com/ccimg/868600/868524-23-6_b.png)
![Thiourea, [2-(4-methoxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/81100/81050-04-6.png)
![Thiourea, [2-(4-methoxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/81100/81050-04-6_b.png)
![2-Cyclohexen-1-one, 3,5,5-trimethyl-4-[(1E)-3-oxo-1-butenyl]-](http://img.cochemist.com/ccimg/79800/79734-43-3.png)
![2-Cyclohexen-1-one, 3,5,5-trimethyl-4-[(1E)-3-oxo-1-butenyl]-](http://img.cochemist.com/ccimg/79800/79734-43-3_b.png)
![L-Alanine, N-[N-[[(2-chloroethyl)nitrosoamino]carbonyl]-L-alanyl]-](http://img.cochemist.com/ccimg/93300/93299-11-7.png)
![L-Alanine, N-[N-[[(2-chloroethyl)nitrosoamino]carbonyl]-L-alanyl]-](http://img.cochemist.com/ccimg/93300/93299-11-7_b.png)
![Thiourea, N-[2-(1H-imidazol-4-yl)ethyl]-N'-phenyl-](http://img.cochemist.com/ccimg/710300/710291-86-4.png)
![Thiourea, N-[2-(1H-imidazol-4-yl)ethyl]-N'-phenyl-](http://img.cochemist.com/ccimg/710300/710291-86-4_b.png)
![Thiourea, N-[2-(1H-imidazol-4-yl)ethyl]-N'-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/717100/717098-39-0.png)
![Thiourea, N-[2-(1H-imidazol-4-yl)ethyl]-N'-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/717100/717098-39-0_b.png)
![Thiourea, N-[2-(1H-imidazol-4-yl)ethyl]-N'-1-naphthalenyl-](http://img.cochemist.com/ccimg/717100/717098-45-8.png)
![Thiourea, N-[2-(1H-imidazol-4-yl)ethyl]-N'-1-naphthalenyl-](http://img.cochemist.com/ccimg/717100/717098-45-8_b.png)
![Thiourea, N-(4-bromophenyl)-N'-[2-(1H-imidazol-4-yl)ethyl]-](http://img.cochemist.com/ccimg/717100/717098-41-4.png)
![Thiourea, N-(4-bromophenyl)-N'-[2-(1H-imidazol-4-yl)ethyl]-](http://img.cochemist.com/ccimg/717100/717098-41-4_b.png)


![L-Alanine, N-[[(2-chloroethyl)nitrosoamino]carbonyl]-](http://img.cochemist.com/ccimg/80700/80687-07-6.png)
![L-Alanine, N-[[(2-chloroethyl)nitrosoamino]carbonyl]-](http://img.cochemist.com/ccimg/80700/80687-07-6_b.png)

![L-Alanine, 3-[[(2-methylphenyl)methyl]seleno]-](http://img.cochemist.com/ccimg/324600/324582-21-0.png)
![L-Alanine, 3-[[(2-methylphenyl)methyl]seleno]-](http://img.cochemist.com/ccimg/324600/324582-21-0_b.png)
![L-Alanine, 3-[(2-methoxyphenyl)seleno]-](http://img.cochemist.com/ccimg/324600/324582-19-6.png)
![L-Alanine, 3-[(2-methoxyphenyl)seleno]-](http://img.cochemist.com/ccimg/324600/324582-19-6_b.png)




![1,4-Cyclohexadiene-1-acetic acid, 6-[(2,6-dichlorophenyl)imino]-3-oxo-](http://img.cochemist.com/ccimg/221600/221555-69-7.png)
![1,4-Cyclohexadiene-1-acetic acid, 6-[(2,6-dichlorophenyl)imino]-3-oxo-](http://img.cochemist.com/ccimg/221600/221555-69-7_b.png)



![L-Alanine, 3-[[(2-nitrophenyl)methyl]seleno]-](http://img.cochemist.com/ccimg/324600/324582-23-2.png)
![L-Alanine, 3-[[(2-nitrophenyl)methyl]seleno]-](http://img.cochemist.com/ccimg/324600/324582-23-2_b.png)
![Cyclopentanone, 2,5-bis[(3,5-diethyl-4-hydroxyphenyl)methylene]-](http://img.cochemist.com/ccimg/195500/195440-25-6.png)
![Cyclopentanone, 2,5-bis[(3,5-diethyl-4-hydroxyphenyl)methylene]-](http://img.cochemist.com/ccimg/195500/195440-25-6_b.png)
![THIOUREA, N-[2-(1H-IMIDAZOL-4-YL)ETHYL]-N'-(4-NITROPHENYL)-](http://img.cochemist.com/ccimg/717100/717098-42-5.png)
![THIOUREA, N-[2-(1H-IMIDAZOL-4-YL)ETHYL]-N'-(4-NITROPHENYL)-](http://img.cochemist.com/ccimg/717100/717098-42-5_b.png)
![BENZOIC ACID, 4-[[(2R)-2-AMINO-2-CARBOXYETHYL]THIO]-3-NITRO-](http://img.cochemist.com/ccimg/680600/680584-46-7.png)
![BENZOIC ACID, 4-[[(2R)-2-AMINO-2-CARBOXYETHYL]THIO]-3-NITRO-](http://img.cochemist.com/ccimg/680600/680584-46-7_b.png)
![1,2-Benzenediol, 4-[2-(ethylamino)propyl]-](http://img.cochemist.com/ccimg/181500/181425-74-1.png)
![1,2-Benzenediol, 4-[2-(ethylamino)propyl]-](http://img.cochemist.com/ccimg/181500/181425-74-1_b.png)
![L-Alanine, 3-[[(4-chlorophenyl)methyl]seleno]-](http://img.cochemist.com/ccimg/176200/176178-20-4.png)
![L-Alanine, 3-[[(4-chlorophenyl)methyl]seleno]-](http://img.cochemist.com/ccimg/176200/176178-20-4_b.png)
![THIOUREA, N-(4-CHLOROPHENYL)-N'-[2-(1H-IMIDAZOL-4-YL)ETHYL]-](http://img.cochemist.com/ccimg/717100/717098-40-3.png)
![THIOUREA, N-(4-CHLOROPHENYL)-N'-[2-(1H-IMIDAZOL-4-YL)ETHYL]-](http://img.cochemist.com/ccimg/717100/717098-40-3_b.png)
![Cyclohexanone, 2,6-bis[(4-hydroxy-3,5-dimethylphenyl)methylene]-](http://img.cochemist.com/ccimg/195500/195440-21-2.png)
![Cyclohexanone, 2,6-bis[(4-hydroxy-3,5-dimethylphenyl)methylene]-](http://img.cochemist.com/ccimg/195500/195440-21-2_b.png)

![L-Alanine, 3-[[(4-methylphenyl)methyl]seleno]-](http://img.cochemist.com/ccimg/176400/176300-64-4.png)
![L-Alanine, 3-[[(4-methylphenyl)methyl]seleno]-](http://img.cochemist.com/ccimg/176400/176300-64-4_b.png)
![L-ALANINE, 3-[[(4-METHOXYPHENYL)METHYL]SELENO]-](http://img.cochemist.com/ccimg/146600/146552-76-3.png)
![L-ALANINE, 3-[[(4-METHOXYPHENYL)METHYL]SELENO]-](http://img.cochemist.com/ccimg/146600/146552-76-3_b.png)



![L-Alanine, 3-[(4-methoxyphenyl)seleno]-](http://img.cochemist.com/ccimg/176200/176178-24-8.png)
![L-Alanine, 3-[(4-methoxyphenyl)seleno]-](http://img.cochemist.com/ccimg/176200/176178-24-8_b.png)
![L-Alanine, 3-[(4-methylphenyl)seleno]-](http://img.cochemist.com/ccimg/176200/176178-22-6.png)
![L-Alanine, 3-[(4-methylphenyl)seleno]-](http://img.cochemist.com/ccimg/176200/176178-22-6_b.png)



![[1,1'-BIPHENYL]-4-OL, 2,2'-DIBROMO-](http://img.cochemist.com/ccimg/868600/868524-22-5.png)
![[1,1'-BIPHENYL]-4-OL, 2,2'-DIBROMO-](http://img.cochemist.com/ccimg/868600/868524-22-5_b.png)
![[1,1'-BIPHENYL]-3-OL, 2,2'-DIBROMO-](http://img.cochemist.com/ccimg/868600/868524-24-7.png)
![[1,1'-BIPHENYL]-3-OL, 2,2'-DIBROMO-](http://img.cochemist.com/ccimg/868600/868524-24-7_b.png)


![L-CYSTEINE, N-ACETYL-S-[2-HYDROXY-3-(2-METHYLPHENOXY)PROPYL]-](http://img.cochemist.com/ccimg/201500/201404-14-0.png)
![L-CYSTEINE, N-ACETYL-S-[2-HYDROXY-3-(2-METHYLPHENOXY)PROPYL]-](http://img.cochemist.com/ccimg/201500/201404-14-0_b.png)





![2-[[2-[[2-chloroethyl(nitroso)carbamoyl]amino]acetyl]amino]acetic Acid](http://img.cochemist.com/ccimg/82300/82219-31-6.png)
![2-[[2-[[2-chloroethyl(nitroso)carbamoyl]amino]acetyl]amino]acetic Acid](http://img.cochemist.com/ccimg/82300/82219-31-6_b.png)








![Thiourea, [2-(4-methylphenyl)ethyl]-](http://img.cochemist.com/ccimg/207600/207596-24-5.png)
![Thiourea, [2-(4-methylphenyl)ethyl]-](http://img.cochemist.com/ccimg/207600/207596-24-5_b.png)





![(2R)-3-benzylsulfanyl-2-[(2,2,2-trideuterioacetyl)amino]propanoic acid](http://img.cochemist.com/ccimg/201500/201404-15-1.png)
![(2R)-3-benzylsulfanyl-2-[(2,2,2-trideuterioacetyl)amino]propanoic acid](http://img.cochemist.com/ccimg/201500/201404-15-1_b.png)
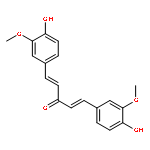
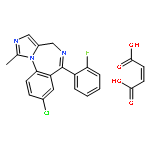
![Cyclohexanone, 2,6-bis[(4-hydroxyphenyl)methylene]-](http://img.cochemist.com/ccimg/19000/18977-35-0.png)
![Cyclohexanone, 2,6-bis[(4-hydroxyphenyl)methylene]-](http://img.cochemist.com/ccimg/19000/18977-35-0_b.png)
![(E)-4-[4-[2-(dimethylamino)ethoxy]phenyl]-3,4-diphenylbut-3-en-2-ol](http://img.cochemist.com/ccimg/97200/97151-02-5.png)
![(E)-4-[4-[2-(dimethylamino)ethoxy]phenyl]-3,4-diphenylbut-3-en-2-ol](http://img.cochemist.com/ccimg/97200/97151-02-5_b.png)


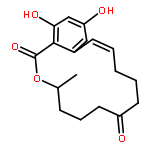
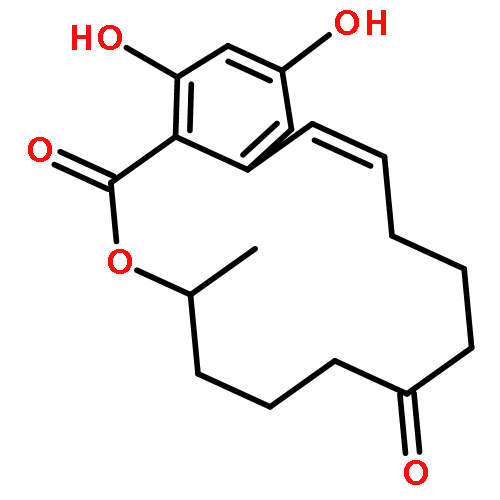






![3-(2-chloroethyl)-2-[(2-chloroethyl)amino]-1,3,2-oxazaphosphinan-4-ol 2-oxide](http://img.cochemist.com/ccimg/50900/50892-10-9.png)
![3-(2-chloroethyl)-2-[(2-chloroethyl)amino]-1,3,2-oxazaphosphinan-4-ol 2-oxide](http://img.cochemist.com/ccimg/50900/50892-10-9_b.png)
![2-[((2',6'-dichloro-4'-hydroxy)phenyl)amino]benzeneacetic Acid](http://img.cochemist.com/ccimg/64200/64118-84-9.png)
![2-[((2',6'-dichloro-4'-hydroxy)phenyl)amino]benzeneacetic Acid](http://img.cochemist.com/ccimg/64200/64118-84-9_b.png)




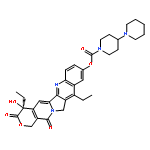
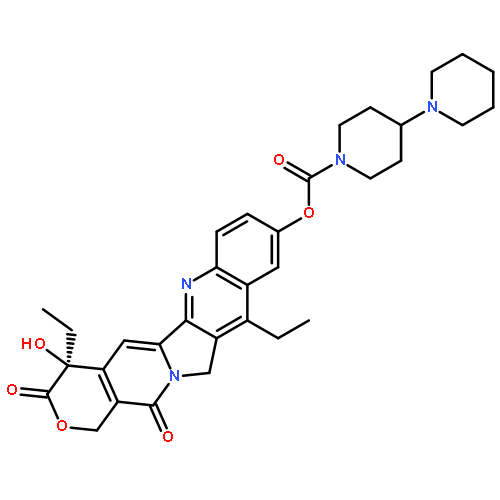




![(8r,9s,10r,13s,14s,16s,17r)-16,17-dihydroxy-10,13-dimethyl-1,2,6,7,8,9,11,12,14,15,16,17-dodecahydrocyclopenta[a]phenanthren-3-one](http://img.cochemist.com/ccimg/17600/17528-90-4.png)
![(8r,9s,10r,13s,14s,16s,17r)-16,17-dihydroxy-10,13-dimethyl-1,2,6,7,8,9,11,12,14,15,16,17-dodecahydrocyclopenta[a]phenanthren-3-one](http://img.cochemist.com/ccimg/17600/17528-90-4_b.png)

![2-[2-(tert-butylamino)-1-hydroxyethyl]-7-ethyl-1-benzofuran-4-ol](http://img.cochemist.com/ccimg/61500/61470-08-4.png)
![2-[2-(tert-butylamino)-1-hydroxyethyl]-7-ethyl-1-benzofuran-4-ol](http://img.cochemist.com/ccimg/61500/61470-08-4_b.png)





![L-gamma-glutamyl-S-[4-(acetylamino)phenyl]-L-cysteinylglycine](http://img.cochemist.com/ccimg/68000/67900-63-4.png)
![L-gamma-glutamyl-S-[4-(acetylamino)phenyl]-L-cysteinylglycine](http://img.cochemist.com/ccimg/68000/67900-63-4_b.png)



![Propanoic acid, 2-[(carboxymethyl)thio]-](http://img.cochemist.com/ccimg/14700/14618-66-7.png)
![Propanoic acid, 2-[(carboxymethyl)thio]-](http://img.cochemist.com/ccimg/14700/14618-66-7_b.png)
















![L-gamma-glutamyl-S-[5-(acetylamino)-2-hydroxyphenyl]-L-cysteinylglycine](http://img.cochemist.com/ccimg/64900/64889-81-2.png)
![L-gamma-glutamyl-S-[5-(acetylamino)-2-hydroxyphenyl]-L-cysteinylglycine](http://img.cochemist.com/ccimg/64900/64889-81-2_b.png)
![1-(4-Fluorobenzyl)-N-(1-(4-methoxyphenethyl)piperidin-4-yl)-1H-benzo[d]imidazol-2-amine](http://img.cochemist.com/ccimg/68900/68844-77-9.png)
![1-(4-Fluorobenzyl)-N-(1-(4-methoxyphenethyl)piperidin-4-yl)-1H-benzo[d]imidazol-2-amine](http://img.cochemist.com/ccimg/68900/68844-77-9_b.png)
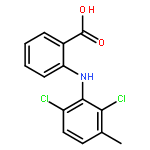
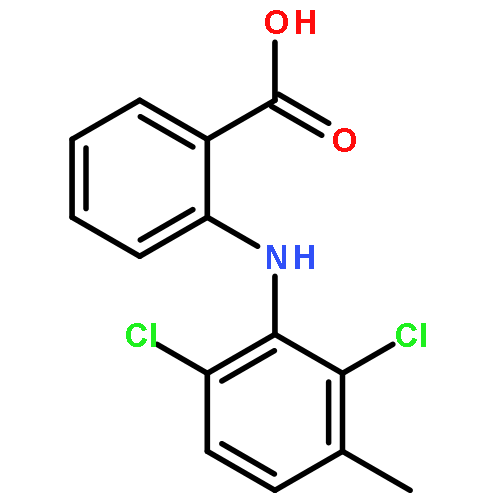
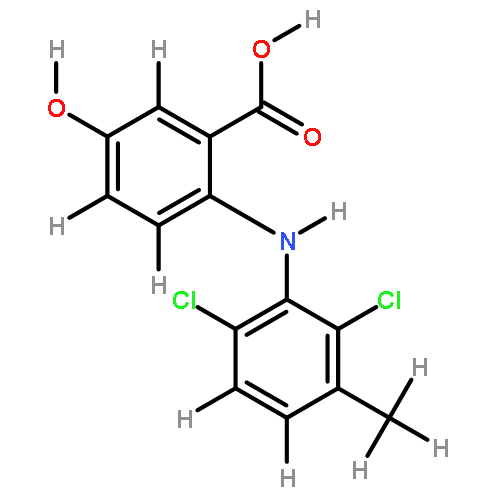
![2-[2,6-dichloro-3-(hydroxymethyl)anilino]benzoic Acid](http://img.cochemist.com/ccimg/67400/67318-61-0.png)
![2-[2,6-dichloro-3-(hydroxymethyl)anilino]benzoic Acid](http://img.cochemist.com/ccimg/67400/67318-61-0_b.png)


![6H-Pyrido[4,3-b]carbazole,5,11-dimethyl-](http://img.cochemist.com/ccimg/600/519-23-3.png)
![6H-Pyrido[4,3-b]carbazole,5,11-dimethyl-](http://img.cochemist.com/ccimg/600/519-23-3_b.png)
![6H-Pyrido[4,3-b]carbazol-9-ol,5,11-dimethyl-](http://img.cochemist.com/ccimg/51200/51131-85-2.png)
![6H-Pyrido[4,3-b]carbazol-9-ol,5,11-dimethyl-](http://img.cochemist.com/ccimg/51200/51131-85-2_b.png)






![2-[3-(hydroxymethyl)-2-methylanilino]benzoic Acid](http://img.cochemist.com/ccimg/5200/5129-20-4.png)
![2-[3-(hydroxymethyl)-2-methylanilino]benzoic Acid](http://img.cochemist.com/ccimg/5200/5129-20-4_b.png)
![[1,1'-Biphenyl]-4-ol,3,4'-dibromo-](http://img.cochemist.com/ccimg/59500/59452-49-2.png)
![[1,1'-Biphenyl]-4-ol,3,4'-dibromo-](http://img.cochemist.com/ccimg/59500/59452-49-2_b.png)
![2,6-bis[4-(dimethylamino)benzylidene]cyclohexanone](http://img.cochemist.com/ccimg/19000/18977-38-3.png)
![2,6-bis[4-(dimethylamino)benzylidene]cyclohexanone](http://img.cochemist.com/ccimg/19000/18977-38-3_b.png)


![(1aS,2S,3R,11cR)-1a,2,3,11c-tetrahydrotetrapheno[1,2-b]oxirene-2,3-diol](http://img.cochemist.com/ccimg/80500/80446-23-7.png)
![(1aS,2S,3R,11cR)-1a,2,3,11c-tetrahydrotetrapheno[1,2-b]oxirene-2,3-diol](http://img.cochemist.com/ccimg/80500/80446-23-7_b.png)














![Phenanthro[9,10-b]oxirene,1a,9b-dihydro-](http://img.cochemist.com/ccimg/600/585-08-0.png)
![Phenanthro[9,10-b]oxirene,1a,9b-dihydro-](http://img.cochemist.com/ccimg/600/585-08-0_b.png)


![2H-1-Benzopyran-2-one, 4-[(dimethylamino)methyl]-7-methoxy-](http://img.cochemist.com/ccimg/250300/250210-93-6.png)
![2H-1-Benzopyran-2-one, 4-[(dimethylamino)methyl]-7-methoxy-](http://img.cochemist.com/ccimg/250300/250210-93-6_b.png)
![2H-1-Benzopyran-2-one, 7-methoxy-4-[(methylamino)methyl]-](http://img.cochemist.com/ccimg/250300/250210-92-5.png)
![2H-1-Benzopyran-2-one, 7-methoxy-4-[(methylamino)methyl]-](http://img.cochemist.com/ccimg/250300/250210-92-5_b.png)



![1a,9b-Dihydrooxireno[2,3-f][1,10]phenanthroline](http://img.cochemist.com/ccimg/65200/65115-91-5.png)
![1a,9b-Dihydrooxireno[2,3-f][1,10]phenanthroline](http://img.cochemist.com/ccimg/65200/65115-91-5_b.png)






![b-D-Glucopyranuronic acid,1-[2-[(2,6-dichlorophenyl)amino]benzeneacetate]](http://img.cochemist.com/ccimg/64200/64118-81-6.png)
![b-D-Glucopyranuronic acid,1-[2-[(2,6-dichlorophenyl)amino]benzeneacetate]](http://img.cochemist.com/ccimg/64200/64118-81-6_b.png)
![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-](/data/chemimg/935900/525-66-6.png)
![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-](/data/chemimg/935900/525-66-6_b.png)
![2H-1-Benzopyran-2-one, 4-[(ethylamino)methyl]-7-methoxy-](http://img.cochemist.com/ccimg/323600/323577-22-6.png)
![2H-1-Benzopyran-2-one, 4-[(ethylamino)methyl]-7-methoxy-](http://img.cochemist.com/ccimg/323600/323577-22-6_b.png)






![2H-1-Benzopyran-2-one, 4-[(butylamino)methyl]-7-methoxy-](http://img.cochemist.com/ccimg/323600/323577-24-8.png)
![2H-1-Benzopyran-2-one, 4-[(butylamino)methyl]-7-methoxy-](http://img.cochemist.com/ccimg/323600/323577-24-8_b.png)
![2H-1-Benzopyran-2-one, 7-methoxy-4-[(propylamino)methyl]-](http://img.cochemist.com/ccimg/323600/323577-23-7.png)
![2H-1-Benzopyran-2-one, 7-methoxy-4-[(propylamino)methyl]-](http://img.cochemist.com/ccimg/323600/323577-23-7_b.png)






![L-Alanine, 3-[(phenylmethyl)seleno]-](http://img.cochemist.com/ccimg/2600/2575-74-8.png)
![L-Alanine, 3-[(phenylmethyl)seleno]-](http://img.cochemist.com/ccimg/2600/2575-74-8_b.png)
![Cyclohexanone,2,6-bis[(4-methylphenyl)methylene]-](http://img.cochemist.com/ccimg/19000/18989-35-0.png)
![Cyclohexanone,2,6-bis[(4-methylphenyl)methylene]-](http://img.cochemist.com/ccimg/19000/18989-35-0_b.png)
![3-Buten-2-one,4-[(1S)-2,6,6-trimethyl-2-cyclohexen-1-yl]-, (3E)-](http://img.cochemist.com/ccimg/14400/14398-36-8.png)
![3-Buten-2-one,4-[(1S)-2,6,6-trimethyl-2-cyclohexen-1-yl]-, (3E)-](http://img.cochemist.com/ccimg/14400/14398-36-8_b.png)



![Glycinamide, N-[[(2-chloroethyl)nitrosoamino]carbonyl]glycyl-N-methyl-](http://img.cochemist.com/ccimg/103500/103419-72-3.png)
![Glycinamide, N-[[(2-chloroethyl)nitrosoamino]carbonyl]glycyl-N-methyl-](http://img.cochemist.com/ccimg/103500/103419-72-3_b.png)




![Benzoic acid,3-[(2-carboxyphenyl)amino]-2-methyl-](http://img.cochemist.com/ccimg/190400/190379-82-9.png)
![Benzoic acid,3-[(2-carboxyphenyl)amino]-2-methyl-](http://img.cochemist.com/ccimg/190400/190379-82-9_b.png)







![b-D-Glucopyranuronic acid,1-[2-[[3-(hydroxymethyl)-2-methylphenyl]amino]benzoate]](http://img.cochemist.com/ccimg/152900/152832-29-6.png)
![b-D-Glucopyranuronic acid,1-[2-[[3-(hydroxymethyl)-2-methylphenyl]amino]benzoate]](http://img.cochemist.com/ccimg/152900/152832-29-6_b.png)








![3-ETHYL-1-(4-FLUOROPHENYL)-1,6-DIHYDRO-7H-PYRAZOLO[3,4-C]PYRIDIN-<WBR />7-ONE](http://img.cochemist.com/ccimg/73800/73774-29-5.png)
![3-ETHYL-1-(4-FLUOROPHENYL)-1,6-DIHYDRO-7H-PYRAZOLO[3,4-C]PYRIDIN-<WBR />7-ONE](http://img.cochemist.com/ccimg/73800/73774-29-5_b.png)


![1,2-Benzenediol, 4-[2-(propylamino)propyl]- (9CI)](http://img.cochemist.com/ccimg/82100/82004-90-8.png)
![1,2-Benzenediol, 4-[2-(propylamino)propyl]- (9CI)](http://img.cochemist.com/ccimg/82100/82004-90-8_b.png)


![N'-(1,8-DIMETHYLIMIDAZO[1,2-A]QUINOXALIN-4-YL)ETHANE-1,2-DIAMINE;HYDROCHLORIDE](http://img.cochemist.com/ccimg/547800/547757-23-3.png)
![N'-(1,8-DIMETHYLIMIDAZO[1,2-A]QUINOXALIN-4-YL)ETHANE-1,2-DIAMINE;HYDROCHLORIDE](http://img.cochemist.com/ccimg/547800/547757-23-3_b.png)








![5H-Dibenzo[b,e][1,4]diazepine,8-chloro-11-(1-piperazinyl)-](http://img.cochemist.com/ccimg/6200/6104-71-8.png)
![5H-Dibenzo[b,e][1,4]diazepine,8-chloro-11-(1-piperazinyl)-](http://img.cochemist.com/ccimg/6200/6104-71-8_b.png)







![2,6-BIS[(3-CHLOROPHENYL)METHYLIDENE]CYCLOHEXAN-1-ONE](http://img.cochemist.com/ccimg/138200/138197-91-8.png)
![2,6-BIS[(3-CHLOROPHENYL)METHYLIDENE]CYCLOHEXAN-1-ONE](http://img.cochemist.com/ccimg/138200/138197-91-8_b.png)
![Cyclohexanone,2,6-bis[(4-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/6300/6275-32-7.png)
![Cyclohexanone,2,6-bis[(4-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/6300/6275-32-7_b.png)






![Phenol,4-[(1Z)-1-[[4-[2-(dimethylamino)ethoxy]phenyl]phenylmethylene]propyl]-](http://img.cochemist.com/ccimg/82500/82413-23-8.png)
![Phenol,4-[(1Z)-1-[[4-[2-(dimethylamino)ethoxy]phenyl]phenylmethylene]propyl]-](http://img.cochemist.com/ccimg/82500/82413-23-8_b.png)


![METHYL (2S)-2-{[{[(2R,3S,5R)-5-{5-[(E)-2-BROMOVINYL]-2,4-DIOXO-3,<WBR />4-DIHYDRO-1(2H)-PYRIMIDINYL}-3-HYDROXYTETRAHYDRO-2-FURANYL]METHOX<WBR />Y}(PHENOXY)PHOSPHORYL]AMINO}PROPANOATE (NON-PREFERRED NAME)](http://img.cochemist.com/ccimg/400/302-33-0.png)
![METHYL (2S)-2-{[{[(2R,3S,5R)-5-{5-[(E)-2-BROMOVINYL]-2,4-DIOXO-3,<WBR />4-DIHYDRO-1(2H)-PYRIMIDINYL}-3-HYDROXYTETRAHYDRO-2-FURANYL]METHOX<WBR />Y}(PHENOXY)PHOSPHORYL]AMINO}PROPANOATE (NON-PREFERRED NAME)](http://img.cochemist.com/ccimg/400/302-33-0_b.png)
![b-D-Glucopyranuronic acid,1-[2-[(3-carboxy-2-methylphenyl)amino]benzoate]](http://img.cochemist.com/ccimg/152900/152832-30-9.png)
![b-D-Glucopyranuronic acid,1-[2-[(3-carboxy-2-methylphenyl)amino]benzoate]](http://img.cochemist.com/ccimg/152900/152832-30-9_b.png)




![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)




![2-Phenylbenzo[d][1,2]selenazol-3(2H)-one](http://img.cochemist.com/ccimg/61000/60940-34-3.png)
![2-Phenylbenzo[d][1,2]selenazol-3(2H)-one](http://img.cochemist.com/ccimg/61000/60940-34-3_b.png)









![2H-1-Benzopyran-2-one,4-hydroxy-3-[(1R)-3-oxo-1-phenylbutyl]-](http://img.cochemist.com/ccimg/5600/5543-58-8.png)
![2H-1-Benzopyran-2-one,4-hydroxy-3-[(1R)-3-oxo-1-phenylbutyl]-](http://img.cochemist.com/ccimg/5600/5543-58-8_b.png)




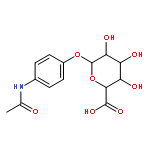
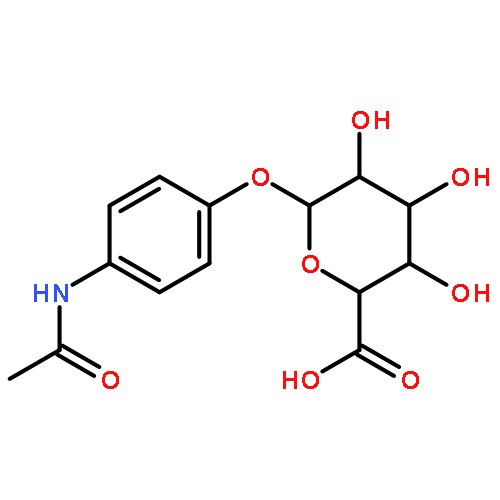






![L-gamma-glutamyl-N-[S-(2-hydroxyethyl)-L-cysteinyl]glycine](http://img.cochemist.com/ccimg/28800/28747-20-8.png)
![L-gamma-glutamyl-N-[S-(2-hydroxyethyl)-L-cysteinyl]glycine](http://img.cochemist.com/ccimg/28800/28747-20-8_b.png)


![Cyclopentanone, 2,5-bis[(4-hydroxyphenyl)methylene]-](http://img.cochemist.com/ccimg/27100/27060-66-8.png)
![Cyclopentanone, 2,5-bis[(4-hydroxyphenyl)methylene]-](http://img.cochemist.com/ccimg/27100/27060-66-8_b.png)
















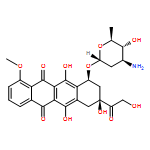
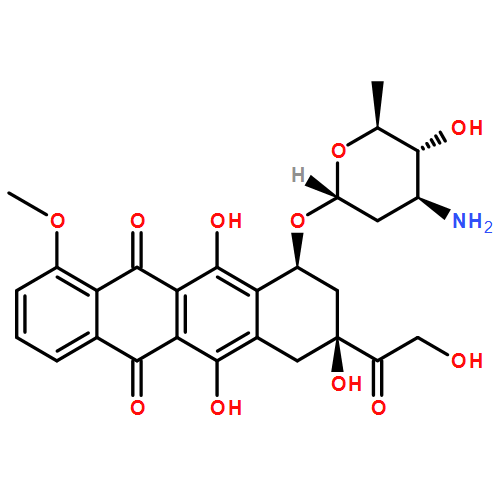
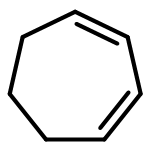
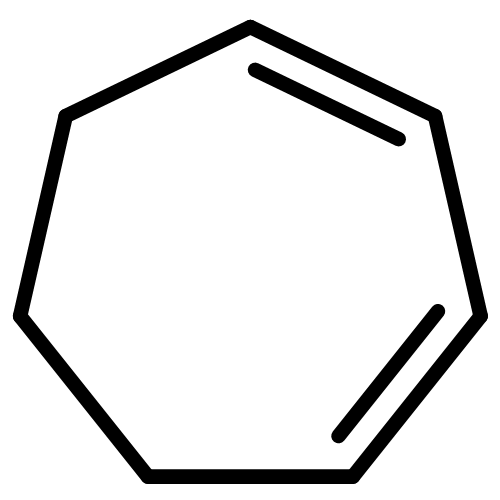


![3-[AMINO-[BIS(2-CHLOROETHYL)AMINO]PHOSPHORYL]OXYPROPANAL](http://img.cochemist.com/ccimg/35200/35144-64-0.png)
![3-[AMINO-[BIS(2-CHLOROETHYL)AMINO]PHOSPHORYL]OXYPROPANAL](http://img.cochemist.com/ccimg/35200/35144-64-0_b.png)


![N-[4-[2-ETHYL-4-(3-METHYLPHENYL)-1,3-THIAZOL-5-YL]PYRIDIN-2-YL]BENZAMIDE](http://img.cochemist.com/ccimg/303200/303162-79-0.png)
![N-[4-[2-ETHYL-4-(3-METHYLPHENYL)-1,3-THIAZOL-5-YL]PYRIDIN-2-YL]BENZAMIDE](http://img.cochemist.com/ccimg/303200/303162-79-0_b.png)
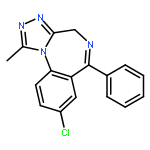

![Thiourea, [2-(4-chlorophenyl)ethyl]-](http://img.cochemist.com/ccimg/207600/207596-23-4.png)
![Thiourea, [2-(4-chlorophenyl)ethyl]-](http://img.cochemist.com/ccimg/207600/207596-23-4_b.png)
![11-Cyclopropyl-5,11-dihydro-4-(hydroxymethyl)-6H-dipyrido[3,2-b:2',3'-e][1,4]diazepin-6-one](http://img.cochemist.com/ccimg/133700/133627-24-4.png)
![11-Cyclopropyl-5,11-dihydro-4-(hydroxymethyl)-6H-dipyrido[3,2-b:2',3'-e][1,4]diazepin-6-one](http://img.cochemist.com/ccimg/133700/133627-24-4_b.png)






















![3-[(carboxymethyl)sulfanyl]-2-oxopropanoic acid](http://img.cochemist.com/ccimg/51800/51783-05-2.png)
![3-[(carboxymethyl)sulfanyl]-2-oxopropanoic acid](http://img.cochemist.com/ccimg/51800/51783-05-2_b.png)
![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-, (2S)-](/data/chemimg/55200/4199-09-1.png)
![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-, (2S)-](/data/chemimg/55200/4199-09-1_b.png)
![(7r,11s)-7,15,17-trihydroxy-11-methyl-12-oxabicyclo[12.4.0]octadeca-1(14),15,17-trien-13-one](http://img.cochemist.com/ccimg/26600/26538-44-3.png)
![(7r,11s)-7,15,17-trihydroxy-11-methyl-12-oxabicyclo[12.4.0]octadeca-1(14),15,17-trien-13-one](http://img.cochemist.com/ccimg/26600/26538-44-3_b.png)
![3-Aminobenzo[e][1,2,4]triazine 1,4-dioxide](http://img.cochemist.com/ccimg/27400/27314-97-2.png)
![3-Aminobenzo[e][1,2,4]triazine 1,4-dioxide](http://img.cochemist.com/ccimg/27400/27314-97-2_b.png)










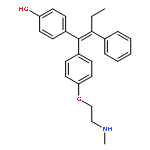
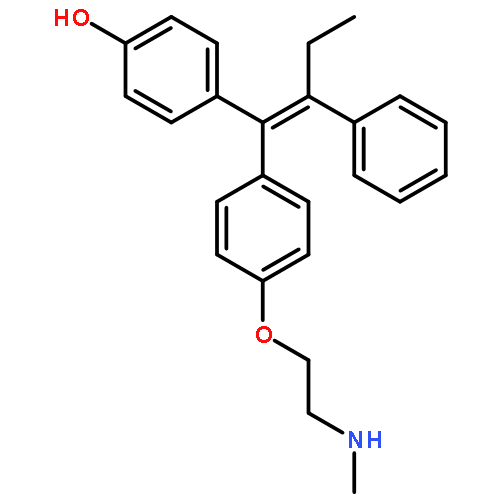


![8-(4-(4-(Pyrimidin-2-yl)piperazin-1-yl)butyl)-8-azaspiro[4.5]decane-7,9-dione](http://img.cochemist.com/ccimg/36600/36505-84-7.png)
![8-(4-(4-(Pyrimidin-2-yl)piperazin-1-yl)butyl)-8-azaspiro[4.5]decane-7,9-dione](http://img.cochemist.com/ccimg/36600/36505-84-7_b.png)
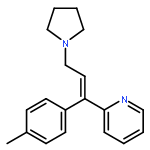



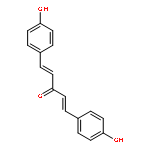


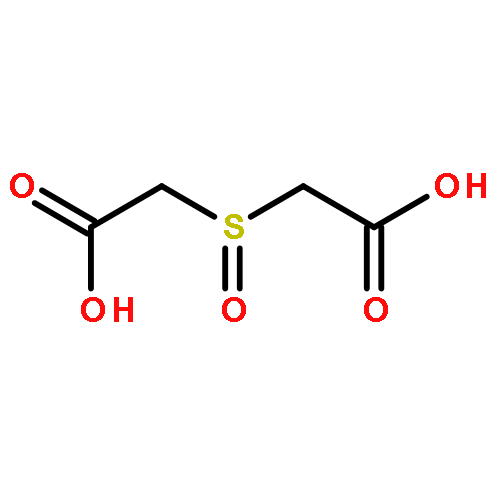

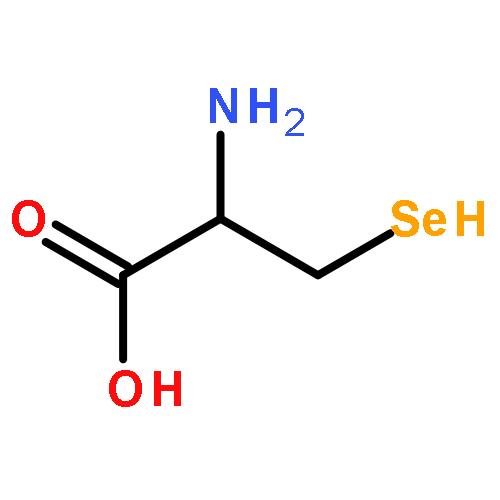









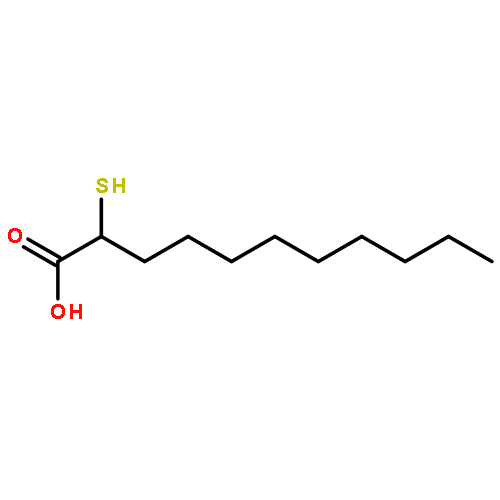






















![5H-Dibenzo[b,e][1,4]diazepine,8-chloro-11-(4-methyl-4-oxido-1-piperazinyl)-](http://img.cochemist.com/ccimg/34300/34233-69-7.png)
![5H-Dibenzo[b,e][1,4]diazepine,8-chloro-11-(4-methyl-4-oxido-1-piperazinyl)-](http://img.cochemist.com/ccimg/34300/34233-69-7_b.png)




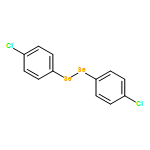
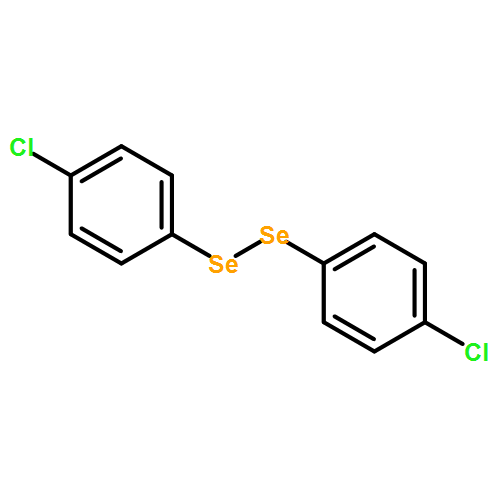
![Oxirane,2,2'-[oxybis(methylene)]bis-](http://img.cochemist.com/ccimg/2300/2238-07-5.png)
![Oxirane,2,2'-[oxybis(methylene)]bis-](http://img.cochemist.com/ccimg/2300/2238-07-5_b.png)
![Propanoic acid, 3-[(2-hydroxyethyl)thio]-2-oxo-](http://img.cochemist.com/ccimg/106000/105930-97-0.png)
![Propanoic acid, 3-[(2-hydroxyethyl)thio]-2-oxo-](http://img.cochemist.com/ccimg/106000/105930-97-0_b.png)


![b-D-Glucopyranuronic acid,1-[2-[(2,3-dimethylphenyl)amino]benzoate]](http://img.cochemist.com/ccimg/102700/102623-18-7.png)
![b-D-Glucopyranuronic acid,1-[2-[(2,3-dimethylphenyl)amino]benzoate]](http://img.cochemist.com/ccimg/102700/102623-18-7_b.png)


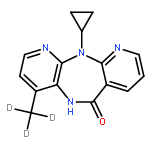
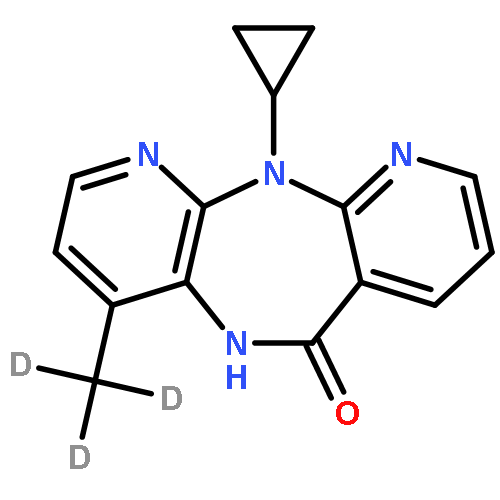
![(2S,8R,9S,10R,13S,14S,17S)-2,17-DIHYDROXY-10,13-DIMETHYL-1,2,6,7,8,9,11,12,14,15,16,17-DODECAHYDROCYCLOPENTA[A]PHENANTHREN-3-ONE](http://img.cochemist.com/ccimg/10400/10390-14-4.png)
![(2S,8R,9S,10R,13S,14S,17S)-2,17-DIHYDROXY-10,13-DIMETHYL-1,2,6,7,8,9,11,12,14,15,16,17-DODECAHYDROCYCLOPENTA[A]PHENANTHREN-3-ONE](http://img.cochemist.com/ccimg/10400/10390-14-4_b.png)




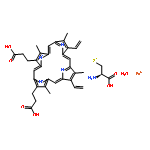
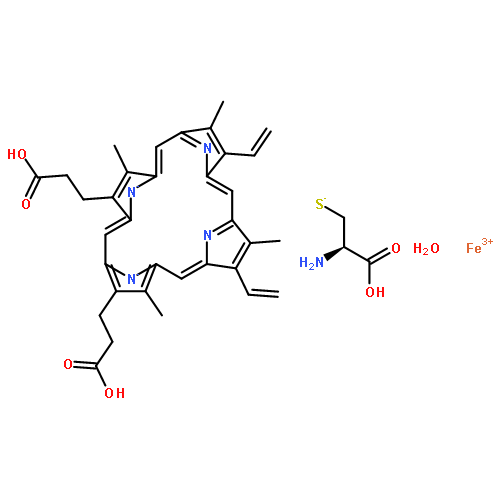


























![3-Buten-2-one,4-[(1R)-2,6,6-trimethyl-2-cyclohexen-1-yl]-, (3E)-](http://img.cochemist.com/ccimg/24200/24190-29-2.png)
![3-Buten-2-one,4-[(1R)-2,6,6-trimethyl-2-cyclohexen-1-yl]-, (3E)-](http://img.cochemist.com/ccimg/24200/24190-29-2_b.png)