Co-reporter:Hirokazu Hara, Dai Kimoto, Miho Kajita, Chisato Takada, Tetsuro Kamiya, Tetsuo Adachi
European Journal of Pharmacology 2017 Volume 795(Volume 795) pp:
Publication Date(Web):15 January 2017
DOI:10.1016/j.ejphar.2016.12.014
Inflammation has been reported to be closely related to exaggeration of cerebral ischemia and neurodegenerative diseases. Microglia, resident immune cells in the central nervous system, can be activated in response to neuronal injury and produce proinflammatory cytokines, resulting in further aggravation of neuronal injury. Interleukin (IL)-23, which consists of p19 and IL-12 p40 subunits, has been shown to be involved in brain injury associated with neuroinflammation. Apomorphine (Apo), a nonselective dopamine receptor agonist, has been used for clinical therapy of Parkinson's disease. Besides the pharmacological effect, Apo is known to have pleiotropic biological functions. In this study, to elucidate the effect of Apo on lipopolysaccharide (LPS)-induced IL-23 p19 mRNA expression in microglial cell line HAPI cells, we pretreated cells with various concentrations of Apo (10 – 30 μM) for 8, 16, and 24 h, followed by exposure to LPS (100 ng/ml). Pretreatment with Apo dose- and time-dependently suppressed the induction of IL-23 p19 mRNA. However, this effect of Apo was exerted independently of dopamine receptors. JNK and ATF4, an endoplasmic reticulum (ER) stress-inducible transcription factor, were involved in expression of LPS-induced IL-23 p19 mRNA. Pretreatment with Apo (30 μM) for 24 h inhibited LPS-induced activation of JNK and the nuclear accumulation of ATF4. Thapsigargin (Tg), an ER stress inducer, stimulated IL-23 p19 mRNA expression via an ATF4 dependent mechanism. We also found that Apo inhibited Tg-induced ATF4 accumulation and IL-23 p19 mRNA expression. Taken together, our findings suggest that Apo exerts anti-inflammatory effects through inhibition of JNK and ATF4 signaling pathways.
Co-reporter:Takuya Doi;Miho Kajita;Tetsuro Kamiya;Tetsuo Adachi
BioMetals 2015 Volume 28( Issue 5) pp:891-902
Publication Date(Web):2015 October
DOI:10.1007/s10534-015-9874-4
Zinc (Zn2+) is considered to be one of the factors aggravating brain damage after cerebral ischemia. Since Zn2+ activates microglia, immune cells in the brain, this metal is proposed to modulate neuroinflammatory responses in the post-ischemic brain. Interleukin (IL)-23 is a heterodimeric cytokine composed of the p19 subunit unique to IL-23 and the p40 subunit common to IL-12. IL-23 has been shown to play a critical role in the progression of ischemic brain injury. However, whether Zn2+ participates in the expression of IL-23 in microglia remains unknown. In this study, we examined the effect of Zn2+ on IL-23 p19 mRNA expression using rat immortalized microglia HAPI cells. Exposure to Zn2+ dose- and time-dependently induced the expression of IL-23 p19 mRNA in HAPI cells. Inhibitors of MAPK and NF-κB pathways failed to suppress this induction. Interestingly, we found that Zn2+ stimulated the phosphorylation of eIF2α and promoted the nuclear accumulation of activating transcription factor 4 (ATF4). Treatment with salubrinal, an eIF2α dephosphorylation inhibitor, enhanced Zn2+-induced ATF4 accumulation and IL-23 p19 mRNA expression. In addition, reporter assay using the IL-23 p19 promoter region revealed that ATF4 directly transactivated IL-23 p19 promoter and that dominant-negative ATF4 suppressed Zn2+-induced activation of IL-23 p19 promoter. Taken together, these findings suggest that Zn2+ up-regulates expression of the IL-23 p19 gene via the eIF2α/ATF4 axis in HAPI cells.
Co-reporter:Hirokazu Hara, Yoko Nakamura, Masayuki Ninomiya, Ryosuke Mochizuki, Tetsuro Kamiya, Elias Aizenman, Mamoru Koketsu, Tetsuo Adachi
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 18) pp:5559-5568
Publication Date(Web):15 September 2011
DOI:10.1016/j.bmc.2011.07.036
Activation of microglia induces the production of various inflammatory mediators including nitric oxide (NO), leading to neurodegeneration in many central nervous system diseases. In this study, we examined the effects of chalcone glycosides isolated from Brassica rapa L. ‘hidabeni’ on lipopolysaccharide (LPS)-induced NO production using rat immortalized microglia HAPI cells. 4′-O-β-d-Glucopyranosyl-3′,4-dimethoxychalcone (A2) inhibited LPS-induced inducible NO synthase (iNOS) expression and NO production. However, A2 did not affect nuclear factor-κB and mitogen-activated protein kinase pathways. The signal transduction and activator of transcription 1 (STAT1), which is activated via production of IFN-β by LPS, is an important transcription factor responsible for LPS-induced iNOS expression. A2 suppressed LPS-induced phosphorylation and nuclear translocation of STAT1, although it had no effects on LPS-induced IFN-β expression. These results indicate that the inhibitory effect of A2 is due to the prevention of STAT signaling. Moreover, structure–activity relationship studies on newly synthesized ‘hidabeni’ chalcone derivatives showed that 4′-O-β-d-glucopyranosyl-3′-methoxychalcone (A11), which has no functional groups in the B-ring, inhibits LPS-induced NO production more potently than A2.
Co-reporter:Hirokazu Hara;Tetsuro Kamiya;Tetsuo Adachi
Neurochemical Research 2009 Volume 34( Issue 8) pp:1498-1506
Publication Date(Web):2009 August
DOI:10.1007/s11064-009-9937-4
Accumulating evidence suggests that zinc (Zn2+) contributes to neuronal death in pathologic states such as ischemia. p53-upregulated modulator of apoptosis (PUMA), which is a BH3-only protein, is known to promote apoptosis through a tumor suppressor p53-dependent and -independent mechanism. In this study, we examined the effect of Zn2+ on the induction of the PUMA gene in human neuroblastoma SH-SY5Y cells. The expression of PUMA was induced by Zn2+ in a dose- and time-dependent manner. A reporter assay revealed that Zn2+ activated the PUMA promoter. In addition, the mutation of the p53 binding site in the PUMA promoter region reduced promoter activation by Zn2+. These findings suggest that p53 participates in Zn2+-induced PUMA expression. Furthermore, we also demonstrated here that Zn2+ stimulates the phosphorylation of ERK and that the MEK-ERK pathway inhibitor, U0126, suppressed Zn2+-induced PUMA expression. Taken together, these results indicate that Zn2+ regulates the induction of PUMA through p53 and ERK pathways.
Co-reporter:Hirokazu Hara;Hideaki Hiramatsu;Tetsuo Adachi
Neurochemical Research 2007 Volume 32( Issue 3) pp:489-495
Publication Date(Web):2007 March
DOI:10.1007/s11064-006-9257-x
Pyrroloquinoline quinone (PQQ), which is an essential nutrient, has been shown to act as an antioxidant. Reactive oxygen species (ROS) are thought to be responsible for neurotoxicity caused by the neurotoxin 6-hydroxydopamine (6-OHDA). In this study, we investigated the ability of PQQ to protect against 6-OHDA-induced neurotoxicity using human neuroblastoma SH-SY5Y. When SH-SY5Y cells were exposed to 6-OHDA in the presence of PQQ, PQQ prevented 6-OHDA-induced cell death and DNA fragmentation. Flow cytometry analysis using the ROS-sensitive fluorescence probe, dihydroethidium, revealed that PQQ reduced elevation of 6-OHDA-induced intracellular ROS. In contrast to PQQ, antioxidant vitamins, ascorbic acid and α-tocopherol, had no protective effect. Moreover, we showed that PQQ effectively scavenged superoxide, compared to the antioxidant vitamins. Therefore, our results suggest the protective effect of PQQ on 6-OHDA-induced neurotoxicity is involved, at least in part, in its function as a scavenger of ROS, especially superoxide.
Co-reporter:Hirokazu Hara, Tetsuro Kamiya, Tetsuo Adachi
Neurochemistry International (January 2011) Volume 58(Issue 1) pp:35-43
Publication Date(Web):1 January 2011
DOI:10.1016/j.neuint.2010.10.006
6-Hydroxydopamine (6-OHDA) is a neurotoxin used to establish experimental models of Parkinson's disease. Exposure to 6-OHDA results in cell death associated with oxidative stress. Pretreatments with sublethal oxidative stress and some pharmacological drugs have been shown to exert preconditioning effects on cytotoxicity caused by 6-OHDA. In this study, we investigated whether endoplasmic reticulum (ER) stress exerts preconditioning effects on 6-OHDA-induced cytotoxicity in human neuroblastoma SH-SY5Y cells. Pretreatment with ER stress inducers, thapsigargin (Tg) and tunicamycin (Tm), promoted GRP78 mRNA induction and ATF4 translation, which are ER stress markers, under our experimental conditions and protected against the cytotoxicity. The protective effect of Tg was more potent than that of Tm. We also found that Tg induced the expression of the antioxidant gene heme oxygenase-1 (HO-1) in a dose-dependent manner, whereas Tm had a weak effect on HO-1 induction. Flow cytometric analysis revealed that reactive oxygen species generated by 6-OHDA were more effectively suppressed in cells pretreated with Tg than with Tm. Therefore, it is likely that Tg enhances antioxidative defenses in SH-SY5Y cells compared with Tm. Because actinomycin D inhibited HO-1 induction by Tg, the induction of HO-1 may be regulated at the transcriptional level. Moreover, the specific eIF2α phosphatase inhibitor salubrinal augmented Tg-induced HO-1 expression. Therefore, the downstream signaling pathway of eIF2α might be involved in Tg-induced HO-1 expression. On the other hand, the reporter assay revealed that Tg stimulated the antioxidant response element (ARE) that is located in regulatory regions of antioxidant genes such as HO-1. Taken together, our data suggest that preconditioning effects induced by Tg mediate an adaptive response to 6-OHDA-induced cytotoxicity via phosphorylation of eIF2α and activation of the ARE.Research highlights▶ Pretreatment with thapsigargin (Tg) protects against 6-OHDA-induced cytotoxicity. ▶ Tg induces the expression of heme oxygenase-1 (HO-1) in a dose-dependent manner. ▶ The specific eIF2α phosphatase inhibitor augments Tg-induced HO-1 expression. ▶ Tg also stimulates the Nrf2-ARE pathway. ▶ Tg-preconditioning may be mediated via the PERK-eIF2α and Nrf2-ARE pathways.
Co-reporter:Hirokazu Hara, Miko Taniguchi, Mari Kobayashi, Tetsuro Kamiya, Tetsuo Adachi
Archives of Biochemistry and Biophysics (15 October 2015) Volume 584() pp:51-60
Publication Date(Web):15 October 2015
DOI:10.1016/j.abb.2015.08.014





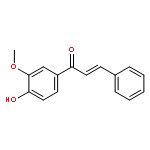
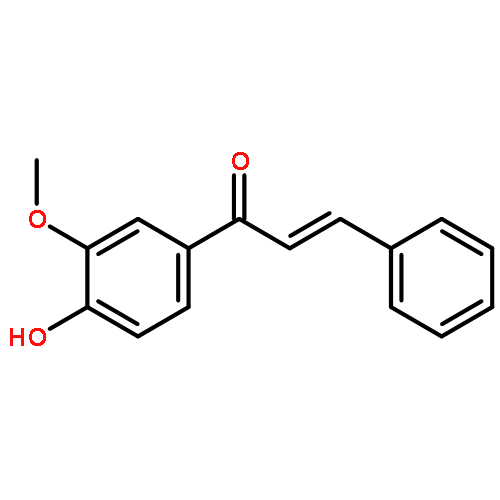
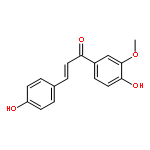
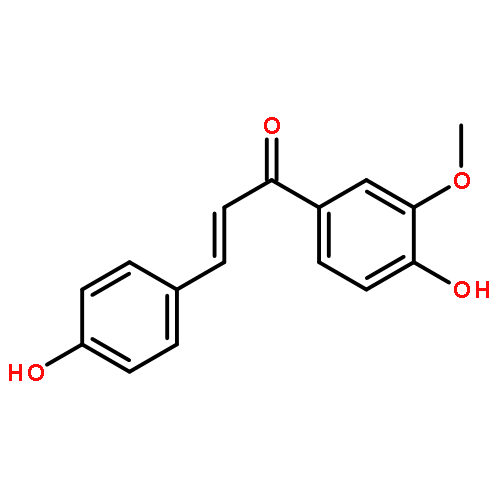









![L-Ornithine,N5-[imino(nitroamino)methyl]-, methyl ester](http://img.cochemist.com/ccimg/51000/50903-99-6.png)
![L-Ornithine,N5-[imino(nitroamino)methyl]-, methyl ester](http://img.cochemist.com/ccimg/51000/50903-99-6_b.png)


![[5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl [hydroxy-[(3,4,5-trihydroxyoxolan-2-yl)methoxy]phosphoryl] Hydrogen Phosphate](http://img.cochemist.com/ccimg/20800/20762-30-5.png)
![[5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl [hydroxy-[(3,4,5-trihydroxyoxolan-2-yl)methoxy]phosphoryl] Hydrogen Phosphate](http://img.cochemist.com/ccimg/20800/20762-30-5_b.png)




![Glycine, N-[(3a,5b,7a)-3,7-dihydroxy-24-oxocholan-24-yl]-](http://img.cochemist.com/ccimg/700/640-79-9.png)
![Glycine, N-[(3a,5b,7a)-3,7-dihydroxy-24-oxocholan-24-yl]-](http://img.cochemist.com/ccimg/700/640-79-9_b.png)









