Co-reporter:Raphael C. Mordi, John C. Walton
Food Chemistry 2016 Volume 197(Part A) pp:836-840
Publication Date(Web):15 April 2016
DOI:10.1016/j.foodchem.2015.11.053
•We examine the oxidation of canthaxanthin.•Canthaxanthin oxidation gave 4-oxo-β-carotenals and 4-oxo-β-carotenones.•We suggest that free radical mechanism is involved in the oxidation of canthaxanthin.Canthaxanthin is a carotenoid that lacks pro-vitamin A activity but is known to have antioxidant activity. The products of its oxidation in oxygen were found to be mainly substituted apo-carotenals and apo-carotenones. The product profile resembles that obtained in the oxidation of β-carotene, except that with canthaxanthin these products are the 4-oxo-β-apo-carotenals and 4-oxo-β-apo-carotenones. Epoxides and diepoxides were clearly identified from β-carotene oxidation but in contrast, with canthaxanthin, apart from 5,6-epoxy-canthaxanthin, which was detected at the early stage of oxidation and minor quantities of 5,6-epoxy-β-ionone and 5,6-epoxy-4-oxo-β-apo-11-carotenal, no other epoxides were detected. The identities of these products lead us to suggest that the mechanism of canthaxanthin oxidation bears significant similarity to that of β-carotene.
Co-reporter: John C. Walton
Angewandte Chemie International Edition 2016 Volume 55( Issue 25) pp:7034-7036
Publication Date(Web):
DOI:10.1002/anie.201602891
Co-reporter: John C. Walton
Angewandte Chemie 2016 Volume 128( Issue 25) pp:7148-7150
Publication Date(Web):
DOI:10.1002/ange.201602891
Co-reporter:John C. Walton
Accounts of Chemical Research 2014 Volume 47(Issue 4) pp:1406
Publication Date(Web):March 21, 2014
DOI:10.1021/ar500017f
Selective syntheses are now available for compounds of many classes, based on C-centered radicals, exploiting a diverse range of mechanisms. The prospect for chemistry based around N- and O-centered radicals is probably more favorable because of the importance of heterocycles as biologically active materials. Heteroradical chemistry is still comparatively underdeveloped due to the need for safe and easy ways of generating them. Oxime esters appeared promising candidates to meet this need because literature reports and our EPR spectroscopic examinations showed they readily dissociated on photolysis with production of a pair of N- and O-centered radicals. It soon became apparent that a whole suite of benign oxime-containing molecules could be pressed into service. The bimodality of the oxime motif meant that by suitable choice of functionality the reactions could be directed to yield selectively products from either the N-centered radicals or from the O-centered radicals.We found that on one hand photolyses of acetophenone oxime esters of carboxylic acids yielded alicyclics. On the other hand, aromatic and heteroaromatic acyl oximes (as well as dioxime oxalates) afforded good yields of phenanthridines and related heterocycles. Easily prepared oxime oxalate amides released carbamoyl radicals, and pleasingly, β-lactams were thereby obtained. Oxime carbonates and oxime carbamates, available via our novel 1,1'-carbonyldiimidazole (CDI)-based preparations, were accessible alternatives for iminyl radicals and hence for phenanthridine preparations. In their second modes, these compounds proved their value as precursors for exotic alkoxycarbonyloxyl and carbamoyloxyl radicals.Microwave-assistance was shown to be a particularly convenient procedure with O-phenyl oxime ethers. The iminyl radicals generated from such precursors with alkene, alkyne, and aromatic acceptor substituents furnished pyrrole, quinoline, phenanthridine, benzonaphthiridine, indolopyridine, and other systems. Microwave irradiations with 2-(aminoaryl)alkanone O-phenyl oximes enabled either dihydroquinazolines or quinazolines to be obtained in very good yields.The fine quality of the EPR spectra, acquired during photolyses of all the O-carbonyl oxime types, marked this as an important complement to existing ways of obtaining such spectra in solution. Quantifications enabled SARs to be obtained for key reaction types of N- and O-centered radicals, thus putting mechanistic chemistry in this area on a much firmer footing. Surprises included the inverse gem-dimethyl effect in 5-exo-cyclizations of iminyls and the interplay of spiro- with ortho-cyclization onto aromatics. Insights into unusual 4-exo-cyclizations of carbamoyl radicals showed the process to be more viable than pent-4-enyl 4-exo-ring closure. Another surprise was the magnitude of the difference in CO2 loss rate from alkoxycarbonyloxyl radicals as compared with acyloxyl radicals. Their rapid 5-exo-cyclization was charted, as was their preferred spiro-cyclization onto aromatics. The first evidence that N-monosubstituted carbamoyloxyls had finite lifetimes was also forthcoming.It is evident that oxime derivatives have excellent credentials as reagents for radical generation and that there is ample room to extend their applications to additional radical types and for further heterocycle syntheses. There is also clear scope for the development of preparative procedures based around the alkoxyl and aminyl radicals that emerge downstream from oxime carbonate and oxime carbamate dissociations.
Co-reporter:David W. Manley and John C. Walton
Organic Letters 2014 Volume 16(Issue 20) pp:5394-5397
Publication Date(Web):October 7, 2014
DOI:10.1021/ol502625w
A titania photoredox catalysis protocol was developed for the homocoupling of C-centered radicals derived from carboxylic acids. Intermolecular reactions were generally efficient and selective, furnishing the desired dimers in good yields under mild neutral conditions. Selective cross-coupling with two acids proved unsuccessful. An intramolecular adaptation enabled macrocycles to be prepared, albeit in modest yields.
Co-reporter:David W. Manley, Roy T. McBurney, Phillip Miller, and John C. Walton, Andrew Mills and Christopher O’Rourke
The Journal of Organic Chemistry 2014 Volume 79(Issue 3) pp:1386-1398
Publication Date(Web):January 17, 2014
DOI:10.1021/jo4027929
Photochemical reactions employing TiO2 and carboxylic acids under dry anaerobic conditions led to several types of C–C bond-forming processes with electron-deficient alkenes. The efficiency of alkylation varied appreciably with substituents in the carboxylic acids. The reactions of aryloxyacetic acids with maleimides resulted in a cascade process in which a pyrrolochromene derivative accompanied the alkylated succinimide. The selectivity for one or other of these products could be tuned to some extent by employing the photoredox catalyst under different conditions. Aryloxyacetic acids adapted for intramolecular ring closures by inclusion of 2-alkenyl, 2-aryl, or 2-oximinyl functionality reacted rather poorly. Profiles of reactant consumption and product formation for these systems were obtained by an in situ NMR monitoring technique. An array of different catalyst forms were tested for efficiency and ease of use. The proposed mechanism, involving hole capture at the TiO2 surface by the carboxylates followed by CO2 loss, was supported by EPR spectroscopic evidence of the intermediates. Deuterium labeling indicated that the titania likely donates protons from surface hydroxyl groups as well as supplying electrons and holes, thus acting as both a catalyst and a reaction partner.
Co-reporter:Roy T. McBurney
Journal of the American Chemical Society 2013 Volume 135(Issue 19) pp:7349-7354
Publication Date(Web):April 21, 2013
DOI:10.1021/ja402833w
A set of oxime carbamates having N-alkyl and N,N-dialkyl substituents were prepared via carbonyldiimidazole intermediates. It was shown by EPR spectroscopy that they underwent clean homolysis of their N–O bonds upon UV photolysis. During photolysis of acetophenone O-allylcarbamoyl oxime, the corresponding oxazolidin-2-onylmethyl radical was detected by EPR spectroscopy, providing the first evidence that N-monosubstituted carbamoyloxyl radicals can hold their structural integrity. N,N-Disubstituted carbamoyloxyl radicals dissociated rapidly at the lowest accessible temperatures. Above room temperature, both types of oxime carbamate acted as selective new precursors for aminyl and iminyl radicals. Rate parameters were measured for 5-exo cyclization of N-benzyl-N-pent-4-enylaminyl radicals; the rate constant was smaller than for C-centered and O-centered analogues. Oxime carbamates derived from the volatile diethylamine afforded aryliminyl radicals that proved convenient for phenanthridine preparations.
Co-reporter:Roy T. McBurney, Annika Eisenschmidt, Alexandra M. Z. Slawin and John C. Walton
Chemical Science 2013 vol. 4(Issue 5) pp:2028-2035
Publication Date(Web):15 Mar 2013
DOI:10.1039/C3SC50500F
Substituted benzyloxycarbonyloxyl radicals were generated by sensitised photolyses of benzyl oxime carbonates. EPR spectroscopy showed they ring closed exclusively by spiro-cyclisation onto the ipso-C-atoms of the aromatic rings. β-Scission of the alkoxycarbonyloxyls to CO2 and benzyloxyl radicals increasingly competed and became dominant above 270 K. The first rate parameters for spiro-cyclisations of O-centred radicals onto aromatics were obtained by the steady-state kinetic EPR method. Pentafluoro-substitution of the ring substantially reduced the spiro-cyclisation rate. Activation barriers of the spiro-cyclisations were computed by DFT to be about half those of the alternative ortho-cyclisations. Consideration of the TS structures suggested the preference for spiro- over ortho-cyclisation resulted from better overlap of the oxyl SOMO with the aromatic π-system during spiro closure.
Co-reporter:David W. Manley ; Roy T. McBurney ; Phillip Miller ; Russell F. Howe ; Shona Rhydderch
Journal of the American Chemical Society 2012 Volume 134(Issue 33) pp:13580-13583
Publication Date(Web):August 7, 2012
DOI:10.1021/ja306168h
Under dry, anaerobic conditions, TiO2 photocatalysis of carboxylic acid precursors resulted in carbon–carbon bond-forming processes. High yields of dimers were obtained from TiO2 treatment of carboxylic acids alone. On inclusion of electron-deficient alkenes, efficient alkylations were achieved with methoxymethyl and phenoxymethyl radicals. In reactions with maleic anhydride or maleimides, phenoxyacetic acid produced chromenedione derivatives in addition to adducts. These photocatalytic reactions are simple and cheap to perform, and the TiO2 is easily removed by filtration. The anaerobic photocatalysis strategy offers a range of synthetic possibilities.
Co-reporter:Giorgio Bencivenni, Riccardo Cesari, Daniele Nanni, Hassane El Mkami and John C. Walton
Organic & Biomolecular Chemistry 2010 vol. 8(Issue 22) pp:5097-5104
Publication Date(Web):24 Aug 2010
DOI:10.1039/C0OB00084A
The reactions of gallium trichloride with phenyl and deuterio-phenyl azides, as well as with 4-methoxyphenyl azide and deuterium isotopomers, were examined by product analysis, CW EPR spectroscopy and pulsed ENDOR spectroscopy. The products included the corresponding anilines together with 4-aminodiphenylamine type dimers, and polyanilines. Complex CW EPR spectra of the radical cations of the dimers [ArNHC6H4NH2]+˙ and trimers [ArNHC6H4NHC6H4NH2]+˙ were obtained. These EPR spectra were analysed with the help of data from the deuterium-substituted analogues as well as the pulse Davies ENDOR spectra. DFT computations of the radical cations provided corroborating evidence and suggested the unpaired electrons were accommodated in extensive π-delocalised orbitals. A mechanism to account for the reductive conversion of aromatic azides to the corresponding anilines and thence to the dimers and trimers is proposed.
Co-reporter:JohnC. Walton Dr.Dr.
Angewandte Chemie 2009 Volume 121( Issue 10) pp:1754-1756
Publication Date(Web):
DOI:10.1002/ange.200805362
Co-reporter:JohnC. Walton Dr.Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 10) pp:1726-1728
Publication Date(Web):
DOI:10.1002/anie.200805362
Co-reporter:Fernando Portela-Cubillo, Jackie S. Scott and John C. Walton
The Journal of Organic Chemistry 2009 Volume 74(Issue 14) pp:4934-4942
Publication Date(Web):May 18, 2009
DOI:10.1021/jo900629g
A wide range of biologically active compounds contain the quinazoline ring system. A new free-radical-based method of making functionalized quinazolines is described, which relies on microwave-promoted reactions of O-phenyl oximes with aldehydes. A small set of 2-aminoaryl alkanone O-phenyl oximes was prepared and shown to produce dihydroquinazolines when mixed with an aldehyde in toluene and subjected to microwave heating. When ZnCl2 was included in the reaction mixture, fully aromatic quinazolines were produced in high yields by a rapid and convenient process. The method worked well with alkyl, aryl, and heterocyclic aldehydes and for a variety of substituents in the benzenic part of the molecule. Similar reactions employing ketones instead of aldehydes were less efficient. Although some dihydroquinazolines did form, they were accompanied by several byproducts. Surprisingly, in each case, one of the byproducts was a quinoline derivative, and a plausible mechanism to account for this rearrangement is proposed.
Co-reporter:Fernando Portela-Cubillo, Rafael Alonso-Ruiz, Diego Sampedro and John C. Walton
The Journal of Physical Chemistry A 2009 Volume 113(Issue 37) pp:10005-10012
Publication Date(Web):August 25, 2009
DOI:10.1021/jp9047902
This paper describes how the rates of 5-exo-ring closures of unsaturated iminyl radicals to pyrrolomethyl radicals respond to substituents in the pentenyl chain and at the C═N bond. Benzyl- and acyl oxime esters, as well as dioxime oxalates, were identified as suitable iminyl radical sources for electron paramagnetic resonance (EPR) spectroscopy. Pentenyliminyl radicals with aryl substituents at their C═N bonds, and one with an alkyl substituent at its C═N bond, were studied in solution by steady-state continuous wave EPR spectroscopy. All the pentenyliminyls selectively ring closed in the 5-exo-mode rather than the 6-endo-mode. EPR monitoring of the decay of the 2,2-dimethyl-1-phenylpent-4-enyliminyl radical showed that it underwent bimolecular combination at about the diffusion controlled limit (2kt ∼ 3 × 108 M−1 s−1 at 245 K). The rate constant for 5-exo-ring closure of phenylpentenyliminyl (8.8 × 103 s−1 at 300 K) was a factor of 25 smaller than the rate constant for hex-5-enyl radical cyclization. The rate of cyclization was slower for an iminyl having a Me group at the site of 5-exo-cyclization but faster for an iminyl with an Et substituent at the terminus of the C═C double bond. Surprisingly, the 2,2-dimethyl-1-phenylpent-4-enyliminyl radical, with a bismethyl group in its pentenyl chain, ring closed more slowly than the unsubstituted analogue. DFT computations were in accord with this inverse gem-dimethyl effect and suggested it resulted from steric interaction of the Ph and bis-Me groups which forced the aromatic ring out of the plane of the imine moiety. To check on the role of the Ph substituent, pentenyliminyls lacking this group were sought. A pentenyliminyl radical with an alkyl group in place of the Ph group, and a single Me group in its pentenyl chain, was generated by means of an unsymmetrical dioxime oxalate precursor. The kc for this species was a factor of 2.5 larger than kc for the original pentenyliminyl, suggesting that the normal positive gem-dimethyl effect does operate for pentenyliminyls lacking the aromatic substituent at the C═N bond. DFT computations also successfully reproduced this trend for model iminyls. It appears that for alkenyliminyl radicals positive or negative gem-dimethyl effects on the cyclization can be induced by appropriate choice of the second substituent on the C═N bond.
Co-reporter:Fernando Portela-Cubillo, Eoin M. Scanlan, Jackie S. Scott and John C. Walton
Chemical Communications 2008 (Issue 35) pp:4189-4191
Publication Date(Web):01 Aug 2008
DOI:10.1039/B808625G
Dioxime oxalates are useful precursors for the clean generation of iminyl radicals by sensitised UV photolysis and can be adapted for serviceable preparations of 3,4-dihydro-2H-pyrroles and phenanthridines.
Co-reporter:Fernando Portela-Cubillo, Jackie S. Scott and John C. Walton
Chemical Communications 2008 (Issue 25) pp:2935-2937
Publication Date(Web):17 Apr 2008
DOI:10.1039/B803630F
Microwave irradiations of 2-(aminoaryl)alkanone O-phenyl oximes and carbonyl compounds generate iminyl radicals in company with imines; iminyl on imine ring closure yields dihydroquinazolines or quinazolines when ZnCl2 is included in the mixture.
Co-reporter:Fernando Portela-Cubillo, Jackie S. Scott and John C. Walton
Chemical Communications 2007 (Issue 39) pp:4041-4043
Publication Date(Web):11 Sep 2007
DOI:10.1039/B712582H
Microwave irradiation of alkenone O-phenyl oximes produces iminyl radicals that ring close to yield dihydropyrrole derivatives; pyrroles and pyridines can be obtained from related precursors.
Co-reporter:Eoin M. Scanlan;John C. Walton
Helvetica Chimica Acta 2006 Volume 89(Issue 10) pp:2133-2143
Publication Date(Web):25 OCT 2006
DOI:10.1002/hlca.200690202
The 4-exo cyclizations of two types of carbamoyl radicals onto O-alkyloxime acceptor groups were studied as potential routes to 3-amino-substituted azetidinones and hence to penicillins. A general synthetic route to ‘benzaldehyde oxime oxalate amides’ (= 2-[(benzylideneamino)oxy]-2-oxoacetamides; see, e.g., 10c) of 2-{[(benzyloxy)imino]methyl}-substituted thiazolidine-4-carboxylic acid methyl esters 9 was developed (Scheme 3). It was shown by EPR spectroscopy that these compounds underwent sensitized photodissociation to the corresponding carbamoyl radicals but that these did not ring close. An analogous open-chain precursor, benzaldehyde O-(benzylaminoacetaldehyde-O-benzyloxalyl)oxime, 15, lacking the 5-membered thiazolidine ring, was shown by EPR spectroscopy to release the corresponding carbamoyl radical (Scheme 4). The latter underwent 4-exo cyclization onto its CNOBn bond in non-H-atom donor solvents. The rate constant for this cyclization was determined by the steady-state EPR method. Spectroscopic evidence indicated that the reverse ring-opening process was slower than cyclization.
Co-reporter:Mark D. Roydhouse and John C. Walton
Chemical Communications 2005 (Issue 35) pp:4453-4455
Publication Date(Web):04 Aug 2005
DOI:10.1039/B506391D
ω-(2-Halophenyl)alkyl-2-oxazolines were prepared and reacted via base promoted intramolecular coupling of radical with carbanionic centres to yield 1-phenyl-1-oxazolino-indan and -tetralin derivatives containing quaternary C-atoms.
Co-reporter:Patricia L. Minin and John C. Walton
Organic & Biomolecular Chemistry 2004 vol. 2(Issue 17) pp:2471-2475
Publication Date(Web):04 Aug 2004
DOI:10.1039/B405735J
The idea that ring closures of C-centred radicals onto isocyanates could be made permanent by designing the cyclised radical to undergo a rapid onward β-scission, was investigated for the 2-(2-isocyanato)cyclopropylphenyl and 2-(2-isocyanato)oxiranylphenyl radicals. The radical precursors, trans- and cis-1-bromo-(2-isocyanatocyclopropyl)benzene and (2-bromophenyl)-3-isocyanatooxirane, were prepared from the corresponding bromophenylcyclopropane and bromophenyloxirane carboxylic acids via Curtius rearrangements of the derived azides. The structure of the trans-2-(2-isocyanato)cyclopropylphenyl radical prevents cyclization, however, it was shown that isomerisation to the analogous cis-radical occurred, probably by scission of the disubstituted cyclopropane bond followed by internal rotation of the resulting resonance stabilised diradical. It was found, however, that the main product from homolytic reactions of both trans- and cis-isocyanatocyclopropyl compounds, with tributyltin hydride and tris(trimethylsilyl)silane, was the direct reduction product, trans-(2-isocyanatocyclopropyl)benzene. Only traces of cyclised products, that were probably 4,5-dihydrobenzo[c]azepin-1-one from the cyclopropane precursor and 5H-6-oxa-8-azabenzocyclohepten-9-one from the oxirane precursor, were detected. We conclude, therefore, that the rate of cyclization onto isocyanate acceptor groups must be slower in these systems than hex-5-enyl cyclization or that the reverse ring-opening process must be faster than for analogous radicals.
Co-reporter:Eoin M. Scanlan, Alexandra M. Z. Slawin and John C. Walton
Organic & Biomolecular Chemistry 2004 vol. 2(Issue 5) pp:716-724
Publication Date(Web):03 Feb 2004
DOI:10.1039/B315223E
A general synthetic route to oxime oxalate amides was developed and applied to the preparation of molecules incorporating N-benzyl-N-alkenyl amides linked with acetone oxime or benzaldoxime units. In addition, 2-substituted-thiazolidine-4-carboxylic acid methyl ester amides of oxalyl benzaldoxime were also prepared. It was shown by EPR spectroscopy that the oxalyl benzaldoxime amides dissociated to produce benziminyl and carbamoyl (aminoacyl) radicals when photolysed with 4-methoxyacetophenone as a photosensitizer. Carbamoyl radicals derived from N-alk-3-enyl oxime oxalate amides underwent ring closure to afford pyrrolidin-2-ones. The analogous N-alk-2-enyl precursors afforded azetidin-2-ones. Reactions of the cyclohexenyl and cinnamyl oxime oxalate amides afforded a bicyclic β-lactam and a 3-benzyl-substituted β-lactam respectively. Interestingly, both products were isolated as hydroxylated compounds. A thiazolidine-derived oxime oxalate amide containing an isobutenyl side chain also dissociated with production of the corresponding thiazolidinyl-carbamoyl radical, as shown by EPR spectroscopy. GC-MS evidence indicated that this radical cyclised to afford some of the corresponding penicillin derivative
Co-reporter:A. Franco Bella, Leon V. Jackson and John C. Walton
Organic & Biomolecular Chemistry 2004 vol. 2(Issue 3) pp:421-428
Publication Date(Web):14 Jan 2004
DOI:10.1039/B313932H
1-Carbamoyl-1-methylcyclohexa-2,5-dienes produced the corresponding delocalised 1-carbamoyl-1-methylcyclohexa-2,5-dienyl radicals on treatment with radical initiators. At temperatures above ca. 300 K dissociation to produce toluene and aminoacyl (carbamoyl) radicals took place. The alternative dissociation of the 1-carbamoyl-1-methylcyclohexa-2,5-dienyl radicals to release methyl radicals and an aromatic amide did not compete. Aminoacyl radicals with allyl, butenyl or similar side chains underwent cyclisations. Moderate yields of N-benzyl-azetidin-2-ones and N-benzyl-pyrrolidin-2-ones were isolated for a range of substituents. The main by-products were N-benzyl-N-alkenylformamides. Ring closure did not take place to a significant extent for precursors with alk-2-ynyl or 2-cyanoalkyl side chains. An improved yield of 1,3-dibenzylazetidin-2-one was obtained by use of lauroyl peroxide as initiator and by inclusion of methyl thioglycolate as polarity reversal catalyst.
Co-reporter:Jessie A. Blake, Keith U. Ingold, Shuqiong Lin, Peter Mulder, Derek A. Pratt, Brad Sheeller and John C. Walton
Organic & Biomolecular Chemistry 2004 vol. 2(Issue 3) pp:415-420
Publication Date(Web):12 Jan 2004
DOI:10.1039/B313491A
Thermolyses of seven dialkyl, two alkyl-aryl and two diaryl O-benzyl ketoxime ethers, R1R2CNOCH2Ph, have been examined in three hydrogen donor solvents: tetralin, 9,10-dihydrophenanthrene, and 9,10-dihydroanthracene. All the oxime ethers gave the products expected from homolytic scission of both the O–C bond (viz., R1R2CNOH and PhCH3) and N–O bond (viz., R1R2CNH and PhCH2OH). The yields of these products depended on which solvent was used and the rates of decomposition of the O-benzyl oxime ethers were greater in 9,10-dihydrophenanthrene and 9,10-dihydroanthracene than in tetralin. These results indicated that a reverse radical disproportionation reaction in which a hydrogen atom was transferred from the solvent to the oxime ether, followed by β-scission of the resultant aminoalkyl radical, must be important in the latter two solvents. Benzaldehyde was found to be an additional product from thermolyses conducted in tetralin. This, and other evidence, indicated that another induced decomposition mode involving abstraction of a benzylic hydrogen atom, followed by β-scission of the resulting benzyl radical, became important for some substrates. Participation by minor amounts of enamine tautomers of the oxime ethers was shown to be negligible by comparison of thermolysis data for the O-benzyloxime of bicyclo[3.3.1]nonan-9-one, which cannot give an enamine tautomer, with that of the O-benzyloxime of cyclohexanone.
Co-reporter:Eoin M. Scanlan and John C. Walton
Chemical Communications 2002 (Issue 18) pp:2086-2087
Publication Date(Web):16 Aug 2002
DOI:10.1039/B205962B
Photosensitised decomposition of oxime oxalate amides is a useful new route to carbamoyl radicals that may cyclise to afford β- or γ-lactams.
Co-reporter:A. Franco Bella, Leon V. Jackson and John C. Walton
Organic & Biomolecular Chemistry 2002 (Issue 11) pp:1839-1843
Publication Date(Web):08 Oct 2002
DOI:10.1039/B206768D
Hydrogen atom abstraction from 1-carbamoyl-1-methylcyclohexa-2,5-dienes generated the corresponding delocalised 1-carbamoyl-1-methylcyclohexa-2,5-dienyl radicals at temperatures below ca. 300 K. At higher temperatures suitably substituted examples dissociated to produce toluene and aminoacyl (carbamoyl) radicals. Both types of intermediate were detected and characterised in solution by EPR spectroscopy. From measurements of the concentrations of the initial and released radicals, rate constants and Arrhenius parameters for dissociation of 1-carbamoyl-1-methylcyclohexa-2,5-dienyl radicals were determined. It was found that dissociation to give a secondary aminoacyl radical [˙C(O)N(R)CH2Ph] took place with a rate constant in the range 50 to 90 s−1 at 300 K. The alternative dissociation of the 1-carbamoyl-1-methylcyclohexa-2,5-dienyl radicals to release methyl radicals and an aromatic amide was much slower and did not compete. Analogous dissociations giving primary aminoacyl radicals [˙C(O)NHR] were significantly slower. Aminoacyl radicals with allyl, butenyl or similar side chains underwent cyclisation and, in the case of the 1,2,2-trimethylbut-3-enyl derivative, cyclisation was faster than dissociation of the parent cyclohexadienyl radical throughout the accessible temperature range. Semi-empirical AM1 and ab initio DFT computations indicated the decarbonylations of the aminoacyl radicals did not compete with cyclisations.
Co-reporter:Leon V. Jackson and John C. Walton
Chemical Communications 2000 (Issue 23) pp:2327-2328
Publication Date(Web):09 Nov 2000
DOI:10.1039/B007454N
Radical induced homolyses of
1-carbamoyl-1-methylcyclohexa-2,5-dienes took place cleanly to yield
aminoacyl radicals, with no competition from the alternative dissociation
to methyl radicals: β- and γ-lactams were obtained from ring
closures of suitably unsaturated model compounds.
Co-reporter:Andrew J. McCarroll and John C. Walton
Chemical Communications 2000 (Issue 5) pp:351-352
Publication Date(Web):21 Feb 2000
DOI:10.1039/A910346P
Arylmethaniminyl and alkyl radicals were generated from di-
and tri-methoxyphenyl aldoxime esters, by photolysis in the presence of
4-methoxyacetophenone, and were detected by EPR spectroscopy: good yields
of cyclised products were isolated from suitably unsaturated alkyl
substituents.
Co-reporter:Andrew J. McCarroll and John C. Walton
Organic & Biomolecular Chemistry 2000 (Issue 12) pp:2399-2409
Publication Date(Web):2000/11/15
DOI:10.1039/B007212P
Photolyses of aldoxime esters, containing a considerable range of alkyl groups, lead to cleavage of their N–O bonds and formation of aryliminyl and alkyl radicals. The process was found to be favoured by 4-methoxyacetophenone as a photosensitiser and by methoxy substituents in the aryl rings. 4-Nitro- and pentafluoro-substitutions of the aryl rings were, on the other hand, deleterious. The intermediate iminyl radicals, together with primary, secondary and tertiary alkyl radicals were characterised by 9 GHz EPR spectroscopy. Cyclopropyl, CF3, and CCl3 radicals were probably also formed, but were too reactive for direct EPR spectroscopic detection. Photosensitised reaction of benzophenone oxime O-nonanoyl ester produced the diphenylmethaniminoxyl, as well as the expected n-octyl and iminyl radicals. This indicated that O–C bond scission accompanied O–N scission for this ketoxime ester.
At higher temperatures the C-centred radicals added to the starting oxime esters to produce alkoxyaminyl radicals that were also spectroscopically detected in some cases. No evidence for abstraction of the iminyl hydrogen by tert-butoxyl radicals was obtained. Instead, the t-BuO˙ radicals added to the CN double bonds of the oxime esters. Similarly, chlorine abstraction from alkylbenzohydroximoyl chlorides by trimethyltin radicals did not take place. Preparative scale experiments with oxime esters containing suitably unsaturated alkyl groups showed that good yields of cyclised products could be obtained in the presence of the photosensitiser. This process constitutes a general method by which carboxylic acids or acid chlorides can be converted into alkyl radicals and hence to cyclised derivatives.
p
Co-reporter:Paul A. Baguley
Angewandte Chemie International Edition 1998 Volume 37(Issue 22) pp:
Publication Date(Web):17 DEC 1998
DOI:10.1002/(SICI)1521-3773(19981204)37:22<3072::AID-ANIE3072>3.0.CO;2-9
An urgent search is being made for generic reagents that promote free radical synthetic transformations as means of banishing the toxic threat of organotin hydrides. Although second-generation tin reagents are beguiling, organosilanes and a range of thiocarbonyl compounds are more intrinsically benign. Metal-free radical chain sequences based around cyclohexadiene derivatives are being developed (see reaction scheme), and tetrathiafulvalenes mimic metals and allow a crossover from homolytic to ionic chemistry. Z=alkene.
Co-reporter:Paul A. Baguley;John C. Walton
Angewandte Chemie 1998 Volume 110(Issue 22) pp:
Publication Date(Web):12 MAR 1999
DOI:10.1002/(SICI)1521-3757(19981116)110:22<3272::AID-ANGE3272>3.0.CO;2-M
Dringend gesucht werden neue Reagentien, die Reaktionen freier Radikale vermitteln, um den Problemen ein Ende zu setzen, die mit Organozinnhydriden einhergehen (vor allem deren hohe Toxizität). Zwar wurden schon Fortschritte bei zinnorganischen Reagentien erzielt, z. B. hinsichtlich der Abtrennbarkeit von den Produkten, doch genießen Organosiliciumverbindungen und etliche Thiocarbonylverbindungen ein weitaus besseres Ansehen. Radikalische Reaktionssequenzen ausgehend von Cyclohexadienderivaten als „proaromatischen” Verbindungen wurden entwickelt (siehe Schema), und mit Tetrathiafulvalenen lassen sich Umwandlungen im Grenzgebiet zwischen radikalischen und ionischen Reaktionen durchführen. Z=Alken
Co-reporter:Fernando Portela-Cubillo, Jackie S. Scott and John C. Walton
Chemical Communications 2007(Issue 39) pp:NaN4043-4043
Publication Date(Web):2007/09/11
DOI:10.1039/B712582H
Microwave irradiation of alkenone O-phenyl oximes produces iminyl radicals that ring close to yield dihydropyrrole derivatives; pyrroles and pyridines can be obtained from related precursors.
Co-reporter:Fernando Portela-Cubillo, Jackie S. Scott and John C. Walton
Chemical Communications 2008(Issue 25) pp:NaN2937-2937
Publication Date(Web):2008/04/17
DOI:10.1039/B803630F
Microwave irradiations of 2-(aminoaryl)alkanone O-phenyl oximes and carbonyl compounds generate iminyl radicals in company with imines; iminyl on imine ring closure yields dihydroquinazolines or quinazolines when ZnCl2 is included in the mixture.
Co-reporter:Roy T. McBurney, Annika Eisenschmidt, Alexandra M. Z. Slawin and John C. Walton
Chemical Science (2010-Present) 2013 - vol. 4(Issue 5) pp:NaN2035-2035
Publication Date(Web):2013/03/15
DOI:10.1039/C3SC50500F
Substituted benzyloxycarbonyloxyl radicals were generated by sensitised photolyses of benzyl oxime carbonates. EPR spectroscopy showed they ring closed exclusively by spiro-cyclisation onto the ipso-C-atoms of the aromatic rings. β-Scission of the alkoxycarbonyloxyls to CO2 and benzyloxyl radicals increasingly competed and became dominant above 270 K. The first rate parameters for spiro-cyclisations of O-centred radicals onto aromatics were obtained by the steady-state kinetic EPR method. Pentafluoro-substitution of the ring substantially reduced the spiro-cyclisation rate. Activation barriers of the spiro-cyclisations were computed by DFT to be about half those of the alternative ortho-cyclisations. Consideration of the TS structures suggested the preference for spiro- over ortho-cyclisation resulted from better overlap of the oxyl SOMO with the aromatic π-system during spiro closure.
Co-reporter:Giorgio Bencivenni, Riccardo Cesari, Daniele Nanni, Hassane El Mkami and John C. Walton
Organic & Biomolecular Chemistry 2010 - vol. 8(Issue 22) pp:NaN5104-5104
Publication Date(Web):2010/08/24
DOI:10.1039/C0OB00084A
The reactions of gallium trichloride with phenyl and deuterio-phenyl azides, as well as with 4-methoxyphenyl azide and deuterium isotopomers, were examined by product analysis, CW EPR spectroscopy and pulsed ENDOR spectroscopy. The products included the corresponding anilines together with 4-aminodiphenylamine type dimers, and polyanilines. Complex CW EPR spectra of the radical cations of the dimers [ArNHC6H4NH2]+˙ and trimers [ArNHC6H4NHC6H4NH2]+˙ were obtained. These EPR spectra were analysed with the help of data from the deuterium-substituted analogues as well as the pulse Davies ENDOR spectra. DFT computations of the radical cations provided corroborating evidence and suggested the unpaired electrons were accommodated in extensive π-delocalised orbitals. A mechanism to account for the reductive conversion of aromatic azides to the corresponding anilines and thence to the dimers and trimers is proposed.
Co-reporter:Fernando Portela-Cubillo, Eoin M. Scanlan, Jackie S. Scott and John C. Walton
Chemical Communications 2008(Issue 35) pp:
Publication Date(Web):
DOI:10.1039/B808625G

![[1]Benzopyrano[3,4-c]pyrrole-1,3(2H,4H)-dione, 3a,9b-dihydro-2-phenyl-, (3aR,9bS)-rel-](/data/chemimg/3766200/1393661-29-4.png)
![[1]Benzopyrano[3,4-c]pyrrole-1,3(2H,4H)-dione, 3a,9b-dihydro-2-phenyl-, (3aR,9bS)-rel-](/data/chemimg/3766200/1393661-29-4_b.png)
![[1]Benzopyrano[3,4-c]pyrrole-1,3(2H,4H)-dione, 3a,9b-dihydro-2-methyl-, (3aR,9bS)-rel-](/data/chemimg/3766200/1393661-27-2.png)
![[1]Benzopyrano[3,4-c]pyrrole-1,3(2H,4H)-dione, 3a,9b-dihydro-2-methyl-, (3aR,9bS)-rel-](/data/chemimg/3766200/1393661-27-2_b.png)
![[1]Benzopyrano[3,4-c]pyrrole-1,3(2H,4H)-dione, 3a,9b-dihydro-, (3aR,9bS)-rel-](/data/chemimg/3766200/1393661-26-1.png)
![[1]Benzopyrano[3,4-c]pyrrole-1,3(2H,4H)-dione, 3a,9b-dihydro-, (3aR,9bS)-rel-](/data/chemimg/3766200/1393661-26-1_b.png)
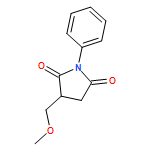
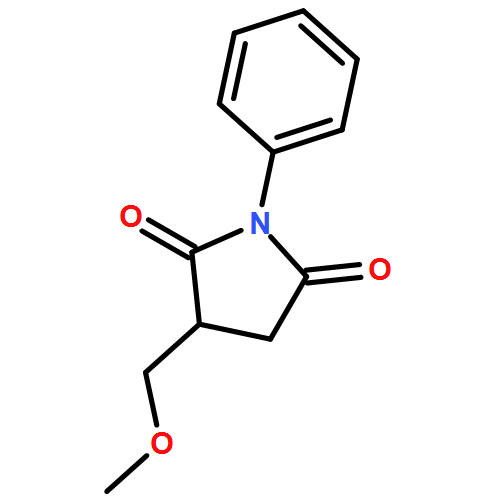

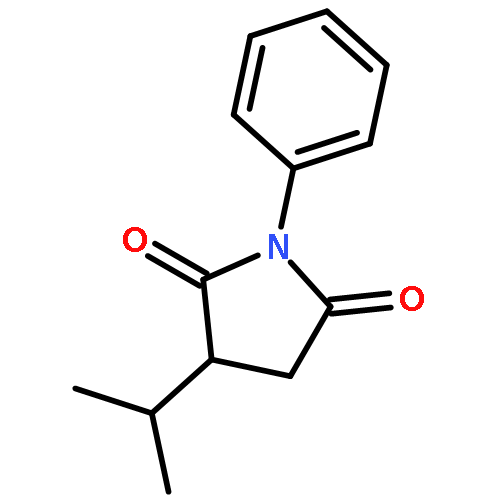

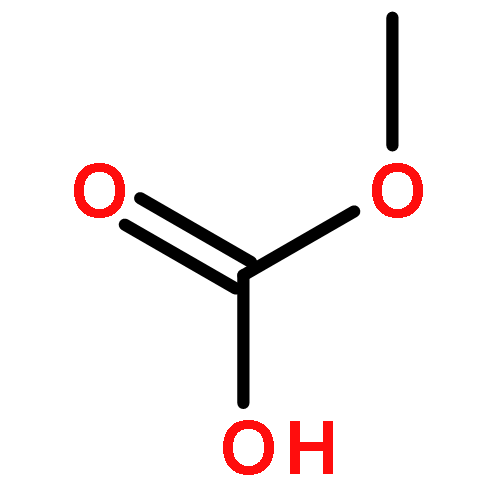


![1,2-Ethanediol, 1,2-bis[4-(trifluoromethyl)phenyl]-, (1R,2S)-rel-](http://img.cochemist.com/ccimg/173100/173035-63-7.png)
![1,2-Ethanediol, 1,2-bis[4-(trifluoromethyl)phenyl]-, (1R,2S)-rel-](http://img.cochemist.com/ccimg/173100/173035-63-7_b.png)
![1,2-Ethanediol, 1,2-bis[4-(trifluoromethyl)phenyl]-, (1R,2R)-rel-](/data/chemimg/3746500/173035-62-6.png)
![1,2-Ethanediol, 1,2-bis[4-(trifluoromethyl)phenyl]-, (1R,2R)-rel-](/data/chemimg/3746500/173035-62-6_b.png)


![4'-(Trifluoromethyl)-[1,1'-biphenyl]-2-ol](http://img.cochemist.com/ccimg/122900/122801-61-0.png)
![4'-(Trifluoromethyl)-[1,1'-biphenyl]-2-ol](http://img.cochemist.com/ccimg/122900/122801-61-0_b.png)

















![Benzene, 1,1'-[(1R,2R)-1,2-difluoro-1,2-ethanediyl]bis-, rel-](http://img.cochemist.com/ccimg/52800/52795-54-7.png)
![Benzene, 1,1'-[(1R,2R)-1,2-difluoro-1,2-ethanediyl]bis-, rel-](http://img.cochemist.com/ccimg/52800/52795-54-7_b.png)




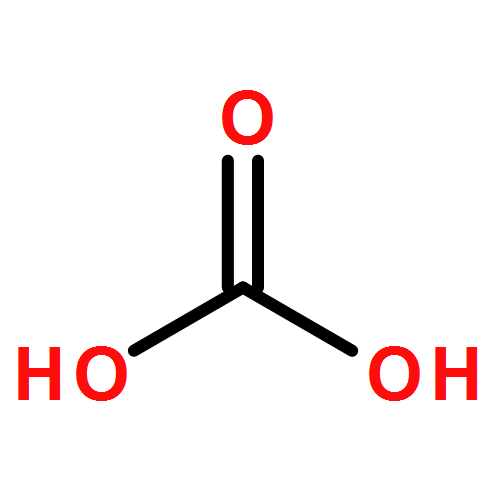
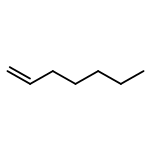
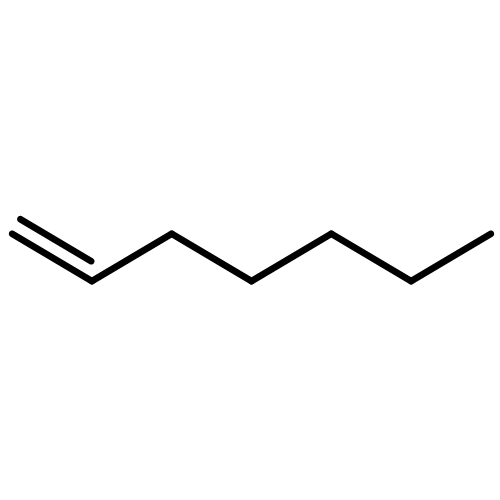
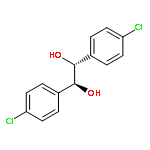



















![Acetic acid,2-[(4-methoxyphenyl)thio]-](http://img.cochemist.com/ccimg/3500/3406-77-7.png)
![Acetic acid,2-[(4-methoxyphenyl)thio]-](http://img.cochemist.com/ccimg/3500/3406-77-7_b.png)






![6H-Dibenzo[b,d]-pyran](http://img.cochemist.com/ccimg/300/229-95-8.png)
![6H-Dibenzo[b,d]-pyran](http://img.cochemist.com/ccimg/300/229-95-8_b.png)



