Co-reporter:Jin-Yi Zhu, Rebecca A. Cuellar, Norbert Berndt, Hee Eun Lee, Sanne H. Olesen, Mathew P. Martin, Jeffrey T. Jensen, Gunda I. Georg, and Ernst Schönbrunn
Journal of Medicinal Chemistry September 28, 2017 Volume 60(Issue 18) pp:7863-7863
Publication Date(Web):August 9, 2017
DOI:10.1021/acs.jmedchem.7b00996
Members of the Wee family of kinases negatively regulate the cell cycle via phosphorylation of CDK1 and are considered potential drug targets. Herein, we investigated the structure–function relationship of human Wee1, Wee2, and Myt1 (PKMYT1). Purified recombinant full-length proteins and kinase domain constructs differed substantially in phosphorylation states and catalytic competency, suggesting complex mechanisms of activation. A series of crystal structures reveal unique features that distinguish Wee1 and Wee2 from Myt1 and establish the structural basis of differential inhibition by the widely used Wee1 inhibitor MK-1775. Kinome profiling and cellular studies demonstrate that, in addition to Wee1 and Wee2, MK-1775 is an equally potent inhibitor of the polo-like kinase PLK1. Several previously unrecognized inhibitors of Wee kinases were discovered and characterized. Combined, the data provide a comprehensive view on the catalytic and structural properties of Wee kinases and a framework for the rational design of novel inhibitors thereof.
Co-reporter:Dr. David S. Huang; Dr. Henry L. Wong; Dr. Gunda I. Georg
ChemMedChem 2017 Volume 12(Issue 7) pp:520-528
Publication Date(Web):2017/04/06
DOI:10.1002/cmdc.201700084
AbstractPironetin is a natural product with potent antiproliferative activity that forms a covalent adduct with α-tubulin via conjugate addition into the natural product's α,β-unsaturated lactone. Although pironetin's α,β-unsaturated lactone is involved in its binding to tubulin, the structure–activity relationship at different positions of the lactone have not been thoroughly evaluated. For a systematic evaluation of the structure–activity relationships at the C4 and C5 positions of the α,β-unsaturated lactone of pironetin, twelve analogues of the natural product were prepared by total synthesis. Modifying the stereochemistry at the C4 and/or C5 positions of the α,β-unsaturated lactone of pironetin resulted in loss of antiproliferative activity in OVCAR5 ovarian cancer cells. While changing the C4 ethyl substituent with groups such as methyl, propyl, cyclopropyl, and isobutyl were tolerated, groups with larger steric properties such as an isopropyl and benzyl groups were not.
Co-reporter:Adwait R. Ranade; LeeAnn Higgins; Todd W. Markowski; Nicole Glaser; Dmitry Kashin; Ruoli Bai; Kwon Ho Hong; Ernest Hamel; Gerhard Höfle;Gunda I. Georg
Journal of Medicinal Chemistry 2016 Volume 59(Issue 7) pp:3499-3514
Publication Date(Web):March 17, 2016
DOI:10.1021/acs.jmedchem.6b00188
Photoaffinity labeling with an epothilone A photoprobe led to the identification of the β-tubulin peptides TARGSQQY and TSRGSQQY as targets of the photoprobe for polymerized tubulin. These peptides represent residues 274–281 in different β-tubulin isotypes. Placing the carbene producing 21-diazo/triazolo moiety of the photoprobe in the vicinity of the TARGSQQY peptide in a homology model of TBB3 predicted a binding pose and conformation of the photoprobe that are very similar to the ones reported for 1) the high resolution cocrystal structure of epothilone A with an α,β-tubulin complex and for 2) a saturation transfer difference NMR and transferred NOESY NMR study of dimeric and polymerized tubulin. Our findings thus provide additional support for these models as physiologically the most relevant among several modes of binding that have been proposed for epothilone A in the taxane pocket of β-tubulin.
Co-reporter:Shameem Sultana Syeda, Erick J. Carlson, Melissa R. Miller, Rawle Francis, David E. Clapham, Polina V. Lishko, Jon E. Hawkinson, Derek Hook, and Gunda I. Georg
ACS Chemical Biology 2016 Volume 11(Issue 2) pp:452
Publication Date(Web):December 16, 2015
DOI:10.1021/acschembio.5b00748
The basal fungus Allomyces macrogynus (A. macrogynus) produces motile male gametes displaying well-studied chemotaxis toward their female counterparts. This chemotaxis is driven by sirenin, a sexual pheromone released by the female gametes. The pheromone evokes a large calcium influx in the motile gametes, which could proceed through the cation channel of sperm (CatSper) complex. Herein, we report the total synthesis of sirenin in 10 steps and 8% overall yield and show that the synthetic pheromone activates the CatSper channel complex, indicated by a concentration-dependent increase in intracellular calcium in human sperm. Sirenin activation of the CatSper channel was confirmed using whole-cell patch clamp electrophysiology with human sperm. Based on this proficient synthetic route and confirmed activation of CatSper, analogues of sirenin can be designed as blockers of the CatSper channel that could provide male contraceptive agents.
Co-reporter:Shameem S. Syeda;Daren Rice;Derek J. Hook;Leslie L. Heckert;Gunda I. Georg
Archiv der Pharmazie 2016 Volume 349( Issue 4) pp:233-241
Publication Date(Web):
DOI:10.1002/ardp.201500440
Two photo-crosslinking biarsenical (CrAsH-EDT2)-modified probes were synthesized that are expected to be useful tools for tetracysteine-labeled proteins to facilitate the co-affinity purification of their DNA binding sequences and interacting proteins. In addition, improvements for the synthesis of CrAsH-EDT2 and N1-(4-azido-2-nitrophenyl)hexane-1,6-diamine are reported. Both photoprobes effectively entered HeLa cells (and the nucleus) and were dependent on the tetracysteine motif in recombinant DMRT1 (doublesex and Mab3-related transcription factor) to induce fluorescence, suggesting that their crosslinking abilities can be exploited for the identification of nucleic acids and proteins associated with a protein of interest.
Co-reporter:Dr. Christopher M. Schneider;Dr. Wei Li;Dr. Kriangsak Khownium;Dr. Gerald H. Lushington;Dr. Gunda I. Georg
ChemMedChem 2016 Volume 11( Issue 15) pp:1600-1616
Publication Date(Web):
DOI:10.1002/cmdc.201600024
Abstract
Analogues of the anticancer natural product oximidine II were prepared and evaluated for cytotoxicity. One analogue of oximidine II that carries a C15 allylic amide side chain as well as two analogues with C15 vinyl sulfone side chains were found to lack cytotoxicity against the cancer cell line SK-Mel-5, thereby confirming the necessity of the C15 enamide side chain of oximidine II for cytotoxicity. Four analogues, designed by comparative molecular similarity index analysis (CoMSIA), that feature a less complex macrolactone scaffold were prepared and tested. The two analogues carrying a C15 vinyl sulfone group and the two analogues with a C15 oximidine II enamide side chain showed weak cytotoxicity against the SK-Mel-5 cell line and other cell lines, indicating that the designed simplified macrocycles cannot replace the oximidine II macrocycle.
Co-reporter:Dr. Christopher M. Schneider;Dr. Wei Li;Dr. Kriangsak Khownium;Dr. Gerald H. Lushington;Dr. Gunda I. Georg
ChemMedChem 2016 Volume 11( Issue 15) pp:
Publication Date(Web):
DOI:10.1002/cmdc.201600376
Co-reporter:Wei Li, Christopher M. Schneider, and Gunda I. Georg
Organic Letters 2015 Volume 17(Issue 15) pp:3902-3905
Publication Date(Web):July 15, 2015
DOI:10.1021/acs.orglett.5b01892
A copper-mediated macrocyclization involving the reaction of a vinyl iodide and a terminal alkyne followed by an in situ reduction of the enyne intermediate is reported. The reaction generates a conjugated Z-double bond within a strained medium-size lactone, lactam, or ether macrocycle. A variety of macrocyclic compounds bearing different ring sizes and functionalities were synthesized. A complementary stepwise procedure was also developed for less strained ring systems.
Co-reporter:Wei Li and Gunda I. Georg
Chemical Communications 2015 vol. 51(Issue 41) pp:8634-8636
Publication Date(Web):22 Apr 2015
DOI:10.1039/C5CC02571K
A formal total synthesis of the cytotoxic natural product lactimidomycin has been achieved in nine steps from (E)-2-methyl-2-pentenoic acid. The 12-membered lactone was efficiently formed via a copper-catalyzed ene–yne coupling/alkyne reduction tandem reaction.
Co-reporter:Satish Patil; Lev G. Lis; Robert J. Schumacher; Beverly J. Norris; Monique L. Morgan; Rebecca A. D. Cuellar; Bruce R. Blazar; Raj Suryanarayanan; Vadim J. Gurvich;Gunda I. Georg
Journal of Medicinal Chemistry 2015 Volume 58(Issue 23) pp:9334-9344
Publication Date(Web):November 24, 2015
DOI:10.1021/acs.jmedchem.5b01329
A disodium phosphonooxymethyl prodrug of the antitumor agent triptolide was prepared from the natural product in three steps (39% yield) and displayed excellent aqueous solubility at pH 7.4 (61 mg/mL) compared to the natural product (17 μg/mL). The estimated shelf life (t90) for hydrolysis of the prodrug at 4 °C and pH 7.4 was found to be two years. In a mouse model of human colon adenocarcinoma (HT-29), the prodrug administered intraperitoneally was effective in reducing or eliminating xenograft tumors at dose levels as low as 0.3 mg/kg when given daily and at 0.9 mg/kg when given less frequently. When given via intraperitoneal and oral routes at daily doses of 0.6 and 0.9 mg/kg, the prodrug was also effective and well tolerated in a mouse model of human ovarian cancer (A2780).
Co-reporter:Xingxian Gu, Gunda I. Georg
Tetrahedron Letters 2015 Volume 56(Issue 43) pp:5874-5877
Publication Date(Web):21 October 2015
DOI:10.1016/j.tetlet.2015.09.004
Reactions of N-alkyl-2,3-dihydro-4-pyridones and 4-(pyrrolidin-1-yl)furan-2(5H)-one with aldehydes and triethylsilane in a one-flask procedure provided C5 and C3 alkylated derivatives, respectively. Mannich-type reactions with formaldehyde and carbamates in the presence of lithium perchlorate furnished C5/C3 methylcarbamates.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Yong Wook Kim and Gunda I. Georg
Organic Letters 2014 Volume 16(Issue 6) pp:1574-1577
Publication Date(Web):March 3, 2014
DOI:10.1021/ol500105d
An oxidative boron–Heck reaction of cyclic enaminones with arylboronic acids is reported. This protocol provides a regioselective arylation at the C6 position of cyclic enaminones. When an N-carbamylated cyclic enaminone was employed, a switch to a conjugate addition reaction occurred in the presence of acid.
Co-reporter:Yi-Yun Yu ;Gunda I. Georg
Advanced Synthesis & Catalysis 2014 Volume 356( Issue 6) pp:1359-1369
Publication Date(Web):
DOI:10.1002/adsc.201300904
Co-reporter:Yi-Yun Yu;Adwait R. Ranade;Gunda I. Georg
Advanced Synthesis & Catalysis 2014 Volume 356( Issue 17) pp:3510-3518
Publication Date(Web):
DOI:10.1002/adsc.201400417
Co-reporter:Yi-Yun Yu ;Gunda I. Georg
Advanced Synthesis & Catalysis 2014 Volume 356( Issue 6) pp:
Publication Date(Web):
DOI:10.1002/adsc.201400050
Co-reporter:Hajime Seki ;Gunda I. Georg
European Journal of Organic Chemistry 2014 Volume 2014( Issue 18) pp:3777-3783
Publication Date(Web):
DOI:10.1002/ejoc.201402232
Abstract
An enantiospecific synthesis of a nuphar alkaloid was achieved in nine steps from N-Boc-(L)-proline. The alkaloid is a minor component of castoreum, which is secreted from the dried scent glands of a beaver. During the course of our study, the stereochemistry of three synthetic intermediates was verified by X-ray crystal structure analysis, which helped to resolve the existing discrepancies among reports regarding the structure of this particular compound. On the basis of our synthesized alkaloid, we proposed the structure of the natural product. Also, intrigued by the therapeutic uses of castoreum, which was employed for gynecological purposes in ancient Greece and Rome, we conducted a biological screening and found that the alkaloid has affinity for the oxytocin receptor.
Co-reporter:Hajime Seki ;Gunda I. Georg
European Journal of Organic Chemistry 2014 Volume 2014( Issue 18) pp:
Publication Date(Web):
DOI:10.1002/ejoc.201490048
Co-reporter:Adwait R. Ranade and Gunda I. Georg
The Journal of Organic Chemistry 2014 Volume 79(Issue 3) pp:984-992
Publication Date(Web):January 13, 2014
DOI:10.1021/jo402445r
The synthesis of 3,4-dihydro-1,2-oxazepin-5(2H)-ones and 2,3-dihydropyridin-4(1H)-ones from β-substituted β-hydroxyaminoaldehydes is reported. The β-hydroxyaminoaldehydes were prepared by enantioselective organocatalytic 1,4-addition of N-tert-butyl (tert-butyldimethylsilyl)oxycarbamate to α,β-unsaturated aldehydes (MacMillan protocol). Alkyne addition to the aldehydes followed by alcohol oxidation furnished N-Boc O-TBS-protected β-aminoynones. Removal of the TBS protecting group initiated a 7-endo-dig cyclization to yield previously unknown 3,4-dihydro-1,2-oxazepin-5(2H)-ones. Reductive cleavage of the N–O bond of the oxazepinones and Boc-deprotection provided 2-substituted 2,3-dihydropyridin-4(1H)-ones via 6-endo-trig cyclization. 2,3-Dihydropyridin-4(1H)-ones are versatile intermediates that have been used for the synthesis of many alkaloids. The new protocol allows the synthesis of 3-dihydropyridin-4(1H)-ones carrying an array of substituents at C2 that cannot be prepared from commercial β-amino acids or by one-carbon homologation of proteinogenic amino acids. The use of readily available β-hydroxylaminoaldehydes expands the utility of our previously reported method to prepare 2,3-dihydropyridin-4(1H)-ones from β-amino acids as the source of diversity and chirality. A broad substrate scope is possible because β-aminoaldehydes can be prepared from α,β-unsaturated aldehydes by an enantioselective organocatalytic process.
Co-reporter:Oliver E. Hutt, Trinh L. Doan, and Gunda I. Georg
Organic Letters 2013 Volume 15(Issue 7) pp:1602-1605
Publication Date(Web):March 26, 2013
DOI:10.1021/ol400385w
We have applied a diversity-oriented approach for the synthesis of skeletally diverse and stereochemically complex templates for small-molecule library production by performing Beckmann rearrangement and Beckmann fragmentation reactions on the bicyclo[3.2.1]octane rings of steviol and isosteviol, aglycones derived from the diterpene natural product stevioside. The optimization of these two reaction pathways is presented along with the successful application of a photo-Beckmann rearrangement. This work also led to the discovery of cyano-Prins-type and Thorpe–Ziegler-type cyclization reactions.
Co-reporter:Yi-Yun Yu and Gunda I. Georg
Chemical Communications 2013 vol. 49(Issue 35) pp:3694-3696
Publication Date(Web):25 Mar 2013
DOI:10.1039/C3CC41130C
A regioselective Pd-catalyzed cross-dehydrogenative coupling between uracils and alkenes is reported. This protocol provides easy access to a variety of 5-alkenyluracil structural motifs.
Co-reporter:Ernst Schonbrunn ; Stephane Betzi ; Riazul Alam ; Mathew P. Martin ; Andreas Becker ; Huijong Han ; Rawle Francis ; Ramappa Chakrasali ; Sudhakar Jakkaraj ; Aslamuzzaman Kazi ; Said M. Sebti ; Christopher L. Cubitt ; Anthony W. Gebhard ; Lori A. Hazlehurst ; Joseph S. Tash ;Gunda I. Georg
Journal of Medicinal Chemistry 2013 Volume 56(Issue 10) pp:3768-3782
Publication Date(Web):April 19, 2013
DOI:10.1021/jm301234k
Cyclin-dependent kinases (CDKs) are serine/threonine protein kinases that act as key regulatory elements in cell cycle progression. We describe the development of highly potent diaminothiazole inhibitors of CDK2 (IC50 = 0.0009–0.0015 μM) from a single hit compound with weak inhibitory activity (IC50 = 15 μM), discovered by high-throughput screening. Structure-based design was performed using 35 cocrystal structures of CDK2 liganded with distinct analogues of the parent compound. The profiling of compound 51 against a panel of 339 kinases revealed high selectivity for CDKs, with preference for CDK2 and CDK5 over CDK9, CDK1, CDK4, and CDK6. Compound 51 inhibited the proliferation of 13 out of 15 cancer cell lines with IC50 values between 0.27 and 6.9 μM, which correlated with the complete suppression of retinoblastoma phosphorylation and the onset of apoptosis. Combined, the results demonstrate the potential of this new inhibitors series for further development into CDK-specific chemical probes or therapeutics.
Co-reporter:Yi-Yun Yu, Lei Bi, and Gunda I. Georg
The Journal of Organic Chemistry 2013 Volume 78(Issue 12) pp:6163-6169
Publication Date(Web):June 10, 2013
DOI:10.1021/jo400830t
A ligand-free method for the Pd-catalyzed direct arylation of cyclic enaminones using aryl iodides was developed. This method can be applied to a wide range of cyclic enaminones and aryl iodides with excellent C5-regioselectivity. Using widely available aryl iodides, the generality of this transformation provides easy access to a variety of 3-arylpiperidine structural motifs.
Co-reporter:Xingxian Gu, Gunda I. Georg
Tetrahedron 2013 69(45) pp: 9406-9416
Publication Date(Web):
DOI:10.1016/j.tet.2013.08.081
Co-reporter:Micah J. Niphakis, Bryant C. Gay, Kwon Ho Hong, Nicholas P. Bleeker, Gunda I. Georg
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 19) pp:5893-5900
Publication Date(Web):1 October 2012
DOI:10.1016/j.bmc.2012.07.044
Tylophorine and many related phenanthropiperidine alkaloids are extraordinarily potent anti-proliferative agents. Despite their impressive anti-cancer activity, clinical development of these alkaloids has been hampered by their poor solubility and neurological side effects. Although it has been suggested that developing polar phenanthropiperidines will mitigate these undesired properties, the lack of practical methods for the synthesis of such analogues has limited this effort. Here, we present a concise synthetic approach to N-substituted phenanthropiperidines, which enabled a systematic investigation of structure-activity relationships at an underexplored region of the tylophorine scaffold. This work suggests that ring E of tylophorine is essential for the anti-proliferative activity of the 6,7,10,11-tetramethoxy-1,2,3,4-tetrahydrodibenzo[f,h]isoquinoline core scaffold.
Co-reporter:Jae Chul Lee, Subhashree Francis, Dinah Dutta, Vijayalaxmi Gupta, Yan Yang, Jin-Yi Zhu, Joseph S. Tash, Ernst Schönbrunn, and Gunda I. Georg
The Journal of Organic Chemistry 2012 Volume 77(Issue 7) pp:3082-3098
Publication Date(Web):March 20, 2012
DOI:10.1021/jo202054g
Eight- and four-membered analogues of N-butyldeoxynojirimycin (NB-DNJ), a reversible male contraceptive in mice, were prepared and tested. A chiral pool approach was used for the synthesis of the target compounds. Key steps for the synthesis of the eight-membered analogues involve ring-closing metathesis and Sharpless asymmetric dihydroxylation and for the four-membered analogues Sharpless epoxidation, epoxide ring-opening (azide), and Mitsunobu reaction to form the four-membered ring. (3S,4R,5S,6R,7R)-1-Nonylazocane-3,4,5,6,7-pentaol (6) was moderately active against rat-derived ceramide-specific glucosyltransferase, and four of the other eight-membered analogues were weakly active against rat-derived β-glucosidase 2. Among the four-membered analogues, ((2R,3S,4S)-3-hydroxy-1-nonylazetidine-2,4-diyl)dimethanol (25) displayed selective inhibitory activity against mouse-derived ceramide-specific glucosyltransferase and was about half as potent as NB-DNJ against the rat-derived enzyme. ((2S,4S)-3-Hydroxy-1-nonylazetidine-2,4-diyl)dimethanol (27) was found to be a selective inhibitor of β-glucosidase 2, with potency similar to NB-DNJ. Additional glycosidase assays were performed to identify potential other therapeutic applications. The eight-membered iminosugars exhibited specificity for almond-derived β-glucosidase, and the 1-nonylazetidine 25 inhibited α-glucosidase (Saccharomyces cerevisiae) with an IC50 of 600 nM and β-glucosidase (almond) with an IC50 of 20 μM. Only N-nonyl derivatives were active, emphasizing the importance of a long lipophilic side chain for inhibitory activity of the analogues studied.
Co-reporter:Yong Wook Kim, Micah J. Niphakis, and Gunda I. Georg
The Journal of Organic Chemistry 2012 Volume 77(Issue 21) pp:9496-9503
Publication Date(Web):October 22, 2012
DOI:10.1021/jo301531k
Described herein is a palladium(II)-catalyzed direct arylation of cyclic enaminones with arylboronic acids. The versatility of this method is that both electron-rich and electron-poor boronic acids can be coupled in high yields. A mixture of two Cu(II) additives was crucial for efficient cross-coupling. The role of each Cu(II) reagent appears to be distinct and complementary serving to assist catalyst reoxidation and transmetalation through a putative arylcopper intermediate.
Co-reporter:Micah J. Niphakis and Gunda I. Georg
Organic Letters 2011 Volume 13(Issue 2) pp:196-199
Publication Date(Web):December 14, 2010
DOI:10.1021/ol1023954
A highly convergent strategy to prepare phenanthroindolizidines is reported involving three consecutive C−C coupling reactions. This sequence features a novel VOF3-mediated aryl-alkene coupling in the final step, which enables regioselective preparation of C5-substituted phenanthroindolizidines for the first time. This strategy has been applied to the synthesis of eight natural and unnatural members in this class to investigate the scope of this chemistry and to explore structure−activity relationships.
Co-reporter:Hajime Seki and Gunda I. Georg
Organic Letters 2011 Volume 13(Issue 9) pp:2147-2149
Publication Date(Web):April 1, 2011
DOI:10.1021/ol200358h
Cyclic six-membered enaminones were synthesized from three components (bromodiazoacetone, primary amine, and alkyne) in high yields via aza-Michael addition, Wolff rearrangement, and nucleophilic ketene cyclization.
Co-reporter:Yi-Yun Yu, Micah J. Niphakis, and Gunda I. Georg
Organic Letters 2011 Volume 13(Issue 21) pp:5932-5935
Publication Date(Web):October 13, 2011
DOI:10.1021/ol202677g
A new Pd(II)-catalyzed dehydrogenative alkenylation reaction involving two alkenes was developed. A variety of nonaromatic, cyclic enaminones were successfully coupled to primary and secondary alkenes yielding a series of unique 1,3-dienes. The generality of this transformation presents a useful strategy for directly cross-coupling alkenes and offers an attractive new approach to functionalize enaminones.
Co-reporter:Matthew W. Leighty and Gunda I. Georg
ACS Medicinal Chemistry Letters 2011 Volume 2(Issue 4) pp:313
Publication Date(Web):February 23, 2011
DOI:10.1021/ml1003074
Both enantiomers of boehmeriasin A were synthesized in seven steps each using a chiral pool approach. Key steps in the syntheses are a one-flask, two-step protocol to generate the quinolizine core and a C−H functionalization reaction between tetrahydroquinolizinones and an aryltrifluoroborate. The natural product (R)-boehmeriasin A demonstrated potent cytotoxicity against several cancer cell lines, whereas the unnatural (+)-(S)-isomer was significantly less potent.Keywords: boehmeriasin A; cytotoxicity; drug resistance; Natural product synthesis; phenanthroquinolizidine
Co-reporter:Antonella Pepe, Michael Pamment, Gunda I. Georg, and Sanjay V. Malhotra
The Journal of Organic Chemistry 2011 Volume 76(Issue 9) pp:3527-3530
Publication Date(Web):March 31, 2011
DOI:10.1021/jo102558u
Monocyclic as well as fused bicyclic systems with a nitrogen 10 atom at the bridgehead, including indolizidines and quinolizidines, can be prepared in four steps from N-Boc β-lactams. These easily prepared, highly robust, and flexible building blocks allow the incorporation of chirality and structural diversity, rendering the method feasible for diversity- as well as target-oriented synthesis.
Co-reporter:Dr. Christopher M. Schneider;Dr. Kriangsak Khownium;Wei Li;Dr. Jared T. Spletstoser;Dr. Torsten Haack;Dr. Gunda I. Georg
Angewandte Chemie International Edition 2011 Volume 50( Issue 34) pp:7855-7857
Publication Date(Web):
DOI:10.1002/anie.201103081
Co-reporter:Dr. Christopher M. Schneider;Dr. Kriangsak Khownium;Wei Li;Dr. Jared T. Spletstoser;Dr. Torsten Haack;Dr. Gunda I. Georg
Angewandte Chemie 2011 Volume 123( Issue 34) pp:8001-8003
Publication Date(Web):
DOI:10.1002/ange.201103081
Co-reporter:Hajime Seki ;Gunda I. Georg
Journal of the American Chemical Society 2010 Volume 132(Issue 44) pp:15512-15513
Publication Date(Web):October 19, 2010
DOI:10.1021/ja107329k
Cyclic enaminones were synthesized in high yields from amino acids in two steps via Wolff rearrangement. The cyclization represents a rare 6-exo-dig cyclization involving a ketene as an electrophile. No racemization was observed during this reaction.
Co-reporter:Wei Li and Gunda I. Georg
Chemical Communications 2015 - vol. 51(Issue 41) pp:NaN8636-8636
Publication Date(Web):2015/04/22
DOI:10.1039/C5CC02571K
A formal total synthesis of the cytotoxic natural product lactimidomycin has been achieved in nine steps from (E)-2-methyl-2-pentenoic acid. The 12-membered lactone was efficiently formed via a copper-catalyzed ene–yne coupling/alkyne reduction tandem reaction.
Co-reporter:Yi-Yun Yu and Gunda I. Georg
Chemical Communications 2013 - vol. 49(Issue 35) pp:NaN3696-3696
Publication Date(Web):2013/03/25
DOI:10.1039/C3CC41130C
A regioselective Pd-catalyzed cross-dehydrogenative coupling between uracils and alkenes is reported. This protocol provides easy access to a variety of 5-alkenyluracil structural motifs.

![(R)-(-)-tert-Butyl 2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetate](http://img.cochemist.com/ccimg/1268600/1268524-71-5.png)
![(R)-(-)-tert-Butyl 2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetate](http://img.cochemist.com/ccimg/1268600/1268524-71-5_b.png)
![(S)-tert-Butyl 2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetate](http://img.cochemist.com/ccimg/1268600/1268524-70-4.png)
![(S)-tert-Butyl 2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetate](http://img.cochemist.com/ccimg/1268600/1268524-70-4_b.png)
![(S)-tert-butyl 2-(5-(4-chlorophenyl)-6,7-dimethyl-2-oxo-2,3-dihydro-1H-thieno[2,3-e][1,4]diazepin-3-yl)acetate](http://img.cochemist.com/ccimg/1268600/1268524-67-9.png)
![(S)-tert-butyl 2-(5-(4-chlorophenyl)-6,7-dimethyl-2-oxo-2,3-dihydro-1H-thieno[2,3-e][1,4]diazepin-3-yl)acetate](http://img.cochemist.com/ccimg/1268600/1268524-67-9_b.png)













![2-Oxazolidinone, 3-[(2E,4S,5R)-5-hydroxy-2,4-dimethyl-1-oxo-2-hexen-1-yl]-4-(1-methylethyl)-, (4S)-](/data/chemimg/182900/1044263-52-6.png)
![2-Oxazolidinone, 3-[(2E,4S,5R)-5-hydroxy-2,4-dimethyl-1-oxo-2-hexen-1-yl]-4-(1-methylethyl)-, (4S)-](/data/chemimg/182900/1044263-52-6_b.png)


![Benzoic acid, 2-[(1E)-2-iodoethenyl]-, methyl ester](http://img.cochemist.com/ccimg/887000/886986-52-3.png)
![Benzoic acid, 2-[(1E)-2-iodoethenyl]-, methyl ester](http://img.cochemist.com/ccimg/887000/886986-52-3_b.png)





![4',5'-Bis(1,3,2-dithiarsolan-2-yl)-3',6'-dihydroxy-3-oxospiro[isobenzofuran-1(3H),9'-[9H]xanthene]-5-carboxylic Acid](/data/chemimg/1019300/439791-28-3.png)
![4',5'-Bis(1,3,2-dithiarsolan-2-yl)-3',6'-dihydroxy-3-oxospiro[isobenzofuran-1(3H),9'-[9H]xanthene]-5-carboxylic Acid](/data/chemimg/1019300/439791-28-3_b.png)
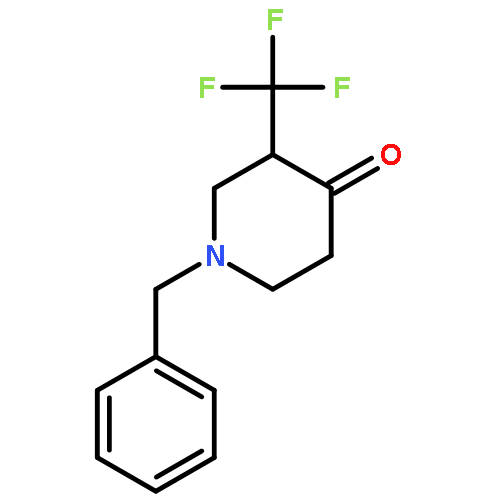
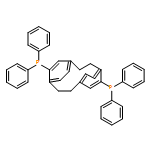
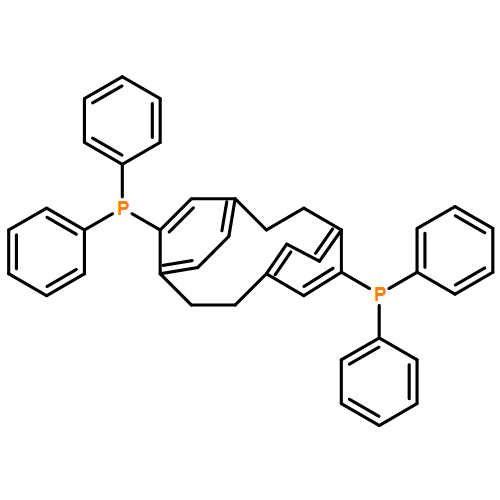


![4,17-Dioxabicyclo[14.1.0]heptadecane-5,9-dione,7,11-dihydroxy-3-[(1E)-2-[2-(hydroxymethyl)- 4-thiazolyl]-1-methylethenyl]-8,8,10,- 12-tetramethyl-,(1S,3S,7S,10R,11S,12S,16R)-](http://img.cochemist.com/ccimg/201100/201049-37-8.png)
![4,17-Dioxabicyclo[14.1.0]heptadecane-5,9-dione,7,11-dihydroxy-3-[(1E)-2-[2-(hydroxymethyl)- 4-thiazolyl]-1-methylethenyl]-8,8,10,- 12-tetramethyl-,(1S,3S,7S,10R,11S,12S,16R)-](http://img.cochemist.com/ccimg/201100/201049-37-8_b.png)








![4,17-Dioxabicyclo[14.1.0]heptadecane-5,9-dione,7,11-dihydroxy-8,8,10,12-tetramethyl-3-[(1E)-1-methyl-2-(2-methyl-4-thiazolyl)ethenyl]-,(1S,3S,7S,10R,11S,12S,16R)-](http://img.cochemist.com/ccimg/152100/152044-53-6.png)
![4,17-Dioxabicyclo[14.1.0]heptadecane-5,9-dione,7,11-dihydroxy-8,8,10,12-tetramethyl-3-[(1E)-1-methyl-2-(2-methyl-4-thiazolyl)ethenyl]-,(1S,3S,7S,10R,11S,12S,16R)-](http://img.cochemist.com/ccimg/152100/152044-53-6_b.png)
![Propanal, 3-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/128500/128461-65-4.png)
![Propanal, 3-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/128500/128461-65-4_b.png)



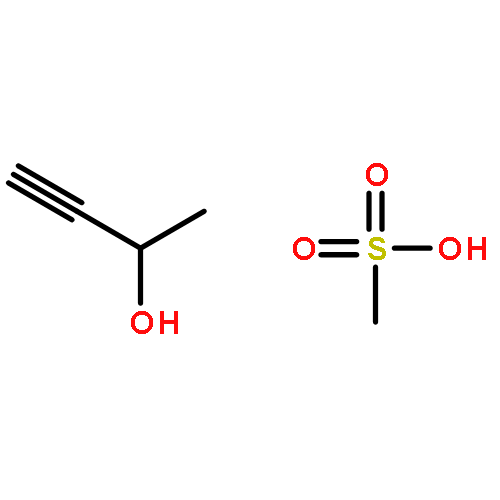


![Butanal, 4-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/119700/119649-27-3.png)
![Butanal, 4-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/119700/119649-27-3_b.png)



![Benzoic acid,4-[3-(trifluoromethyl)-3H-diazirin-3-yl]-](http://img.cochemist.com/ccimg/85600/85559-46-2.png)
![Benzoic acid,4-[3-(trifluoromethyl)-3H-diazirin-3-yl]-](http://img.cochemist.com/ccimg/85600/85559-46-2_b.png)


![Phenol, 2-[(trimethylsilyl)ethynyl]-](/data/chemimg/974000/81787-62-4.png)
![Phenol, 2-[(trimethylsilyl)ethynyl]-](/data/chemimg/974000/81787-62-4_b.png)










![(2E)-5-[(1R,6S,7R)-3-(hydroxymethyl)-7-methylbicyclo[4.1.0]hept-2-en-7-yl]-2-methylpent-2-en-1-ol](http://img.cochemist.com/ccimg/19900/19888-27-8.png)
![(2E)-5-[(1R,6S,7R)-3-(hydroxymethyl)-7-methylbicyclo[4.1.0]hept-2-en-7-yl]-2-methylpent-2-en-1-ol](http://img.cochemist.com/ccimg/19900/19888-27-8_b.png)













![<br>3-(3-methoxyphenyl)-6-(phenoxymethyl)-[1,2,4]triazolo[3,4-b][1,3,4]thiadiaz ole](http://img.cochemist.com/ccimg/934700/934609-13-9.png)
![<br>3-(3-methoxyphenyl)-6-(phenoxymethyl)-[1,2,4]triazolo[3,4-b][1,3,4]thiadiaz ole](http://img.cochemist.com/ccimg/934700/934609-13-9_b.png)
![2,6-Piperidinedione,4-[(2R,5S,6E)-2-hydroxy-5-methyl-7-[(2R,3S,4Z,6E,10E)-3-methyl-12-oxooxacyclododeca-4,6,10-trien-2-yl]-4-oxo-6-octen-1-yl]-](http://img.cochemist.com/ccimg/134900/134869-15-1.png)
![2,6-Piperidinedione,4-[(2R,5S,6E)-2-hydroxy-5-methyl-7-[(2R,3S,4Z,6E,10E)-3-methyl-12-oxooxacyclododeca-4,6,10-trien-2-yl]-4-oxo-6-octen-1-yl]-](http://img.cochemist.com/ccimg/134900/134869-15-1_b.png)






![(13aR)-2,3,5,6-tetramethoxy-9,11,12,13,13a,14-hexahydrodibenzo[f,h]pyrrolo[1,2-b]isoquinoline](http://img.cochemist.com/ccimg/6900/6879-02-3.png)
![(13aR)-2,3,5,6-tetramethoxy-9,11,12,13,13a,14-hexahydrodibenzo[f,h]pyrrolo[1,2-b]isoquinoline](http://img.cochemist.com/ccimg/6900/6879-02-3_b.png)