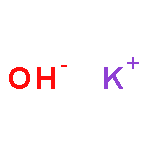
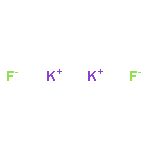
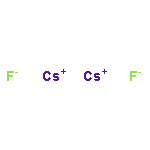
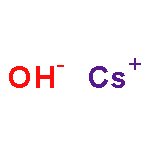
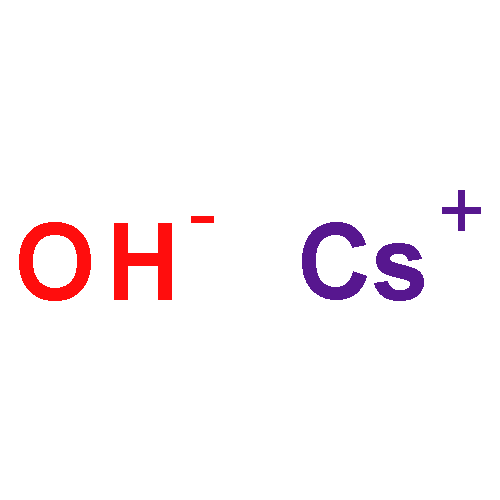
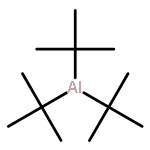
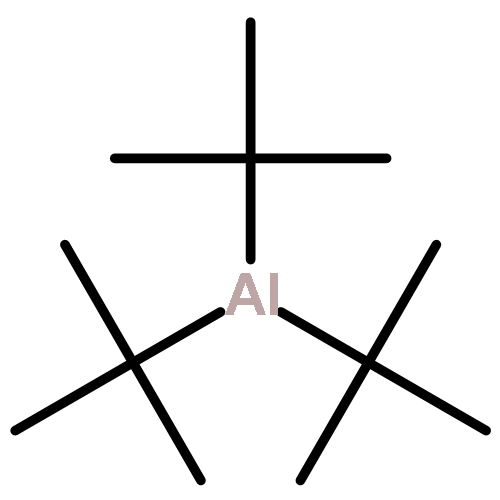
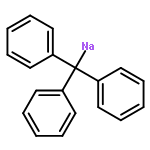
Co-reporter:Benjamin Freitag, Holger Elsen, Jürgen Pahl, Gerd Ballmann, Alberto Herrera, Romano Dorta, and Sjoerd Harder
Organometallics May 8, 2017 Volume 36(Issue 9) pp:1860-1860
Publication Date(Web):April 24, 2017
DOI:10.1021/acs.organomet.7b00200
The dibenzo[b,f]azepinate (DBAP) complexes (DBAP)Li·(THF)3, (DBAP)2Mg·(THF)2, and (DBAP)2Ca·(THF)3 could be isolated as highly air-sensitive compounds in yields of 93%, 72%, and 48%, respectively. Crystal structures of these THF adducts reveal monomeric complexes in which the degree of ring puckering depends on the nature of the metal. The most extreme deviation from planarity is found for the most covalent bound metal, Mg, but in all cases no interaction between the metal and the azepine C═C bond is observed. The THF-free complex [(DBAP)2Mg]2, which could be obtained in 77% yield, crystallizes as an unusual dimer with three bridging and one terminal DBAP ligand. The bridging DBAP ligands are highly bent and span a cavity in which a Mg2+ ion is bound through three alkene–Mg interactions with an average Mg···C distance of 2.794(3) Å. Theoretical calculations support these contacts. A combination of AIM and NPA analyses shows polarization of the alkene π-electron density toward the metal (vertical polarization) but also demonstrates a polarization of electron density toward the C atom closest to Mg (horizontal polarization). Such metal–alkene interactions and implicit C═C bond polarization are key features in main group metal catalyzed alkene conversions.
Co-reporter:Andrea Causero;Gerd Ballmann;Jürgen Pahl;Christian Färber;Julia Intemann
Dalton Transactions 2017 vol. 46(Issue 6) pp:1822-1831
Publication Date(Web):2017/02/14
DOI:10.1039/C6DT04659B
A series of (DIPPnacnac)CaN(SiMe3)2·S complexes (DIPPnacnac = HC[C(Me)N(2,6-iPr-C6H3)]2; S = solvent) could be obtained by the addition of S = THF, DME or N-Me-morpholine (Morph) to (DIPPnacnac)CaN(SiMe3)2·OEt2 or (DIPPnacnac)CaN(SiMe3)2. Crystal structures for complexes with S = DME and Morph are compared to literature-known structures with S = none, THF or Et2O. Bulkier and weaker Lewis bases like the tertiary amines Et3N, TMEDA and DABCO did not interact with (DIPPnacnac)CaN(SiMe3)2. The reaction of (DIPPnacnac)CaN(SiMe3)2 with PhSiH3 gave conversion to a calcium hydride complex that dismutated in (DIPPnacnac)2Ca and CaH2. The reaction of (DIPPnacnac)CaN(SiMe3)2·S with PhSiH3 gave [(DIPPnacnac)CaH·S]2 for S = THF, Et2O or N-Me-morpholine (Morph). For S = DME, high reaction temperatures were needed and dismutation into (DIPPnacnac)2Ca and CaH2 was observed. Extensive NMR investigations (VT-NMR and PGSE) confirm the dimeric nature of [(DIPPnacnac)CaH·THF]2 in aromatic solvents or in THF. Thermal decomposition of [(DIPPnacnac)CaH·THF]2 (release of H2 at 200 °C) is compared to that of Mg and Zn analogues. Weakly coordinating Et2O in [(DIPPnacnac)CaH·OEt2]2 could be replaced by THF, Morph or DABCO but not with Et3N. The addition of TMEDA led to the formation of CaH2 and unidentified products. The addition of DME led to the decomposition of Et2O and complex [(DIPPnacnac)CaOEt]2 was obtained. Crystal structures of the following compounds are presented: (DIPPnacnac)CaN(SiMe3)2·S (S = Morph, DME), [(DIPPnacnac)CaH·S]2 (S = Et2O, Morph and DABCO) and [(DIPPnacnac)CaOEt]2. Although bulky ligands have long been thought to be the key to the stabilization of calcium hydride complexes, the presence of a polar, strongly coordinating, co-solvent is also crucial.
Co-reporter:Harmen S. Zijlstra;Jürgen Pahl;Johanne Penafiel
Dalton Transactions 2017 vol. 46(Issue 11) pp:3601-3610
Publication Date(Web):2017/03/14
DOI:10.1039/C7DT00318H
The reaction of Ph2P(DIPP)NH with AlMe3 cleanly gives an aluminum amide complex that crystallizes as a centrosymmetric dimer with a six-membered Al–N–P–Al–N–P ring. In aromatic solvents the dimer remains intact but the Al–P bond is readily broken upon addition of THF to form Ph2P(DIPP)NAlMe2·THF. Efforts to use [Ph2P(DIPP)NAlMe2]2 as a “masked” Lewis acidic activator for olefin polymerization catalysts were unsuccessful but the complex showed a Frustrated Lewis pair reactivity instead. The P/Al complex reacts with isocyanates to give the CO inserted product that crystallizes as a five-membered ring system Al–O–C(NR)–P–N. The reaction of [Ph2P(DIPP)NAlMe2]2 with CO2, however, gave an insertion in the N–Al bond and the dimeric product [Ph2P(DIPP)NCO2AlMe2]2 was isolated. The dimer [Ph2P(DIPP)NAlMe2]2 is one of the few Al/P FLPs that can activate CC double bonds irreversibly. A reaction with allyl methyl sulfide and 1-hexene led to the clean formation of the structurally similar activated alkene products [(DIPP)N-Ph2P-CH(CH2SMe)CH2]AlMe2 and [(DIPP)N-Ph2P-CH(C4H9)CH2]AlMe2.
Co-reporter:Benjamin Freitag;Christian A. Fischer;Johanne Penafiel;Gerd Ballmann;Holger Elsen;Christian Färber;Dirk F. Piesik
Dalton Transactions 2017 vol. 46(Issue 34) pp:11192-11200
Publication Date(Web):2017/08/29
DOI:10.1039/C7DT02136D
Syntheses and crystal structures of the monomeric bora-amidinate (bam) complexes DIPPNBN-Mg·(THF)3 and DIPPNBN-Ca·(THF)4 are presented; DIPPNBN = HB[N(2,6-iPr2-C6H3)]2. The simplicity of their 1H NMR spectra in THF-d8 suggest that their monomeric solid state structures are retained in solution. DIPPNBN-Mg·(THF)3 in C6D6, however, is in equilibrium with a dimeric species. Calculations (B3PW91/6-311++G**) reveal a very high localized negative charge (NPA: −1.103) on the N atoms in DIPPNBN-Mg. The strongly basic properties of the bam ligand are in agreement with catalytic activity of these complexes in the intramolecular alkene hydroamination. A mechanism is proposed in which the bam ligand is non-innocent and cooperative, playing an active role in substrate deprotonation and product protonation.
Co-reporter:Andrea Causero;Holger Elsen;Gerd Ballmann;Ana Escalona
Chemical Communications 2017 vol. 53(Issue 75) pp:10386-10389
Publication Date(Web):2017/09/19
DOI:10.1039/C7CC05792J
Addition of a calcium hydride complex to diphenylacetylene gave a complex in which the stilbene dianion symmetrically bridges two Ca2+ ions. DFT calculations discuss the effect of the metal stilbene coordination. The stilbene complex reacts as a base (with H2) or an electron donor (with I2) and catalyzes the reduction of diphenylacetylene.
Co-reporter:Dr. Brant Maitl;Michael Wiesinger;Dr. Jens Langer;Gerd Ballmann;Jürgen Pahl;Holger Elsen;Dr. Christian Färber; Dr. Sjoerd Harder
Angewandte Chemie 2017 Volume 129(Issue 39) pp:12042-12046
Publication Date(Web):2017/09/18
DOI:10.1002/ange.201706786
AbstractThe first strontium hydride complex has been obtained by simply treating Sr[N(SiMe3)2]2 with PhSiH3 in the presence of PMDTA. The Sr complex Sr6H9[N(SiMe3)2]3⋅(PMDTA)3 crystallizes as an “inverse cryptand”: an interstitial H− is surrounded by a Sr6H84+ cage decorated with amide and PMDTA ligands. The analogous Ca complex could also be obtained and both retain their solid-state structures in solution: 1H NMR spectra in C6D6 show two doublets and one nonet (4:4:1). Up to 90 °C, no coalescence is observed. The Ca cluster was investigated by DFT calculations and shows atypically low charges on Ca (+1.14) and H (−0.59) which signifies an unexpectedly low ionicity. AIM analysis shows hydride⋅⋅⋅hydride bond paths with considerable electron densities in the bond critical point. The clusters thermally decompose into larger, undefined, metal hydride aggregates.
Co-reporter:Michael Wiesinger;Dr. Brant Maitl;Dr. Christian Färber;Gerd Ballmann;Christian Fischer;Holger Elsen; Dr. Sjoerd Harder
Angewandte Chemie 2017 Volume 129(Issue 52) pp:16881-16886
Publication Date(Web):2017/12/22
DOI:10.1002/ange.201709771
AbstractReaction of Ba[N(SiMe3)2]2 with PhSiH3 in toluene gave simple access to the unique Ba hydride cluster Ba7H7[N(SiMe3)2]7 that can be described as a square pyramid spanned by five Ba2+ ions with two flanking BaH[N(SiMe3)2] units. This heptanuclear cluster is well soluble in aromatic solvents, and the hydride 1H NMR signals and coupling pattern suggests that the structure is stable in solution. At 95 °C, no coalescence of hydride signals is observed but the cluster slowly decomposes to undefined barium hydride species. The complex Ba7H7[N(SiMe3)2]7 is a very strong reducing agent that already at room temperature reacts with Me3SiCH=CH2, norbornadiene, and ethylene. The highly reactive alkyl barium intermediates cannot be observed and deprotonate the (Me3Si)2N− ion, as confirmed by the crystal structure of Ba14H12[N(SiMe3)2]12[(Me3Si)(Me2SiCH2)N]4.
Co-reporter:Andrea Causero;Holger Elsen;Jürgen Pahl; Dr. Sjoerd Harder
Angewandte Chemie International Edition 2017 Volume 56(Issue 24) pp:6906-6910
Publication Date(Web):2017/06/06
DOI:10.1002/anie.201703037
AbstractAn anionic N-heterocyclic olefin ligand was serendipitously obtained by reaction of an amidinate calcium hydride complex with 1,3-dimethyl-2-methyleneimidazole (NHO). Instead of anticipated addition to the polarized C=CH2 bond to form an unstabilized alkylcalcium complex, deprotonation of the NHO ligand in the backbone was observed. Preference for deprotonation versus addition is explained by loss of aromaticity in the latter conversion. Theoretical calculations demonstrate the substantially increased ylidic character of this anionic NHO ligand which, like N-heterocyclic dicarbenes, shows strong bifunctional coordination.
Co-reporter:Michael Wiesinger;Dr. Brant Maitl;Dr. Christian Färber;Gerd Ballmann;Christian Fischer;Holger Elsen; Dr. Sjoerd Harder
Angewandte Chemie International Edition 2017 Volume 56(Issue 52) pp:16654-16659
Publication Date(Web):2017/12/22
DOI:10.1002/anie.201709771
AbstractReaction of Ba[N(SiMe3)2]2 with PhSiH3 in toluene gave simple access to the unique Ba hydride cluster Ba7H7[N(SiMe3)2]7 that can be described as a square pyramid spanned by five Ba2+ ions with two flanking BaH[N(SiMe3)2] units. This heptanuclear cluster is well soluble in aromatic solvents, and the hydride 1H NMR signals and coupling pattern suggests that the structure is stable in solution. At 95 °C, no coalescence of hydride signals is observed but the cluster slowly decomposes to undefined barium hydride species. The complex Ba7H7[N(SiMe3)2]7 is a very strong reducing agent that already at room temperature reacts with Me3SiCH=CH2, norbornadiene, and ethylene. The highly reactive alkyl barium intermediates cannot be observed and deprotonate the (Me3Si)2N− ion, as confirmed by the crystal structure of Ba14H12[N(SiMe3)2]12[(Me3Si)(Me2SiCH2)N]4.
Co-reporter:Dr. Brant Maitl;Michael Wiesinger;Dr. Jens Langer;Gerd Ballmann;Jürgen Pahl;Holger Elsen;Dr. Christian Färber; Dr. Sjoerd Harder
Angewandte Chemie International Edition 2017 Volume 56(Issue 39) pp:11880-11884
Publication Date(Web):2017/09/18
DOI:10.1002/anie.201706786
AbstractThe first strontium hydride complex has been obtained by simply treating Sr[N(SiMe3)2]2 with PhSiH3 in the presence of PMDTA. The Sr complex Sr6H9[N(SiMe3)2]3⋅(PMDTA)3 crystallizes as an “inverse cryptand”: an interstitial H− is surrounded by a Sr6H84+ cage decorated with amide and PMDTA ligands. The analogous Ca complex could also be obtained and both retain their solid-state structures in solution: 1H NMR spectra in C6D6 show two doublets and one nonet (4:4:1). Up to 90 °C, no coalescence is observed. The Ca cluster was investigated by DFT calculations and shows atypically low charges on Ca (+1.14) and H (−0.59) which signifies an unexpectedly low ionicity. AIM analysis shows hydride⋅⋅⋅hydride bond paths with considerable electron densities in the bond critical point. The clusters thermally decompose into larger, undefined, metal hydride aggregates.
Co-reporter:Dr. Jens Langer;Dr. Brant Maitl;Samuel Grams;Alexra Ciucka;Jürgen Pahl;Holger Elsen; Dr. Sjoerd Harder
Angewandte Chemie 2017 Volume 129(Issue 18) pp:5103-5107
Publication Date(Web):2017/04/24
DOI:10.1002/ange.201700719
AbstractWhile magnesium hydride complexes are generally stabilized by hard, bulky N-donor ligands, softer ligands with a broad variety of coordination modes are shown to efficiently adapt themselves to the large variety of Mg2+ centers in a growing magnesium hydride cluster. A P,N-chelating ligand is introduced that displays coordination modes between that of enamide, aza-allyl, and phosphinomethanide. Slight changes in the ligand bite angle have dramatic consequences for the structure type. The hitherto largest neutral magnesium hydride clusters are isolated either in a nonanuclear sheet-structure (brucite-type) or a dodecanuclear ring structure.
Co-reporter:Andrea Causero;Holger Elsen;Jürgen Pahl; Dr. Sjoerd Harder
Angewandte Chemie 2017 Volume 129(Issue 24) pp:7010-7014
Publication Date(Web):2017/06/06
DOI:10.1002/ange.201703037
AbstractAn anionic N-heterocyclic olefin ligand was serendipitously obtained by reaction of an amidinate calcium hydride complex with 1,3-dimethyl-2-methyleneimidazole (NHO). Instead of anticipated addition to the polarized C=CH2 bond to form an unstabilized alkylcalcium complex, deprotonation of the NHO ligand in the backbone was observed. Preference for deprotonation versus addition is explained by loss of aromaticity in the latter conversion. Theoretical calculations demonstrate the substantially increased ylidic character of this anionic NHO ligand which, like N-heterocyclic dicarbenes, shows strong bifunctional coordination.
Co-reporter:Tom E. Stennett and Sjoerd Harder
Chemical Society Reviews 2016 vol. 45(Issue 4) pp:1112-1128
Publication Date(Web):30 Nov 2015
DOI:10.1039/C5CS00544B
Metal amidoborane compounds of the alkali- and alkaline earth metals have in recent years found applications in diverse disciplines, notably as hydrogen storage materials, as reagents for the reduction of organic functional groups and as catalysts and intermediates in dehydrocoupling reactions. These functions are connected by the organometallic chemistry of the MNR2BH3 group. This review focusses on central aspects of the s-block amidoborane compounds – their syntheses, structures and reactivity. Well-defined amidoborane complexes of group 2 metals are now available by a variety of solution-phase routes, which has allowed a more detailed analysis of this functional group, which was previously largely confined to solid-state materials chemistry. Structures obtained from X-ray crystallography have begun to provide increased understanding of the fundamental steps of key processes, including amine–borane dehydrocoupling and hydrogen release from primary and secondary amidoboranes. We review structural parameters and reactivity to rationalise the effects of the metal, nitrogen substituents and supporting ligands on catalytic performance and dehydrogenative decomposition routes. Mechanistic features of key processes involving amidoborane compounds as starting materials or intermediates are discussed, alongside emerging applications such as the use of group 1 metal amidoboranes in synthesis. Finally, the future prospects of this vibrant branch of main group chemistry are evaluated.
Co-reporter:Tom E. Stennett, Jürgen Pahl, Harmen S. Zijlstra, Falk W. Seidel, and Sjoerd Harder
Organometallics 2016 Volume 35(Issue 2) pp:207-217
Publication Date(Web):January 8, 2016
DOI:10.1021/acs.organomet.5b00927
The highly Lewis acidic, cationic aluminum species [DIPP-nacnacAlMe]+[B(C6F5)4]− (1, DIPP-nacnac = [HC{C(Me)N(2,6-iPr2C6H3)}2]−) has been shown to undergo reactions with a wide variety of small molecules, in both the presence and absence of an external weak phosphine base, PPh3. Cycloaddition reactions of unsaturated C–C bonds across the aluminum diketiminate framework are reported, and the first structural confirmation of this type of cycloaddition product is presented. Addition of PPh3 to 1 produces the cationic aluminum phosphine complex [DIPP-nacnacAl(Me)PPh3]+[B(C6F5)4]−, which undergoes fluxional dissociation/coordination of the phosphine in solution. This weak Al–P interaction can be utilized in frustrated Lewis pair type reactions to activate alkenes, alkynes, CO2, propylene oxide, and the C–Cl bonds of CH2Cl2. The CO2 adduct [DIPP-nacnacAl(Me)OC(PPh3)O]+[B(C6F5)4]− undergoes further stoichiometric reduction with Et3SiH to produce an aluminum formate species.
Co-reporter:Andrea Causero, Gerd Ballmann, Jürgen Pahl, Harmen Zijlstra, Christian Färber, and Sjoerd Harder
Organometallics 2016 Volume 35(Issue 19) pp:3350-3360
Publication Date(Web):September 25, 2016
DOI:10.1021/acs.organomet.6b00566
A range of symmetric amidinate ligands RAmAr (R is backbone substituent, Ar is N substituent) have been investigated for their ability to stabilize calcium hydride complexes of the type RAmArCaH. It was found that the precursors of the type RAmArCaN(SiMe3)2 are only stable toward ligand exchange for Ar = DIPP (2,6-diisopropylphenyl). The size of the backbone substituent R determines aggregation and solvation. The following complexes could be obtained: [RAmDIPPCaN(SiMe3)2]2 (R = Me, p-Tol), RAmDIPPCaN(SiMe3)2·Et2O (R = Np, tBu), AdAmDIPPCaN(SiMe3)2·THF, and AdAmDIPPCaN(SiMe3)2. Reaction of these heteroleptic calcium amide complexes with PhSiH3 gave only for larger backbone substituents (R = tBu, Ad) access to the dimeric calcium hydride complexes (RAmArCaH)2. (N,aryl)-coordination of the amidinate ligand seems crucial for the stability of these complexes, and the aryl···Ca interaction is found to be strong (17 kcal/mol). Addition of polar solvents led to a new type of trimeric calcium hydride complex exemplified by the crystal structures of (tBuAmDIPPCaH)3·2Et2O and (AdAmDIPPCaH)3·2THF. The overall conclusion of this work is that minor changes in sterics (tBu vs Ad) or coordinated solvent (THF vs Et2O) can have large consequences for product formation and stability.
Co-reporter:Harmen S. Zijlstra
European Journal of Inorganic Chemistry 2015 Volume 2015( Issue 1) pp:19-43
Publication Date(Web):
DOI:10.1002/ejic.201402978
Abstract
Since its discovery, methylalumoxane (MAO) has become of great importance as a cocatalyst in homogeneous olefin polymerization. The working principles of single-site polymerization catalysts are well-understood, but those of the cocatalyst MAO itself are not. Thus far structural and functional investigations have yielded limited insights and often give contradicting results. MAO's complex nature is due to multiple equilibria between undefined oligomers and “free” trimethylaluminum. Fundamental studies do not clearly portray the molecular structure, and the exact functioning of MAO remains a topic of debate. This comprehensive overview starts with the historical background of MAO and then focuses on its synthesis and large-scale production, structural characterization, properties, and different roles in the activation of olefin polymerization catalysts. Also given is an overview of potential modifications, immobilization on surfaces, and other alternative applications that have been reported for MAO.
Co-reporter:Harmen S. Zijlstra
European Journal of Inorganic Chemistry 2015 Volume 2015( Issue 1) pp:
Publication Date(Web):
DOI:10.1002/ejic.201403168
Abstract
Invited for the cover of this issue is the group of Sjoerd Harder at the University of Erlangen-Nürnberg, Germany. The cover image shows the birth of methylalumoxane MeAlO (MAO), or at least one of the possible MAO species, by reaction of trimethylaluminum with water.
Co-reporter:Harmen S. Zijlstra
European Journal of Inorganic Chemistry 2015 Volume 2015( Issue 1) pp:
Publication Date(Web):
DOI:10.1002/ejic.201590000
Co-reporter:Cédric Boulho;Harmen S. Zijlstra
European Journal of Inorganic Chemistry 2015 Volume 2015( Issue 12) pp:2132-2138
Publication Date(Web):
DOI:10.1002/ejic.201500123
Abstract
A bimetallic aluminum/zirconium complex Cp*2Zr(Me)OAl(DIPH) [DIPH-H2 = 3,3′-bis(2-methylallyl)-(1,1′-biphenyl)-2,2′-diol; Cp* = C5Me5] was prepared in good yield by the reaction of (DIPH)AlMe with Cp*2Zr(Me)OH. In contrast to Roesky's catalyst, Cp2Zr(Me)O(Me)Al(DIPP-nacnac) {DIPP-nacnac = CH[(CMe)(2,6-iPr2C6H3N)]2}, it contains no Al–Me functionality and has increased Lewis acidity at Al. Crystal structures and the NMR spectra of the aluminum complex (DIPH)AlMe and the bimetallic Al/Zr species reveal in both cases dimeric complexes. The crystal structure of [Cp*2Zr(Me)OAl(DIPH)]2 shows a planar Al2O2 core and a terminal Cp*2Zr(Me)O group. Ethylene polymerization in toluene was studied for the bimetallic complex with a variety of scavengers. Under comparable polymerization conditions, the methylalumoxane-activated species show high activities that are similar to those obtained with Cp*2ZrCl2 and Roesky's bimetallic complex Cp2Zr(Me)OAl(Me)(DIPP-nacnac), with a relatively narrow polydispersity index (2.47).
Co-reporter:Johanne Penafiel;Dr. Laurent Maron;Dr. Sjoerd Harder
Angewandte Chemie 2015 Volume 127( Issue 1) pp:203-208
Publication Date(Web):
DOI:10.1002/ange.201408814
Abstract
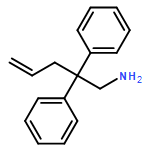
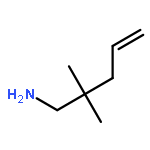
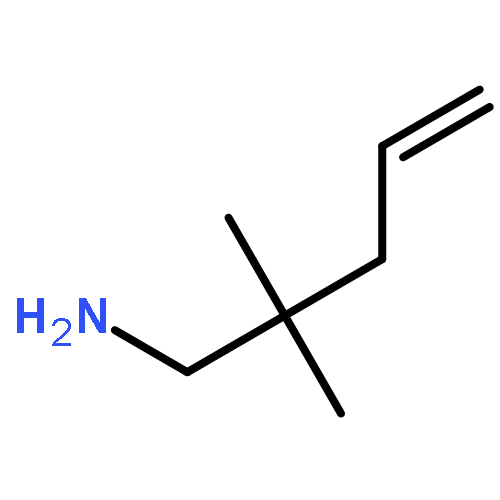
Organocalcium compounds have been reported as efficient catalysts for various alkene transformations. In contrast to transition metal catalysis, the alkenes are not activated by metal–alkene orbital interactions. Instead it is proposed that alkene activation proceeds through an electrostatic interaction with a Lewis acidic Ca2+. The role of the metal was evaluated by a study using the metal-free catalysts: [Ph2N−][Me4N+] and [Ph3C−][Me4N+]. These “naked” amides and carbanions can act as catalysts in the conversion of activated double bonds (CO and CN) in the hydroamination of ArNCO and RNCNR (R=alkyl) by Ph2NH. For the intramolecular hydroamination of unactivated CC bonds in H2CCHCH2CPh2CH2NH2 the presence of a metal cation is crucial. A new type of hybrid catalyst consisting of a strong organic Schwesinger base and a simple metal salt can act as catalyst for the intramolecular alkene hydroamination. The influence of the cation in catalysis is further evaluated by a DFT study.
Co-reporter:Harmen S. Zijlstra, Marc C. A. Stuart, and Sjoerd Harder
Macromolecules 2015 Volume 48(Issue 15) pp:5116-5119
Publication Date(Web):July 28, 2015
DOI:10.1021/acs.macromol.5b00803
Decades after its initial discovery and application as a potent activator for single-site catalysts, methylalumoxane (MAO) still remains a mysterious and poorly understood compound. Using an adapted cryo-TEM setup, for the first time images of dried MAO solutions have been obtained. Particle analysis, size comparison, and decomposition studies yield insights into the structural nature of the observed large MAO particles and can be correlated back to different MAO clusters and their behavior. The studied MAO samples were found to consist of highly aggregated fractals with the radius of the primary particles ranging from ∼10 to 60 nm upon increasing sample age. These findings support a common particle sintering growing mechanism in which primary particles cluster together to form larger secondary particles. A repetition of this process could explain the dramatic change in structure, average molecular weight, and activity of MAO over time.
Co-reporter:Johanne Penafiel;Dr. Laurent Maron;Dr. Sjoerd Harder
Angewandte Chemie International Edition 2015 Volume 54( Issue 1) pp:201-206
Publication Date(Web):
DOI:10.1002/anie.201408814
Abstract
Organocalcium compounds have been reported as efficient catalysts for various alkene transformations. In contrast to transition metal catalysis, the alkenes are not activated by metal–alkene orbital interactions. Instead it is proposed that alkene activation proceeds through an electrostatic interaction with a Lewis acidic Ca2+. The role of the metal was evaluated by a study using the metal-free catalysts: [Ph2N−][Me4N+] and [Ph3C−][Me4N+]. These “naked” amides and carbanions can act as catalysts in the conversion of activated double bonds (CO and CN) in the hydroamination of ArNCO and RNCNR (R=alkyl) by Ph2NH. For the intramolecular hydroamination of unactivated CC bonds in H2CCHCH2CPh2CH2NH2 the presence of a metal cation is crucial. A new type of hybrid catalyst consisting of a strong organic Schwesinger base and a simple metal salt can act as catalyst for the intramolecular alkene hydroamination. The influence of the cation in catalysis is further evaluated by a DFT study.
Co-reporter:Sjoerd Harder, Dominik Naglav, Peter Schwerdtfeger, Israel Nowik, and Rolfe H. Herber
Inorganic Chemistry 2014 Volume 53(Issue 4) pp:2188-2194
Publication Date(Web):February 5, 2014
DOI:10.1021/ic4028546
CpBIG2Sn (CpBIG = (4-n-Bu-C6H4)5cyclopentadienyl), prepared by reaction of 2 equiv of CpBIGNa with SnCl2, crystallized isomorphous to other known metallocenes with this ligand (Ca, Sr, Ba, Sm, Eu, Yb). Similarly, it shows perfect linearity, C–H···C(π) bonding between the CpBIG rings and out-of-plane bending of the aryl substituents toward the metal. Whereas all other CpBIG2M complexes show large disorder in the metal position, the Sn atom in CpBIG2Sn is perfectly ordered. In contrast, 119Sn and 151Eu Mößbauer investigations on the corresponding CpBIG2M metallocenes show that Sn(II) is more dynamic and loosely bound than Eu(II). The large displacement factors in the group 2 and especially in the lanthanide(II) metallocenes CpBIG2M can be explained by static metal disorder in a plane parallel to the CpBIG rings. Despite parallel CpBIG rings, these metallocenes have a nonlinear Cpcenter–M–Cpcenter geometry. This is explained by an ionic model in which metal atoms are polarized by the negatively charged Cp rings. The extent of nonlinearity is in line with trends found in M2+ ion polarizabilities. The range of known calculated dipole polarizabilities at the Douglas–Kroll CCSD(T) level was extended with values (atomic units) for Sn2+ 15.35, Sm2+(4f6 7F) 9.82, Eu2+(4f7 8S) 8.99, and Yb2+(4f14 1S) 6.55. This polarizability model cannot be applied to predominantly covalently bound CpBIG2Sn, which shows a perfectly ordered structure. The bent geometry of Cp*2Sn should therefore not be explained by metal polarizability but is due to van der Waals Cp*···Cp* attraction and (to some extent) to a small p-character component in the Sn lone pair.
Co-reporter:Sjoerd Harder, Jan Spielmann and Julia Intemann
Dalton Transactions 2014 vol. 43(Issue 38) pp:14284-14290
Publication Date(Web):14 Apr 2014
DOI:10.1039/C4DT00835A
Reaction of PYR-(MgnBu)2, in which PYR is 2,6-[(DIPP)NC(Me)CHC(Me)N-]2-pyridine and DIPP is 2,6-iPr2-phenyl, with (DIPP)NH2BH3 gave PYR-[MgNH(DIPP)BH3]2 (56%) which was characterized by crystal structure determination. Addition of THF resulted in β-H elimination and formation of PYR-[MgNH(DIPP)BH3](MgH)·THF (57%), likewise characterized by crystal structure determination. Conversion of the second amidoborane anion in H− could not be achieved. Reaction of PYR-(MgnBu)2 with PhSiH3 gave PYR-(MgH)2, which crystallized as a dimer. The structure of [PYR-(MgH)2]2 shows an 8-membered ring of Mg2+ and H− ions. Thermal decomposition at 130 °C releases one equivalent of H2, i.e. 50% of the expected value. Nucleophilic attack at the para-position and reduction of the pyridylene bridge might explain reduced H2 release.
Co-reporter:Julia Intemann, Martin Lutz, and Sjoerd Harder
Organometallics 2014 Volume 33(Issue 20) pp:5722-5729
Publication Date(Web):June 13, 2014
DOI:10.1021/om500469h
Multinuclear magnesium hydride complexes react with pyridine, forming 1,2- and 1,4-dihydropyridide (DHP) complexes. Reaction of PARA3Mg8H10 with pyridine initially formed 1,2-DHP and 1,4-DHP product mixtures which converted at 60 °C into PARA-[Mg(1,4-DHP)]2·(pyridine)2 (PARA = [(2,6-iPr2C6H3)NC(Me)C(H)C(Me)N]2-(p-C6H4)). Reaction of [NN-(MgH)2]2 with pyridine gave exclusive formation of the 1,2-DHP product NN-[Mg(1,2-DHP)]2·(pyridine)2 (NN = [(2,6-iPr2C6H3)NC(Me)CHC(Me)N−]2). Both products were characterized by crystal structure determinations. The unusual preference for 1,2-addition is likely caused by secondary intramolecular interactions based on mutual communication between the metal coordination geometries: an extended network of C–H···C π-interactions and π-stacking interactions is found. Whereas PARA3Mg8H10 is hardly active in magnesium-catalyzed hydroboration of pyridines with pinacolborane, [NN-(MgH)2]2 shows efficient coupling. However, the regioselectivity of the stoichiometric reaction is not translated to the catalytic regime. This result is taken as an indication for a potential alternative mechanism in which magnesium hydride intermediates do not play a role but the hydride is transferred from an intermediate borate complex.
Co-reporter:Dr. Julia Intemann;Dr. Peter Sirsch; Dr. Sjoerd Harder
Chemistry - A European Journal 2014 Volume 20( Issue 35) pp:11204-11213
Publication Date(Web):
DOI:10.1002/chem.201402141
Abstract
In analogy to the previously reported tetranuclear magnesium hydride cluster with a bridged dianionic bis-β-diketiminate ligand, a related zinc hydride cluster has been prepared. The crystal structures of these magnesium and zinc hydride complexes are similar: the metal atoms are situated at the corners of a tetrahedron in which the vertices are bridged either by dianionic bis-β-diketiminate ligands or hydride ions. Both structures are retained in solution and show examples of H−⋅⋅⋅H− NMR coupling (Mg: 8.5 Hz; Zn: 16.0 Hz). The zinc hydride cluster [NN-(ZnH)2]2 thermally decomposes at 90 °C and releases 1.8 equivalents of H2. In contrast to magnesium hydride clusters, there is no apparent relationship between cluster size and thermal decomposition temperature for the zinc hydrides. DFT calculations reproduced the structure of the zinc hydride cluster reasonably well and charge density analysis showed no bond paths between the hydride ions. This contrasts with calculations on the analogous magnesium hydride cluster in which a counter-intuitive H−⋅⋅⋅H− bond path was observed. Forcing a reduced H−⋅⋅⋅H− distance in the zinc hydride cluster, however, gave rise to a H−⋅⋅⋅H− bond path. Such weak interactions could play a role in H2 desorption. The presumed molecular product after H2 release, a Zn(I) cluster, could not be characterized experimentally but DFT calculations predicted a cluster with two localized ZnZn bonds.
Co-reporter:Dr. Sjoerd Harder;Dominik Naglav;Dr. Christian Ruspic;Dr. Claudia Wickleder;Dr. Matthias Adlung;Dr. Wilfried Hermes;Dr. Matthias Eul;Dr. Rainer Pöttgen;Dr. Daniel B. Rego;Dr. Frederic Poineau;Dr. Kenneth R. Czerwinski;Dr. Rolfe H. Herber;Dr. Israel Nowik
Chemistry - A European Journal 2013 Volume 19( Issue 37) pp:12272-12280
Publication Date(Web):
DOI:10.1002/chem.201302021
Abstract
The superbulky deca-aryleuropocene [Eu(CpBIG)2], CpBIG=(4-nBu-C6H4)5-cyclopentadienyl, was prepared by reaction of [Eu(dmat)2(thf)2], DMAT=2-Me2N-α-Me3Si-benzyl, with two equivalents of CpBIGH. Recrystallizyation from cold hexane gave the product with a surprisingly bright and efficient orange emission (45 % quantum yield). The crystal structure is isomorphic to those of [M(CpBIG)2] (M=Sm, Yb, Ca, Ba) and shows the typical distortions that arise from CpBIG⋅⋅⋅CpBIG attraction as well as excessively large displacement parameter for the heavy Eu atom (Ueq=0.075). In order to gain information on the true oxidation state of the central metal in superbulky metallocenes [M(CpBIG)2] (M=Sm, Eu, Yb), several physical analyses have been applied. Temperature-dependent magnetic susceptibility data of [Yb(CpBIG)2] show diamagnetism, indicating stable divalent ytterbium. Temperature-dependent 151Eu Mössbauer effect spectroscopic examination of [Eu(CpBIG)2] was examined over the temperature range 93–215 K and the hyperfine and dynamical properties of the EuII species are discussed in detail. The mean square amplitude of vibration of the Eu atom as a function of temperature was determined and compared to the value extracted from the single-crystal X-ray data at 203 K. The large difference in these two values was ascribed to the presence of static disorder and/or the presence of low-frequency torsional and librational modes in [Eu(CpBIG)2]. X-ray absorbance near edge spectroscopy (XANES) showed that all three [Ln(CpBIG)2] (Ln=Sm, Eu, Yb) compounds are divalent. The XANES white-line spectra are at 8.3, 7.3, and 7.8 eV, for Sm, Eu, and Yb, respectively, lower than the Ln2O3 standards. No XANES temperature dependence was found from room temperature to 100 K. XANES also showed that the [Ln(CpBIG)2] complexes had less trivalent impurity than a [EuI2(thf)x] standard. The complex [Eu(CpBIG)2] shows already at room temperature strong orange photoluminescence (quantum yield: 45 %): excitation at 412 nm (24270 cm−1) gives a symmetrical single band in the emission spectrum at 606 nm (νmax=16495 cm−1, FWHM: 2090 cm−1, Stokes-shift: 2140 cm−1), which is assigned to a 4f65d14f7 transition of EuII. These remarkable values compare well to those for EuII-doped ionic host lattices and are likely caused by the rigidity of the [Eu(CpBIG)2] complex. Sharp emission signals, typical for EuIII, are not visible.
Co-reporter:Dr. Sjoerd Harder;Dominik Naglav;Dr. Christian Ruspic;Dr. Claudia Wickleder;Dr. Matthias Adlung;Dr. Wilfried Hermes;Dr. Matthias Eul;Dr. Rainer Pöttgen;Dr. Daniel B. Rego;Dr. Frederic Poineau;Dr. Kenneth R. Czerwinski;Dr. Rolfe H. Herber;Dr. Israel Nowik
Chemistry - A European Journal 2013 Volume 19( Issue 37) pp:
Publication Date(Web):
DOI:10.1002/chem.201390139
Co-reporter:Tom E. Stennett and Sjoerd Harder
Chemical Society Reviews 2016 - vol. 45(Issue 4) pp:NaN1128-1128
Publication Date(Web):2015/11/30
DOI:10.1039/C5CS00544B
Metal amidoborane compounds of the alkali- and alkaline earth metals have in recent years found applications in diverse disciplines, notably as hydrogen storage materials, as reagents for the reduction of organic functional groups and as catalysts and intermediates in dehydrocoupling reactions. These functions are connected by the organometallic chemistry of the MNR2BH3 group. This review focusses on central aspects of the s-block amidoborane compounds – their syntheses, structures and reactivity. Well-defined amidoborane complexes of group 2 metals are now available by a variety of solution-phase routes, which has allowed a more detailed analysis of this functional group, which was previously largely confined to solid-state materials chemistry. Structures obtained from X-ray crystallography have begun to provide increased understanding of the fundamental steps of key processes, including amine–borane dehydrocoupling and hydrogen release from primary and secondary amidoboranes. We review structural parameters and reactivity to rationalise the effects of the metal, nitrogen substituents and supporting ligands on catalytic performance and dehydrogenative decomposition routes. Mechanistic features of key processes involving amidoborane compounds as starting materials or intermediates are discussed, alongside emerging applications such as the use of group 1 metal amidoboranes in synthesis. Finally, the future prospects of this vibrant branch of main group chemistry are evaluated.
Co-reporter:Sjoerd Harder, Jan Spielmann and Julia Intemann
Dalton Transactions 2014 - vol. 43(Issue 38) pp:NaN14290-14290
Publication Date(Web):2014/04/14
DOI:10.1039/C4DT00835A
Reaction of PYR-(MgnBu)2, in which PYR is 2,6-[(DIPP)NC(Me)CHC(Me)N-]2-pyridine and DIPP is 2,6-iPr2-phenyl, with (DIPP)NH2BH3 gave PYR-[MgNH(DIPP)BH3]2 (56%) which was characterized by crystal structure determination. Addition of THF resulted in β-H elimination and formation of PYR-[MgNH(DIPP)BH3](MgH)·THF (57%), likewise characterized by crystal structure determination. Conversion of the second amidoborane anion in H− could not be achieved. Reaction of PYR-(MgnBu)2 with PhSiH3 gave PYR-(MgH)2, which crystallized as a dimer. The structure of [PYR-(MgH)2]2 shows an 8-membered ring of Mg2+ and H− ions. Thermal decomposition at 130 °C releases one equivalent of H2, i.e. 50% of the expected value. Nucleophilic attack at the para-position and reduction of the pyridylene bridge might explain reduced H2 release.
Co-reporter:Harmen S. Zijlstra, Jürgen Pahl, Johanne Penafiel and Sjoerd Harder
Dalton Transactions 2017 - vol. 46(Issue 11) pp:NaN3610-3610
Publication Date(Web):2017/02/28
DOI:10.1039/C7DT00318H
The reaction of Ph2P(DIPP)NH with AlMe3 cleanly gives an aluminum amide complex that crystallizes as a centrosymmetric dimer with a six-membered Al–N–P–Al–N–P ring. In aromatic solvents the dimer remains intact but the Al–P bond is readily broken upon addition of THF to form Ph2P(DIPP)NAlMe2·THF. Efforts to use [Ph2P(DIPP)NAlMe2]2 as a “masked” Lewis acidic activator for olefin polymerization catalysts were unsuccessful but the complex showed a Frustrated Lewis pair reactivity instead. The P/Al complex reacts with isocyanates to give the CO inserted product that crystallizes as a five-membered ring system Al–O–C(NR)–P–N. The reaction of [Ph2P(DIPP)NAlMe2]2 with CO2, however, gave an insertion in the N–Al bond and the dimeric product [Ph2P(DIPP)NCO2AlMe2]2 was isolated. The dimer [Ph2P(DIPP)NAlMe2]2 is one of the few Al/P FLPs that can activate CC double bonds irreversibly. A reaction with allyl methyl sulfide and 1-hexene led to the clean formation of the structurally similar activated alkene products [(DIPP)N-Ph2P-CH(CH2SMe)CH2]AlMe2 and [(DIPP)N-Ph2P-CH(C4H9)CH2]AlMe2.
Co-reporter:Andrea Causero, Gerd Ballmann, Jürgen Pahl, Christian Färber, Julia Intemann and Sjoerd Harder
Dalton Transactions 2017 - vol. 46(Issue 6) pp:NaN1831-1831
Publication Date(Web):2017/01/23
DOI:10.1039/C6DT04659B
A series of (DIPPnacnac)CaN(SiMe3)2·S complexes (DIPPnacnac = HC[C(Me)N(2,6-iPr-C6H3)]2; S = solvent) could be obtained by the addition of S = THF, DME or N-Me-morpholine (Morph) to (DIPPnacnac)CaN(SiMe3)2·OEt2 or (DIPPnacnac)CaN(SiMe3)2. Crystal structures for complexes with S = DME and Morph are compared to literature-known structures with S = none, THF or Et2O. Bulkier and weaker Lewis bases like the tertiary amines Et3N, TMEDA and DABCO did not interact with (DIPPnacnac)CaN(SiMe3)2. The reaction of (DIPPnacnac)CaN(SiMe3)2 with PhSiH3 gave conversion to a calcium hydride complex that dismutated in (DIPPnacnac)2Ca and CaH2. The reaction of (DIPPnacnac)CaN(SiMe3)2·S with PhSiH3 gave [(DIPPnacnac)CaH·S]2 for S = THF, Et2O or N-Me-morpholine (Morph). For S = DME, high reaction temperatures were needed and dismutation into (DIPPnacnac)2Ca and CaH2 was observed. Extensive NMR investigations (VT-NMR and PGSE) confirm the dimeric nature of [(DIPPnacnac)CaH·THF]2 in aromatic solvents or in THF. Thermal decomposition of [(DIPPnacnac)CaH·THF]2 (release of H2 at 200 °C) is compared to that of Mg and Zn analogues. Weakly coordinating Et2O in [(DIPPnacnac)CaH·OEt2]2 could be replaced by THF, Morph or DABCO but not with Et3N. The addition of TMEDA led to the formation of CaH2 and unidentified products. The addition of DME led to the decomposition of Et2O and complex [(DIPPnacnac)CaOEt]2 was obtained. Crystal structures of the following compounds are presented: (DIPPnacnac)CaN(SiMe3)2·S (S = Morph, DME), [(DIPPnacnac)CaH·S]2 (S = Et2O, Morph and DABCO) and [(DIPPnacnac)CaOEt]2. Although bulky ligands have long been thought to be the key to the stabilization of calcium hydride complexes, the presence of a polar, strongly coordinating, co-solvent is also crucial.

![Propanimidamide, N,N'-bis[2,6-bis(1-methylethyl)phenyl]-2,2-dimethyl-](http://img.cochemist.com/ccimg/865300/865200-87-9.png)
![Propanimidamide, N,N'-bis[2,6-bis(1-methylethyl)phenyl]-2,2-dimethyl-](http://img.cochemist.com/ccimg/865300/865200-87-9_b.png)



![[1,1'-Biphenyl]-2,2'-diol, 3,3'-bis(2-methyl-2-propenyl)-](http://img.cochemist.com/ccimg/761500/761447-78-3.png)
![[1,1'-Biphenyl]-2,2'-diol, 3,3'-bis(2-methyl-2-propenyl)-](http://img.cochemist.com/ccimg/761500/761447-78-3_b.png)
![ETHANIMIDAMIDE, N,N'-BIS[2,6-BIS(1-METHYLETHYL)PHENYL]-](http://img.cochemist.com/ccimg/582400/582334-08-5.png)
![ETHANIMIDAMIDE, N,N'-BIS[2,6-BIS(1-METHYLETHYL)PHENYL]-](http://img.cochemist.com/ccimg/582400/582334-08-5_b.png)


![Lithium, [2,6-bis(1-methylethoxy)phenyl]-](http://img.cochemist.com/ccimg/215900/215811-03-3.png)
![Lithium, [2,6-bis(1-methylethoxy)phenyl]-](http://img.cochemist.com/ccimg/215900/215811-03-3_b.png)




![Sodium, [1,2,4-tris(1,1-dimethylethyl)-2,4-cyclopentadien-1-yl]-](http://img.cochemist.com/ccimg/158900/158897-87-1.png)
![Sodium, [1,2,4-tris(1,1-dimethylethyl)-2,4-cyclopentadien-1-yl]-](http://img.cochemist.com/ccimg/158900/158897-87-1_b.png)
![Sodium, [1,3-bis(trimethylsilyl)-2,4-cyclopentadien-1-yl]-](http://img.cochemist.com/ccimg/145400/145332-76-9.png)
![Sodium, [1,3-bis(trimethylsilyl)-2,4-cyclopentadien-1-yl]-](http://img.cochemist.com/ccimg/145400/145332-76-9_b.png)






![Sodium, [1,2,3,4-tetrakis(1-methylethyl)-2,4-cyclopentadien-1-yl]-](http://img.cochemist.com/ccimg/123300/123278-30-8.png)
![Sodium, [1,2,3,4-tetrakis(1-methylethyl)-2,4-cyclopentadien-1-yl]-](http://img.cochemist.com/ccimg/123300/123278-30-8_b.png)




![Tricyclo[3.3.1.13,7]decanecarbonyl chloride](http://img.cochemist.com/ccimg/63100/63023-16-5.png)
![Tricyclo[3.3.1.13,7]decanecarbonyl chloride](http://img.cochemist.com/ccimg/63100/63023-16-5_b.png)