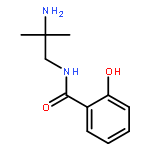
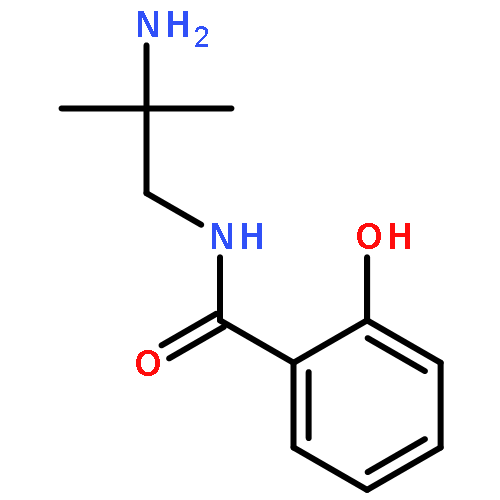
Co-reporter:Ryoji Mitsuhashi, Rina Ogawa, Ryuta Ishikawa, Takayoshi Suzuki, Yukinari Sunatsuki, Satoshi Kawata
Inorganica Chimica Acta 2016 Volume 447() pp:113-120
Publication Date(Web):1 June 2016
DOI:10.1016/j.ica.2016.03.036
•Dinuclear methoxido-bridged MnIII complexes having amine–amidato–phenolato type ligands were characterized.•A tetranuclear methoxido-bridged MnII2MnIII2 complex was determined by X-ray analysis.•Magnetic behavior was changed drastically when the crystals of the Mn/HL3 complex were efflorescent.Complexation of 2-hydroxy-N-(n-aminoalkyl)benzamides, where n-aminoalkyl substituents are 2-amino-2-methylpropyl (H2L2Me2), (R)-2-aminopropyl {(R)-H2L2Me} and 3-aminopropyl (H2L3), with manganese(II) chloride or perchlorate were examined. With the 2-aminopropyl derivatives, dinuclear methoxido-bridged manganese(III) complexes bearing the dianionic ligands, [{Mn(L2Me2 or (R)-L2Me)(MeOH)}2(μ-OMe)2] (2 or 4), were isolated and their crystal structures were determined. There was a weak antiferromagnetic interaction between two axially distorted MnIII centers in 2. Also, with the ligand of (L2Me2)2− a mononuclear manganese(IV) complex, [Mn(L2Me2)2]·DMF (3·DMF) was obtained. In the crystal of 3·DMF a heterochiral dimerization via the intermolecular double hydrogen-bonds was observed. In the case of reactions with H2L3, mononuclear manganese(III) complexes, [Mn(H2L3′)2Cl2]Cl (6) and [Mn(H2L3′)2(MeOH)2]Cl2(ClO4) (7), were afforded, where the ligand (H2L3′) has a phenolate–amide–ammonium type zwitterionic form of H2L3 and coordinates to a MnIII center via the phenolato-O and amide-O atoms to form a six-membered chelate ring. Furthermore, a tetranuclear MnII2MnIII2 double incomplete-cubane type cluster compound, [Mn4(HL3)2Cl2(OMe)6(MeOH)2]·4MeOH (5a·4MeOH) was revealed by the single-crystal X-ray diffraction study, although the bulk crystals (5) obtained from the reaction mixture showed a different PXRD pattern. The elemental analysis of the bulk crystals of 5 suggested that they consist of the same tetranuclear clusters. It is interesting that the bulk crystals of 5 were highly efflorescent, and it was suggested that their coordinated MeO− and MeOH ligands were easily substituted by OH− and H2O, respectively, in atmospheric air. This conversion changed their magnetic behaviors drastically.Complexation of 2-hydroxy-N-(2-amino-2-methylpropyl or 3-aminopropyl)benzamides with manganese(II) chloride or perchlorate were examined, and dinuclear or tetranuclear methoxido-bridged complexes bearing amine–(amidato or amido)–phenolato type ligands were characterized by X-ray analysis.
Co-reporter:Mika Sakate, Ayana Kashima, Haruka Hosoda, Yukinari Sunatsuki, Hiromi Ota, Akira Fuyuhiro, Takayoshi Suzuki
Inorganica Chimica Acta 2016 Volume 452() pp:205-213
Publication Date(Web):1 October 2016
DOI:10.1016/j.ica.2016.01.023
•The reactions of [(Cp∗Rh)2(μ-OH)3]OH and thymine with template salts were examined.•Structures of tetranuclear RhIII complexes bridged by thyminato(2-) are reported.•The thym2− ligand exhibited two kinds of bridging modes.Reactions of [(Cp∗Rh)2(μ-OH)3]OH (Cp∗ = η5-C5Me5−) and thymine (=H2thym) in methanol in the presence or absence of a certain metal salt were examined. From the reaction mixture without any template metal salt, a dinuclear hydroxido-bridged complex bearing monodentate monodeprotonated thyminato ligands, [{Cp∗Rh(Hthym-κN1)}2(μ-OH)2], was isolated after recrystallization from water. In the presence of a metal salt MXn {=NaPF6, NaBF4, NaNO3, Ca(NO3)2, Ca(ClO4)2, La(NO3)3, Eu(NO3)3, Dy(NO3)3 and Er(NO3)3} were deposited orange crystals (1P, 1B, 1N, 2N, 2C, 3N, 4N, 5N and 6N, respectively) which consist of a thyminato(2-)-bridged tetranuclear Cp∗Rh complex incorporating a Mn+ cation, [(Cp∗Rh)4(μ-thym)4M]n+. All complexes were characterized by the single-crystal X-ray diffraction method, which revealed that homochiral aggregations of four RhIII centers were achieved to give metallacalix[4]arene-type clusters. It was also observed that the bridging modes of thym2− ligands were dependent on the template metal ion. In the Na+-incorporated clusters (1X, X = P, B or N) a thym2− ligand bridged two RhIII and the third Na+ ions with a μ3-1κN1:2κ2N3,O2:3κO2 mode, while in the Ln3+ analogs (Ln = La, Eu, Dy or Er: 3N–6N) thym2− exhibited a different bridging mode, μ3-1κN1:2κ2N3,O4:3κO2. In the cases of Ca2+-incorporated clusters, the bridging mode is further dependent on the associated anion; only the μ3-1κN1:2κ2N3,O2:3κO2-type cluster was obtained in the NO3− salt (2N), but the ClO4− salt (2C) afforded both types of the bridging clusters piled up alternatively in the crystals. The tetranuclear structures with a template metal cation, [(Cp∗Rh)4(μ-thym)4M]n+, are maintained in solution. The magnetic behaviors of the Dy3+- and Er3+-incorporated complexes (5N and 6N) are also reported.In the cyclic tetranuclear Cp∗RhIII complexes bridged by thyminato (thym2−) ligands, two bridging modes of thym2− were observed, dependent on the metal ion used as a template: μ3-1κN1:2κ2N3,O2:3κO2 for Na+ and μ3-1κN1:2κ2N3,O4:3κO2 for Ln3+. In the case of a Ca2+-template synthesis both bridging isomers are piled up alternatively in the crystal.
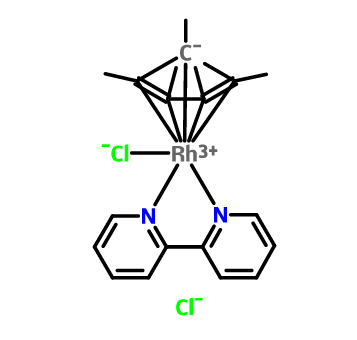
Co-reporter:Ayana Kashima, Mika Sakate, Hiromi Ota, Akira Fuyuhiro, Yukinari Sunatsuki and Takayoshi Suzuki
Chemical Communications 2015 vol. 51(Issue 10) pp:1889-1892
Publication Date(Web):11 Dec 2014
DOI:10.1039/C4CC09087J
In the thyminate(2−)-bridged tetranuclear Cp*RhIII complexes incorporating a Na+, Ca2+ or Ln3+ cation, homochiral aggregation of four RhIII centres was achieved to form metallacalix[4]arene-type clusters. The thyminate(2−) bridged two RhIII and the third metal ion with a μ3-1κN1:2κ2N3,O2:3κO2 mode in the Na+ and Ca2+ complexes, while in the Ln3+ analogues it exhibited a different bridging mode, μ3-1κN1:2κ2N3,O4:3κO2.
Co-reporter:Atsutoshi Yamada, Takuya Mabe, Ryohei Yamane, Kyoko Noda, Yuko Wasada, Masahiko Inamo, Koji Ishihara, Takayoshi Suzuki and Hideo D. Takagi
Dalton Transactions 2015 vol. 44(Issue 31) pp:13979-13990
Publication Date(Web):22 Jun 2015
DOI:10.1039/C5DT01808K
Six-coordinate [Cu(pdt)2(H2O)2]2+ and four-coordinate [Cu(pdt)2]+ complexes were synthesized and the cross redox reactions were studied in acetonitrile (pdt = 3-(2-pyridyl)-5,6-diphenyl-1,2,4-triazine). Single crystal analyses revealed that [Cu(pdt)2(H2O)2](BF4)2 was of pseudo-D2h symmetry with two axial water molecules and two symmetrically coordinated equatorial pdt ligands, while the coordination structure of [Cu(pdt)2]BF4 was a squashed tetrahedron (dihedral angle = 54.87°) with an asymmetric coordination by two pdt ligands: one pdt ligand was coordinated to Cu(I) through pyridine-N and triazine-N2 while another pdt ligand was coordinated through pyridine-N and triazine-N4, and a stacking interaction between the phenyl ring on one pdt ligand and the triazine ring on another pdt ligand caused the squashed structure and non-equivalent Cu–N bond lengths. The cyclic voltammograms for [Cu(pdt)2(H2O)2]2+ and [Cu(pdt)2]+ in acetonitrile were identical to each other and quasi-reversible. The reduction of [Cu(pdt)2(H2O)2]2+ by decamethylferrocene and the oxidation of [Cu(pdt)2]+ by [Co(2,2′-bipyridine)3]3+ in acetonitrile revealed that both cross reactions were sluggish through a gated process (the structural change took place prior to the electron transfer) accompanied by slow direct electron transfer processes. It was found that the triazine ring of the coordinated pdt ligand rotates around the C–C bond between the triazine and pyridine rings with the kinetic parameters k = 51 ± 5 s−1 (297.8 K), ΔH‡ = 6.2 ± 1.1 kJ mol−1 and ΔS‡ = −192 ± 4 J mol−1 K−1. The electron self-exchange process was directly measured using the line-broadening method: kex = (9.9 ± 0.5) × 104 kg mol−1 s−1 (297.8 K) with ΔH‡ = 44 ± 7 kJ mol−1 and ΔS‡ = 0.2 ± 2.6 J mol−1 K−1. By comparing this rate constant with the self-exchange rate constants estimated from the cross reactions using the Marcus cross relation, the non-adiabaticity (electronic) factors, κel, for the direct electron transfer processes between [Cu(pdt)2]+/2+ and non-copper metal (Fe2+ and Co3+) complexes were estimated as ca. 10−7, indicating that the electronic coupling between the d orbitals of copper and of non-copper metals is very small.
Co-reporter:Asami Mori, Takayoshi Suzuki, Yuichi Nakatani, Yukinari Sunatsuki, Masaaki Kojima and Kiyohiko Nakajima
Dalton Transactions 2015 vol. 44(Issue 36) pp:15757-15760
Publication Date(Web):31 Jul 2015
DOI:10.1039/C5DT02345A
A reaction of [PdCl2(cod)] (cod = 1,5-cyclooctadiene) and an E/Z mixture of quinoline-2-carbaldehyde (pyridine-2-carbonyl)hydrazone (HL) gave two kinds of PdII mononuclear complexes, [PdCl(Z-L-κ3N,N′,N′′)] (1) and [PdCl2(E-HL′-κ2N,N′)] (2), where L− is the deprotonated hydrazonate anion and HL′ is the quinolinium–hydrazonate zwitterionic form of HL. Complex 2 is gradually converted to 1 in solution, and complex 1 is a good precursor to prepare a PdII/RuII heterodinuclear complex bridged by hydrazonate, trans(Cl,Cl)-[RuCl2(PPh3)2(μ-L)PdCl] (3).
Co-reporter:Keiko Kihara, Syohei Yamaguchi, Yasuo Kawahata, Masakazu Kita, Takayoshi Suzuki, Yukinari Sunatsuki, Masaaki Kojima, Kazuo Kashiwabara
Polyhedron 2014 Volume 67() pp:111-114
Publication Date(Web):8 January 2014
DOI:10.1016/j.poly.2013.09.001
A series of cobalt(III) complexes containing 2-cyanoethylphosphines, [Co(acac)2{P(CH2CH2CN)nPh3–n}2]BF4 {acac = 2,4-pentanedionate; n = 1 (1), 2 (2), and 3 (3)} have been prepared, and their molecular and crystal structures have been determined. The X-ray diffraction study revealed that these complexes are the trans isomers in the crystals. The Co–P bond lengths are little affected by the number of 2-cyanoethyl substituents on the P atom, which is consistent with a small change of the σ-donicity among these phosphines. The trans configuration of these complexes is maintained in solution, as revealed by 1H NMR spectroscopy. The 2-cyanoethylphosphines could not form stable cis-isomers of [Co(acac)2{P(CH2CH2CN)nPh3–n}2]+, like the corresponding methylphosphines (PMenPh3–n). This fact probably results from a strong electron-withdrawing property of a terminal cyano group of phosphine substituent. In the UV–Vis absorption spectra of complexes 1–3, the LMCT transition band is observed in the range of 20 500–24 000 cm−1, which is comparable to the analogous PMenPh3–n complexes, but their intensity is characteristically weaker than that of the corresponding PMenPh3–n complexes.A series of bis(2,4-pentanedionato)cobalt(III) complexes containing 2-cyanoethylphosphines, trans-[Co(acac)2{P(CH2CH2CN)nPh3–n}2](BF4 or BPh4) (n = 1, 2 or 3) have been prepared, and their crystal structures and spectroscopic properties have been determined. In accord with the weak σ-donor capability of 2-cyanoethylphosphines, the intensity of the lowest energy LMCT transition band of their complexes decreased remarkably, as compared to that of the corresponding PPh3 complex.
Co-reporter:Kyohei Sakano, Syohei Yamaguchi, Takayoshi Suzuki, Yukinari Sunatsuki, Masaaki Kojima, Kazuo Kashiwabara
Inorganica Chimica Acta 2014 410() pp: 122-125
Publication Date(Web):
DOI:10.1016/j.ica.2013.10.028
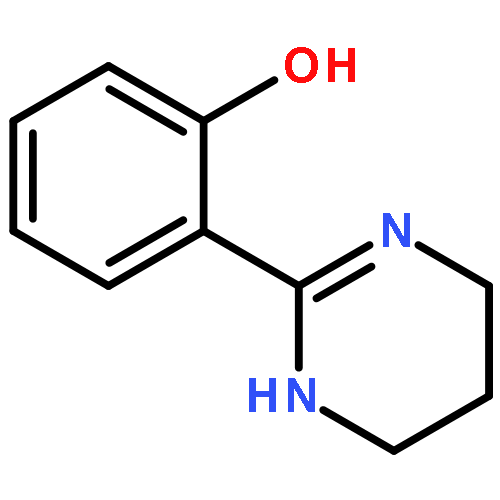
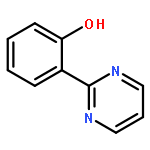
Co-reporter:Ryoji Mitsuhashi, Takayoshi Suzuki, and Yukinari Sunatsuki
Inorganic Chemistry 2013 Volume 52(Issue 17) pp:10183-10190
Publication Date(Web):August 22, 2013
DOI:10.1021/ic401667v
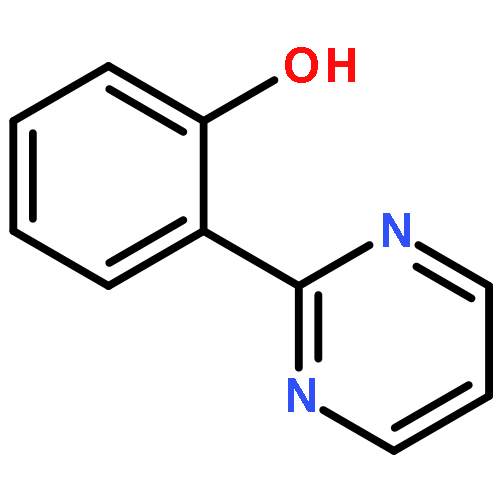
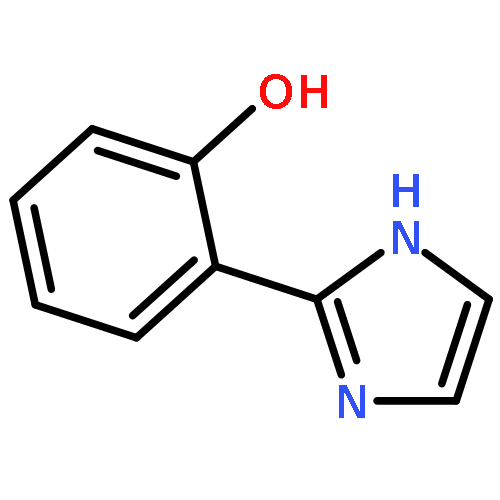
New ruthenium(II or III) complexes with general formula [Ru(O-N)(bpy)2]n+ (O-N = unsymmetrical bidentate phenolate ligand; bpy = 2,2′-bipyridine) were synthesized, and their crystal structures and electrochemical properties were characterized. RuII complexes with 2-(2-imidazolinyl)phenolate (Himn–) or 2-(1,4,5,6-tetrahydropyrimidin-2-yl)phenolate (Hthp–) could be deprotonated by addition of excess KOtBu, although the deprotonated species were easily reprotonated by exposure to air. Unlike these RuII complexes, their RuIII analogs showed interesting ligand oxidation reactions upon addition of bases. With [RuIII(Himn)(bpy)2]2+, two-electron oxidation of Himn– and reduction of the RuIII center resulted in conversion of the 2-imidazolinyl group to a 2-imidazolyl group. On the other hand, the corresponding Hthp– complex exhibited four-electron oxidation of the ligand to form 2-(2-pyrimidyl)phenolate (pym–). These aromatization reactions of imidazolinyl and 1,4,5,6-tetrahydropyrimidyl groups were also achieved by the electrochemically generated RuIII complexes.
Co-reporter:Asuka Takayama, Takayoshi Suzuki, Miyu Ikeda, Yukinari Sunatsuki and Masaaki Kojima
Dalton Transactions 2013 vol. 42(Issue 40) pp:14556-14567
Publication Date(Web):01 Aug 2013
DOI:10.1039/C3DT51667A
Several new iridium(III) and rhodium(III) complexes bearing 5-methyltetrazolate (MeCN4−) have been prepared, and their structures in the crystals and in solution have been determined by X-ray analysis and by NMR spectroscopy, respectively. In the crystals of the mononuclear complexes, κN2-coordination of MeCN4− was observed when the ancillary ligand was 2,2′-bipyridine (bpy): [Cp*M(bpy)(MeCN4-κN2)]PF6 (Cp* = η5-C5Me5; M = Ir: 1 and Rh: 2), while the corresponding complexes with 2-(2′-pyridyl)phenyl (ppy−) were confirmed to have the κN1-coordination of MeCN4−: [Cp*M(ppy)(MeCN4-κN1)] (M = Ir: 3 and Rh: 4). In solution, the IrIII complexes (1 and 3) were robust enough to maintain their molecular structures, but the RhIII complexes (2 and 4) existed as an equilibrium mixture of the κN1- and κN2-isomers. In addition to the IrIII–IrIII and RhIII–RhIII homodinuclear complexes bridged by MeCN4− (5–8), the corresponding heterodinuclear IrIII–RhIII complexes (9–12) were prepared using the mononuclear IrIII complexes (1 and 3) as precursors. The molecular structures of these dinuclear complexes were also characterised. Interestingly, both of the heterodinuclear complexes comprised of Cp*M(bpy)2+ and Cp*M′(ppy)+ fragments, [Cp*M(bpy)(μ-MeCN4)M′(ppy)Cp*](PF6)2 (M = Ir, M′ = Rh: 10 and M = Rh, M′ = Ir: 11), exhibited selective crystallisation of a specific μ-κN1(M′-ppy):κN3(M-bpy) isomer. In solution, the dinuclear complexes with a Rh–N(MeCN4) bond and more than one positive charge (6 and 9–11) showed a dissociation equilibrium, while monocationic MeCN4-bridged complexes (7, 8 and 12) were inactive for dissociation. Furthermore, the heterodinuclear complexes of 9–12, as well as the RhIII–RhIII complex of 8, exhibited a bridging isomerisation, which would proceed via a η2:η2-intermediate without dissociation of Cp*M(bpy or ppy) fragments.
Co-reporter:Ryoji Mitsuhashi, Takayoshi Suzuki, Yukinari Sunatsuki, Masaaki Kojima
Inorganica Chimica Acta 2013 Volume 399() pp:131-137
Publication Date(Web):1 April 2013
DOI:10.1016/j.ica.2013.01.011
Four cobalt(III) complexes with the formula of [Co(Ln)2]− bearing tridentate amine-amidato-phenolato-type ligands (Ln: n = 1–4) were synthesized. All of the complexes were characterized by 1H NMR spectroscopy and X-ray analysis. The geometrical selectivity was found to depend on the flexibility of the amine-amidato chelate in combination with the planar 2-oxybenzamido 6-membered chelate; that is, the amine-amidato 5-membered chelate took the mer-type geometry, and the 6-membered chelate took the fac-type geometry. In most of the mer-type complexes, intermolecular double hydrogen bonds via amidato(O) and amino group were selectively formed between their enantiomeric pairs of mononuclear complexes. In the case of two chiral ligands {L22– = 2-amino-1-(2-oxybenzamido)propane; L32− = trans-1-amino-2-(2-oxybenzamido)cyclohexane}, [Co(L3)2]− showed diastereoselectivity while [Co(L2)2]− did not. Furthermore, PPh4[Co(L1)2] (L12− = 2-amino-1-(2-oxybenzamido)-2-methylpropane) showed an apparent solvatochromic behavior in several solvents. Although the molecular structures of [Co(L2 or L3)2]− are quite similar to that of [Co(L1)2]−, these complexes did not exhibit such a solvatochromic behavior.Graphical abstractFour cobalt(III) complexes with the formula of [Co(Ln)2]− bearing tridentate amine-amidato-phenolato-type ligands (Ln: n = 1–4) were synthesized. One of the complexes having a trans-cyclohexylene backbone, mer-[Co(L3)2]− {L32– = trans-1-amino-2-(2-oxybenzamido)cyclohexane}, afforded violet crystals consisting of a hydrogen-bonded dimer of [Co(RR-L3)(SS-L3)]−; however, in solution the complex slowly isomerized to the corresponding C2-symmetrical species ([Co(RR-L3)2]− and [Co(SS-L3)2]−). The preferential formation of the heterochiral isomer in the crystals would result from the intermolecular double hydrogen bonds between the amino and amidato groups.Highlights► Four cobalt(III) complexes bearing tridentate amine-amidato-phenolato-type ligands were synthesized. ► The geometrical selectivity of the complexes was examined. ► The effect of the hydrogen-bonding interactions on diastereoselectivity was studied. ► An apparent solvatochromic behavior was observed for one of the complexes.
Co-reporter:Jan G. Małecki, Anna Maroń, Iza Gryca, Asami Mori, Takayoshi Suzuki
Polyhedron 2013 Volume 62() pp:188-202
Publication Date(Web):7 October 2013
DOI:10.1016/j.poly.2013.06.042
Ruthenium(II) complexes with the general formulas [Ru(H/Cl)(CO)(PPh3)2(L)] and cis/trans-[Ru(PPh3)2(L)2] (L = isoquinoline-1-carboxylate or quinoline-2-carboxylate) were synthesized and characterized by IR, 1H, 13C and 31P NMR, UV–Vis spectroscopy and X-ray crystallography. The experimental studies were completed by quantum chemical calculations, which were used to describe the nature of the interactions between the ligands and the central ion, and the orbital compositions in the frontier electronic structures. Based on a molecular orbital scheme, the calculated results allowed the interpretation of the UV–Vis spectra obtained at an experimental level. The luminescence properties of the complexes were determined.Ruthenium(II) complexes with the general formulas [Ru(H/Cl)(CO)(PPh3)2(L)] and cis/trans-[Ru(PPh3)2(L)2] (L = isoquinoline-1-carboxylate or quinoline-2-carboxylate) were synthesized and characterized by IR, 1H, 13C and 31P NMR, UV–Vis spectroscopy and X-ray crystallography. The experimental studies were completed by quantum chemical calculations, which were used to describe the nature of the interactions between the ligands and the central ion, and the orbital compositions in the frontier electronic structures. Based on a molecular orbital scheme, the calculated results allowed the interpretation of the UV–Vis spectra obtained at an experimental level. The luminescence properties of the complexes were determined.
Co-reporter:Ayana Kashima, Mika Sakate, Hiromi Ota, Akira Fuyuhiro, Yukinari Sunatsuki and Takayoshi Suzuki
Chemical Communications 2015 - vol. 51(Issue 10) pp:NaN1892-1892
Publication Date(Web):2014/12/11
DOI:10.1039/C4CC09087J
In the thyminate(2−)-bridged tetranuclear Cp*RhIII complexes incorporating a Na+, Ca2+ or Ln3+ cation, homochiral aggregation of four RhIII centres was achieved to form metallacalix[4]arene-type clusters. The thyminate(2−) bridged two RhIII and the third metal ion with a μ3-1κN1:2κ2N3,O2:3κO2 mode in the Na+ and Ca2+ complexes, while in the Ln3+ analogues it exhibited a different bridging mode, μ3-1κN1:2κ2N3,O4:3κO2.
Co-reporter:Asuka Takayama, Takayoshi Suzuki, Miyu Ikeda, Yukinari Sunatsuki and Masaaki Kojima
Dalton Transactions 2013 - vol. 42(Issue 40) pp:NaN14567-14567
Publication Date(Web):2013/08/01
DOI:10.1039/C3DT51667A
Several new iridium(III) and rhodium(III) complexes bearing 5-methyltetrazolate (MeCN4−) have been prepared, and their structures in the crystals and in solution have been determined by X-ray analysis and by NMR spectroscopy, respectively. In the crystals of the mononuclear complexes, κN2-coordination of MeCN4− was observed when the ancillary ligand was 2,2′-bipyridine (bpy): [Cp*M(bpy)(MeCN4-κN2)]PF6 (Cp* = η5-C5Me5; M = Ir: 1 and Rh: 2), while the corresponding complexes with 2-(2′-pyridyl)phenyl (ppy−) were confirmed to have the κN1-coordination of MeCN4−: [Cp*M(ppy)(MeCN4-κN1)] (M = Ir: 3 and Rh: 4). In solution, the IrIII complexes (1 and 3) were robust enough to maintain their molecular structures, but the RhIII complexes (2 and 4) existed as an equilibrium mixture of the κN1- and κN2-isomers. In addition to the IrIII–IrIII and RhIII–RhIII homodinuclear complexes bridged by MeCN4− (5–8), the corresponding heterodinuclear IrIII–RhIII complexes (9–12) were prepared using the mononuclear IrIII complexes (1 and 3) as precursors. The molecular structures of these dinuclear complexes were also characterised. Interestingly, both of the heterodinuclear complexes comprised of Cp*M(bpy)2+ and Cp*M′(ppy)+ fragments, [Cp*M(bpy)(μ-MeCN4)M′(ppy)Cp*](PF6)2 (M = Ir, M′ = Rh: 10 and M = Rh, M′ = Ir: 11), exhibited selective crystallisation of a specific μ-κN1(M′-ppy):κN3(M-bpy) isomer. In solution, the dinuclear complexes with a Rh–N(MeCN4) bond and more than one positive charge (6 and 9–11) showed a dissociation equilibrium, while monocationic MeCN4-bridged complexes (7, 8 and 12) were inactive for dissociation. Furthermore, the heterodinuclear complexes of 9–12, as well as the RhIII–RhIII complex of 8, exhibited a bridging isomerisation, which would proceed via a η2:η2-intermediate without dissociation of Cp*M(bpy or ppy) fragments.
Co-reporter:Asami Mori, Takayoshi Suzuki, Yuichi Nakatani, Yukinari Sunatsuki, Masaaki Kojima and Kiyohiko Nakajima
Dalton Transactions 2015 - vol. 44(Issue 36) pp:NaN15760-15760
Publication Date(Web):2015/07/31
DOI:10.1039/C5DT02345A
A reaction of [PdCl2(cod)] (cod = 1,5-cyclooctadiene) and an E/Z mixture of quinoline-2-carbaldehyde (pyridine-2-carbonyl)hydrazone (HL) gave two kinds of PdII mononuclear complexes, [PdCl(Z-L-κ3N,N′,N′′)] (1) and [PdCl2(E-HL′-κ2N,N′)] (2), where L− is the deprotonated hydrazonate anion and HL′ is the quinolinium–hydrazonate zwitterionic form of HL. Complex 2 is gradually converted to 1 in solution, and complex 1 is a good precursor to prepare a PdII/RuII heterodinuclear complex bridged by hydrazonate, trans(Cl,Cl)-[RuCl2(PPh3)2(μ-L)PdCl] (3).
Co-reporter:Atsutoshi Yamada, Takuya Mabe, Ryohei Yamane, Kyoko Noda, Yuko Wasada, Masahiko Inamo, Koji Ishihara, Takayoshi Suzuki and Hideo D. Takagi
Dalton Transactions 2015 - vol. 44(Issue 31) pp:NaN13990-13990
Publication Date(Web):2015/06/22
DOI:10.1039/C5DT01808K
Six-coordinate [Cu(pdt)2(H2O)2]2+ and four-coordinate [Cu(pdt)2]+ complexes were synthesized and the cross redox reactions were studied in acetonitrile (pdt = 3-(2-pyridyl)-5,6-diphenyl-1,2,4-triazine). Single crystal analyses revealed that [Cu(pdt)2(H2O)2](BF4)2 was of pseudo-D2h symmetry with two axial water molecules and two symmetrically coordinated equatorial pdt ligands, while the coordination structure of [Cu(pdt)2]BF4 was a squashed tetrahedron (dihedral angle = 54.87°) with an asymmetric coordination by two pdt ligands: one pdt ligand was coordinated to Cu(I) through pyridine-N and triazine-N2 while another pdt ligand was coordinated through pyridine-N and triazine-N4, and a stacking interaction between the phenyl ring on one pdt ligand and the triazine ring on another pdt ligand caused the squashed structure and non-equivalent Cu–N bond lengths. The cyclic voltammograms for [Cu(pdt)2(H2O)2]2+ and [Cu(pdt)2]+ in acetonitrile were identical to each other and quasi-reversible. The reduction of [Cu(pdt)2(H2O)2]2+ by decamethylferrocene and the oxidation of [Cu(pdt)2]+ by [Co(2,2′-bipyridine)3]3+ in acetonitrile revealed that both cross reactions were sluggish through a gated process (the structural change took place prior to the electron transfer) accompanied by slow direct electron transfer processes. It was found that the triazine ring of the coordinated pdt ligand rotates around the C–C bond between the triazine and pyridine rings with the kinetic parameters k = 51 ± 5 s−1 (297.8 K), ΔH‡ = 6.2 ± 1.1 kJ mol−1 and ΔS‡ = −192 ± 4 J mol−1 K−1. The electron self-exchange process was directly measured using the line-broadening method: kex = (9.9 ± 0.5) × 104 kg mol−1 s−1 (297.8 K) with ΔH‡ = 44 ± 7 kJ mol−1 and ΔS‡ = 0.2 ± 2.6 J mol−1 K−1. By comparing this rate constant with the self-exchange rate constants estimated from the cross reactions using the Marcus cross relation, the non-adiabaticity (electronic) factors, κel, for the direct electron transfer processes between [Cu(pdt)2]+/2+ and non-copper metal (Fe2+ and Co3+) complexes were estimated as ca. 10−7, indicating that the electronic coupling between the d orbitals of copper and of non-copper metals is very small.




![Dichloro[bis(1,3-diphenylphosphino)propane]palladium(II)](http://img.cochemist.com/ccimg/59900/59831-02-6.png)
![Dichloro[bis(1,3-diphenylphosphino)propane]palladium(II)](http://img.cochemist.com/ccimg/59900/59831-02-6_b.png)








![Chloro(pentamethylcyclopentadienyl)[(2-pyridinyl-kN)phenyl-kC]iridum(III)](http://img.cochemist.com/ccimg/945500/945491-51-0.png)
![Chloro(pentamethylcyclopentadienyl)[(2-pyridinyl-kN)phenyl-kC]iridum(III)](http://img.cochemist.com/ccimg/945500/945491-51-0_b.png)