Co-reporter:Haijun Chen ; Tamara Tsalkova ; Oleg G. Chepurny ; Fang C. Mei ; George G. Holz ; Xiaodong Cheng ;Jia Zhou
Journal of Medicinal Chemistry 2013 Volume 56(Issue 3) pp:952-962
Publication Date(Web):January 3, 2013
DOI:10.1021/jm3014162
EPAC1 and EPAC2, two isoforms of exchange proteins directly activated by cAMP (EPAC), respond to the second messenger cAMP and regulate a wide variety of intracellular processes under physiological and pathophysiological circumstances. Herein, we report the chemical design, synthesis, and pharmacological characterization of three different scaffolds (diarylsulfones, N,N-diarylamines, and arylsulfonamides) as highly potent and selective antagonists of EPAC2. Several selective EPAC2 antagonists have been identified including 20i (HJC0350), which has an apparent IC50 of 0.3 μM for competing with 8-NBD-cAMP binding of EPAC2 and is about 133-fold more potent than cAMP. Compounds 1 (ESI-05), 14c (HJC0338), and 20i, selected from each series, have exhibited no inhibition of EPAC1-mediated Rap1-GDP exchange activity at 25 μM, indicating that they are EPAC2-specific antagonists. Moreover, live-cell imaging studies using EPAC1, EPAC2, or PKA FRET sensor also demonstrate that 20i functions as an EPAC2 specific antagonist.
Co-reporter:Maki Wakamiya;Thomas Shelite;Bin Gong;Paul J. Boor;Tuha Ha;Fang C. Mei;Yaohua Hu;Donald H. Bouyer;Qing Chang;Guang Xu;Thomas G. Ksiazek;David H. Walker;Vsevolod L. Popov;Ju Chen
PNAS 2013 Volume 110 (Issue 48 ) pp:19615-19620
Publication Date(Web):2013-11-26
DOI:10.1073/pnas.1314400110
Rickettsiae are responsible for some of the most devastating human infections. A high infectivity and severe illness after
inhalation make some rickettsiae bioterrorism threats. We report that deletion of the exchange protein directly activated
by cAMP (Epac) gene, Epac1, in mice protects them from an ordinarily lethal dose of rickettsiae. Inhibition of Epac1 suppresses bacterial adhesion and
invasion. Most importantly, pharmacological inhibition of Epac1 in vivo using an Epac-specific small-molecule inhibitor, ESI-09,
completely recapitulates the Epac1 knockout phenotype. ESI-09 treatment dramatically decreases the morbidity and mortality associated with fatal spotted fever
rickettsiosis. Our results demonstrate that Epac1-mediated signaling represents a mechanism for host–pathogen interactions
and that Epac1 is a potential target for the prevention and treatment of fatal rickettsioses.
Co-reporter:Meiling Lu, Yan Huang, Mark A. White, Xuri Wu, Nan Liu, Xiaodong Cheng and Yijun Chen
Chemical Communications 2012 vol. 48(Issue 92) pp:11352-11354
Publication Date(Web):08 Oct 2012
DOI:10.1039/C2CC36334H
Diketoreductase catalyzes a two-step bioreduction on a dicarbonyl substrate through a novel dual catalysis mode, in which random hydride attack simultaneously forms two mono-carbonyl intermediates, and subsequently distinct catalytic sites are responsible for the reductions of respective carbonyl group of the intermediates to yield the final dihydroxy product.
Co-reporter:Haijun Chen, Tamara Tsalkova, Fang C. Mei, Yaohua Hu, Xiaodong Cheng, Jia Zhou
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 12) pp:4038-4043
Publication Date(Web):15 June 2012
DOI:10.1016/j.bmcl.2012.04.082
Exchange proteins directly activated by cAMP (Epac) are a family of guanine nucleotide exchange factors that regulate a wide variety of intracellular processes in response to second messenger cAMP. To explore the structural determinants for Epac antagonist properties of high throughput screening (HTS) hit ESI-08, pyrimidine 1, a series of 5-cyano-6-oxo-1,6-dihydro-pyrimidine analogues have been synthesized and evaluated for their activities for Epac inhibition. Structure–activity relationship (SAR) analysis led to the identification of three more potent Epac antagonists (6b, 6g, and 6h). These inhibitors may serve as valuable pharmacological probes for further elucidation of the physiological functions and mechanisms of Epac regulation. Our SAR results and molecular docking studies have also revealed that further optimization of the moieties at the C-6 position of pyrimidine scaffold may allow us to discover more potent Epac-specific antagonists.
Co-reporter:Fang C. Mei;Tamara Tsalkova;Sheng Li;Tong Liu;Virgil L. Woods, Jr.;George G. Holz;Oleg G. Chepurny;Colin A. Leech
PNAS 2012 Volume 109 (Issue 45 ) pp:18613-18618
Publication Date(Web):2012-11-06
DOI:10.1073/pnas.1210209109
The major physiological effects of cAMP in mammalian cells are transduced by two ubiquitously expressed intracellular cAMP
receptors, protein kinase A (PKA) and exchange protein directly activated by cAMP (EPAC), as well as cyclic nucleotide-gated
ion channels in certain tissues. Although a large number of PKA inhibitors are available, there are no reported EPAC-specific
antagonists, despite extensive research efforts. Here we report the identification and characterization of noncyclic nucleotide
EPAC antagonists that are exclusively specific for the EPAC2 isoform. These EAPC2-specific antagonists, designated as ESI-05
and ESI-07, inhibit Rap1 activation mediated by EAPC2, but not EPAC1, with high potency in vitro. Moreover, ESI-05 and ESI-07
are capable of suppressing the cAMP-mediated activation of EPAC2, but not EPAC1 and PKA, as monitored in living cells through
the use of EPAC- and PKA-based FRET reporters, or by the use of Rap1-GTP pull-down assays. Deuterium exchange mass spectroscopy
analysis further reveals that EPAC2-specific inhibitors exert their isoform selectivity through a unique mechanism by binding
to a previously undescribed allosteric site: the interface of the two cAMP binding domains, which is not present in the EPAC1
isoform. Isoform-specific EPAC pharmacological probes are highly desired and will be valuable tools for dissecting the biological
functions of EPAC proteins and their roles in various disease states.
Co-reporter:Xiaodong Cheng;Zhenyu Ji;Tamara Tsalkova ;Fang Mei
Acta Biochimica et Biophysica Sinica 2008 Volume 40( Issue 7) pp:651-662
Publication Date(Web):
DOI:10.1111/j.1745-7270.2008.00438.x
cAMP-mediated signaling pathways regulate a multitude of important biological processes under both physiological and pathological conditions, including diabetes, heart failure and cancer. In eukaryotic cells, the effects of cAMP are mediated by two ubiquitously expressed intracellular cAMP receptors, the classic protein kinase A (PKA)/cAMP-dependent protein kinase and the recently discovered exchange protein directly activated by cAMP (Epac)/cAMP-regulated guanine nucleotide exchange factors. Like PKA, Epac contains an evolutionally conserved cAMP binding domain that acts as a molecular switch for sensing intracellular second messenger cAMP levels to control diverse biological functions. The existence of two families of cAMP effectors provides a mechanism for a more precise and integrated control of the cAMP signaling pathways in a spatial and temporal manner. Depending upon the specific cellular environments as well as their relative abundance, distribution and localization, Epac and PKA may act independently, converge synergistically or oppose each other in regulating a specific cellular function.
Co-reporter:Fang C. Mei and Xiaodong Cheng
Molecular BioSystems 2005 vol. 1(Issue 4) pp:325-331
Publication Date(Web):26 Sep 2005
DOI:10.1039/B511267B
“Exchange protein directly activated by cAMP” (Epac) is a newly discovered cAMP receptor that mediates the intracellular cAMP actions in addition to the classic cAMP-dependent protein kinase system. In this study, we show that Epac interacts directly with tubulin, co-purifies with cellular microtubules, and co-localizes with the mitotic spindle assembly. Association with microtubules suppresses Epac-mediated Rap1 activation, while the binding of Epac promotes microtubule formation. These results demonstrate that Epac plays an important role in connecting the microtubule cytoskeleton network and intracellular cAMP-signalling.
Co-reporter:Travis Young;Fang Mei;Jinsong Liu;Robert C Bast Jr;Alexander Kurosky;Xiaodong Cheng
Oncogene 2005 24(40) pp:6174-6184
Publication Date(Web):2005-06-06
DOI:10.1038/sj.onc.1208753
RAS is a small GTP binding protein mutated in approximately 30% human cancer. Despite its important role in the initiation and progression of human cancer, the underlying mechanism of RAS-induced human epithelial transformation remains elusive. In this study, we probe the cellular and molecular mechanisms of RAS-mediated transformation, by profiling two human ovarian epithelial cell lines. One cell line was immortalized with SV40 T/t antigens and the human catalytic subunit of telomerase (T29), while the second cell line was transformed with an additional oncogenic rasV12 allele (T29H). In total, 32 proteins associated with RAS-mediated transformation have been identified using peptide mass fingerprinting. These protein targets are involved in several cellular pathways, including metabolism, redox balance, calcium signaling, apoptosis, and cellular methylation. One such target, the 40 kDa procaspase 4 is significantly upregulated at the protein level in RAS-transformed T29H cells, related directly to signaling through MEK, but not PI3 kinase. Cellular caspase 4 activity is, however, suppressed in the T29H cells, suggesting that the maturation process of caspase 4 is abrogated in RAS-transformed T29H cells. Consistent with this notion, transformed T29H cells were less susceptible to the toxic effects of anti-Fas antibody than were immortalized, nontransformed T29 cells, associated with less activation of caspase 4. This study demonstrates that functional proteomic analysis of a genetically defined cancer model provides a powerful approach toward systematically identifying cellular targets associated with oncogenic transformation.
Co-reporter:Meiling Lu, Yan Huang, Mark A. White, Xuri Wu, Nan Liu, Xiaodong Cheng and Yijun Chen
Chemical Communications 2012 - vol. 48(Issue 92) pp:NaN11354-11354
Publication Date(Web):2012/10/08
DOI:10.1039/C2CC36334H
Diketoreductase catalyzes a two-step bioreduction on a dicarbonyl substrate through a novel dual catalysis mode, in which random hydride attack simultaneously forms two mono-carbonyl intermediates, and subsequently distinct catalytic sites are responsible for the reductions of respective carbonyl group of the intermediates to yield the final dihydroxy product.





![Hexanamide,2,6-diamino-N-[[1-(1-oxotridecyl)-2-piperidinyl]methyl]-, (2S)-](http://img.cochemist.com/ccimg/136500/136449-85-9.png)
![Hexanamide,2,6-diamino-N-[[1-(1-oxotridecyl)-2-piperidinyl]methyl]-, (2S)-](http://img.cochemist.com/ccimg/136500/136449-85-9_b.png)
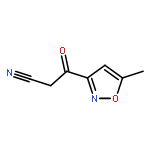
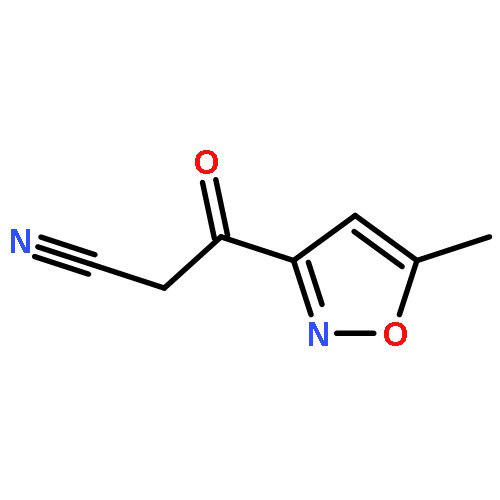
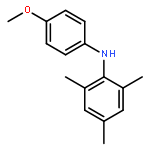
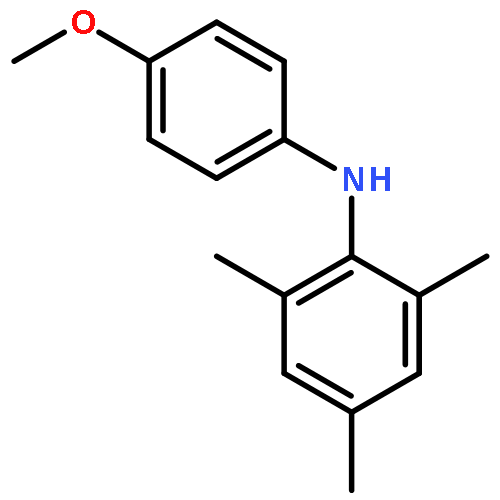


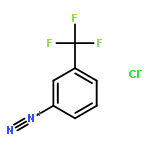



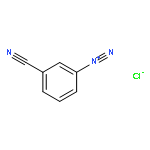
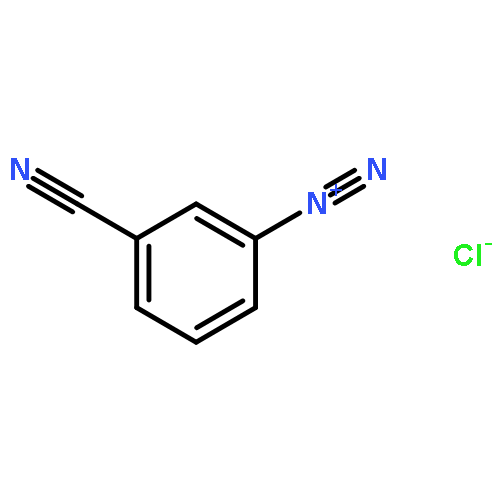
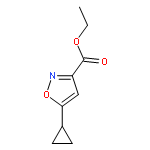



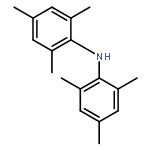
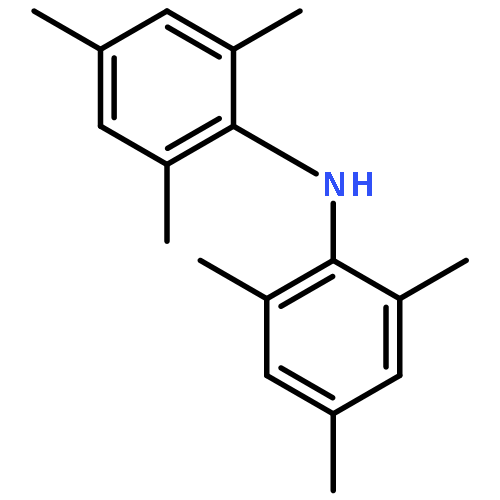
![Benzene,1,3,5-trimethyl-2-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/5200/5184-64-5.png)
![Benzene,1,3,5-trimethyl-2-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/5200/5184-64-5_b.png)

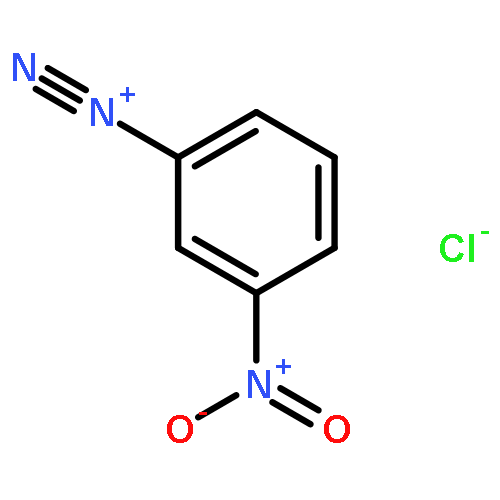


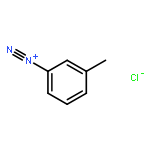



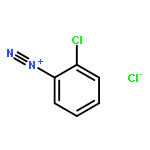
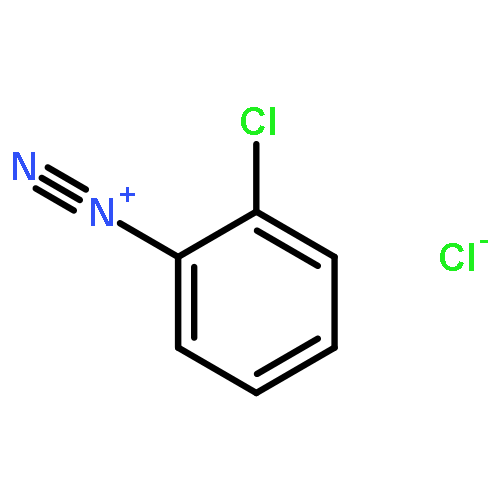


![3-?Isoxazolepropanenitr?ile, α-?[2-?(3-?chlorophenyl)?hydrazinylidene]?-?5-?(1,?1-?dimethylethyl)?-?β-?oxo-](http://img.cochemist.com/ccimg/263800/263707-16-0.png)
![3-?Isoxazolepropanenitr?ile, α-?[2-?(3-?chlorophenyl)?hydrazinylidene]?-?5-?(1,?1-?dimethylethyl)?-?β-?oxo-](http://img.cochemist.com/ccimg/263800/263707-16-0_b.png)