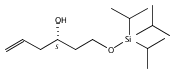
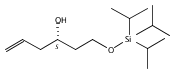
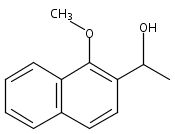
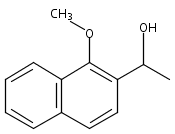
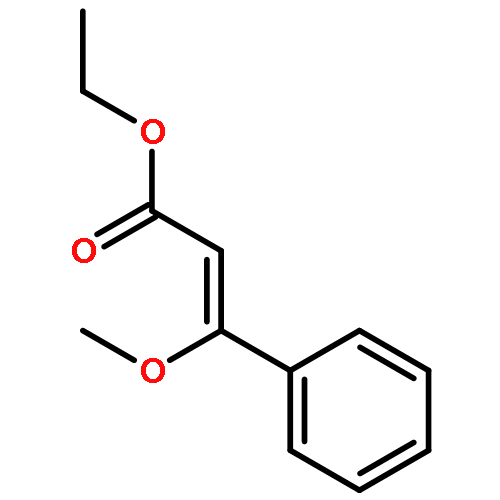
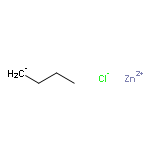
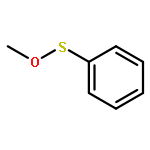
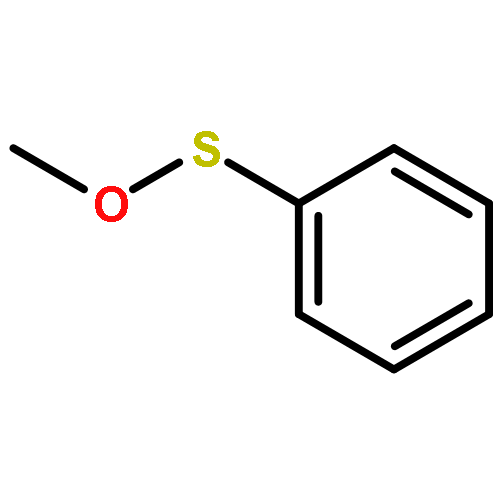
Co-reporter:Stephen W. Laws, Lucas C. Moore, Michael J. Di Maso, Q. Nhu N. Nguyen, Dean J. Tantillo, and Jared T. Shaw
Organic Letters May 19, 2017 Volume 19(Issue 10) pp:
Publication Date(Web):May 5, 2017
DOI:10.1021/acs.orglett.7b00468
A diastereoselective base-catalyzed Mannich reaction of cyclic, enolizable anhydrides and N-sulfonyl imines for the synthesis of δ-lactams is reported. This anhydride Mannich reaction tolerates imines derived from aryl and enolizable aldehydes. A base-catalyzed product epimerization pathway ensures high anti diastereoselectivity in aryl and achiral enolizable imines.
Co-reporter:Katherine A. Hurley;Thiago M. A. Santos;Molly R. Fensterwald;Madhusudan Rajendran;Jared T. Moore;Edward I. Balmond;Brice J. Blahnik;Katherine C. Faulkner;Marie H. Foss;Victoria A. Heinrich;Matthew G. Lammers;Lucas C. Moore;Gregory D. Reynolds;Galen P. Shearn-Nance;Brian A. Stearns;Zi W. Yao;Douglas B. Weibel
MedChemComm (2010-Present) 2017 vol. 8(Issue 5) pp:942-951
Publication Date(Web):2017/05/24
DOI:10.1039/C7MD00012J
Bacterial DNA gyrase is an essential type II topoisomerase that enables cells to overcome topological barriers encountered during replication, transcription, recombination, and repair. This enzyme is ubiquitous in bacteria and represents an important clinical target for antibacterial therapy. In this paper we report the characterization of three exciting new gyramide analogs—from a library of 183 derivatives—that are potent inhibitors of DNA gyrase and are active against clinical strains of Gram-negative bacteria (Escherichia coli, Shigella flexneri, and Salmonella enterica; 3 of 10 wild-type strains tested) and Gram-positive bacteria (Bacillus spp., Enterococcus spp., Staphylococcus spp., and Streptococcus spp.; all 9 of the wild-type strains tested). E. coli strains resistant to the DNA gyrase inhibitors ciprofloxacin and novobiocin display very little cross-resistance to these new gyramides. In vitro studies demonstrate that the new analogs are potent inhibitors of the DNA supercoiling activity of DNA gyrase (IC50s of 47–170 nM) but do not alter the enzyme's ATPase activity. Although mutations that confer bacterial cells resistant to these new gyramides map to the genes encoding the subunits of the DNA gyrase (gyrA and gyrB genes), overexpression of GyrA, GyrB, or GyrA and GyrB together does not suppress the inhibitory effect of the gyramides. These observations support the hypothesis that the gyramides inhibit DNA gyrase using a mechanism that is unique from other known inhibitors.
Co-reporter:Michael J. Di Maso, Gabriella M. Nepomuceno, Michael A. St. Peter, Haley H. Gitre, Kevin S. Martin, and Jared T. Shaw
Organic Letters 2016 Volume 18(Issue 8) pp:1740-1743
Publication Date(Web):March 29, 2016
DOI:10.1021/acs.orglett.6b00413
Bisavenanthramide B-6 (2) is a highly substituted γ-lactam derived from oat leaves. Development of a new base-promoted anhydride Mannich reaction with N-sulfonylated imines that forms the core structure of 2 in a single step is presented. Further elaboration allows for a facile one-pot double Buchwald N-arylation to install the final rings onto the densely substituted γ-lactam core. This route provides the natural product in a longest linear sequence of nine steps.
Co-reporter:Katherine A. Hurley, Thiago M. A. Santos, Gabriella M. Nepomuceno, Valerie Huynh, Jared T. Shaw, and Douglas B. Weibel
Journal of Medicinal Chemistry 2016 Volume 59(Issue 15) pp:6975-6998
Publication Date(Web):January 12, 2016
DOI:10.1021/acs.jmedchem.5b01098
Similar to its eukaryotic counterpart, the prokaryotic cytoskeleton is essential for the structural and mechanical properties of bacterial cells. The essential protein FtsZ is a central player in the cytoskeletal family, forms a cytokinetic ring at mid-cell, and recruits the division machinery to orchestrate cell division. Cells depleted of or lacking functional FtsZ do not divide and grow into long filaments that eventually lyse. FtsZ has been studied extensively as a target for antibacterial development. In this Perspective, we review the structural and biochemical properties of FtsZ, its role in cell biochemistry and physiology, the different mechanisms of inhibiting FtsZ, small molecule antagonists (including some misconceptions about mechanisms of action), and their discovery strategies. This collective information will inform chemists on different aspects of FtsZ that can be (and have been) used to develop successful strategies for devising new families of cell division inhibitors.
Co-reporter:Leslie A. Nickerson, Valerie Huynh, Edward I. Balmond, Stephen P. Cramer, and Jared T. Shaw
The Journal of Organic Chemistry 2016 Volume 81(Issue 22) pp:11404-11408
Publication Date(Web):September 29, 2016
DOI:10.1021/acs.joc.6b01997
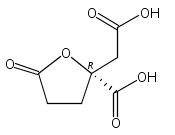
A short, diastereoselective synthesis of homocitric acid lactone is described. The key step is a bioinspired aldol addition to set the stereogenic center in an intermediate that requires only modest oxidation state manipulation to complete the synthesis. This approach enables rapid generation of isotopomers in which carbon and hydrogen can be replaced by heavier nuclei at nearly every position.
Co-reporter:Michael J. DiMaso;Kevin M. Snyder;Fábio DeSouzaFernes;Ommidala Pattawong;Dr. Darlene Q. Tan;Dr. James C. Fettinger; Paul Ha-Yeon Cheong; Jared T. Shaw
Chemistry - A European Journal 2016 Volume 22( Issue 14) pp:4794-4801
Publication Date(Web):
DOI:10.1002/chem.201504424
Abstract
2-Piperidinones are synthesized in a single step from imines and 2-cyano glutaric anhydrides. The reaction provides the products in good diastereoselectivity and generates a quaternary stereogenic center. Substitutions on the anhydride skeleton are well tolerated to provide 2-piperidinones with three stereogenic centers from a single transformation. The pertinent transition structures have also been computed using quantum mechanics and reveal the key interactions controlling the stereochemical outcome of the reaction.
Co-reporter:Richard A. Squitieri, Galen P. Shearn-Nance, Jason E. Hein, and Jared T. Shaw
The Journal of Organic Chemistry 2016 Volume 81(Issue 13) pp:5278-5284
Publication Date(Web):May 18, 2016
DOI:10.1021/acs.joc.6b00408
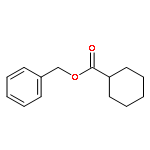
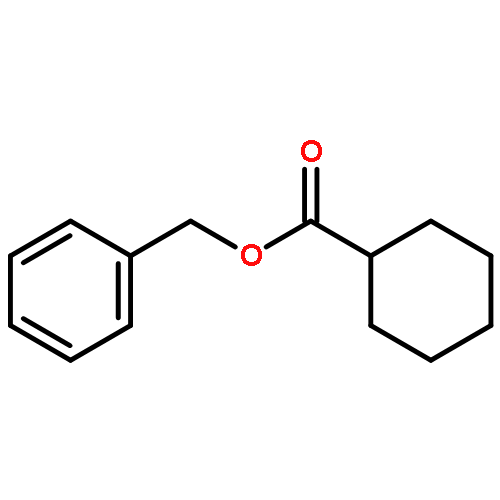
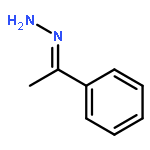
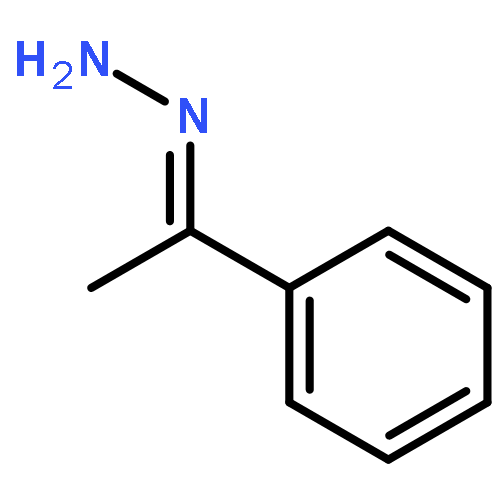
A general method has been developed for the in situ formation and trapping of diazoalkanes by carboxylic acids to form esters. The method is applicable to a large variety of carboxylic acids using diazo compounds that are formed from the hydrazones of benzaldehydes and aryl ketones. In situ reaction monitoring with IR spectroscopy (ReactIR) was used to demonstrate that slow addition of the hydrazone to a mixture of oxidant and carboxylic acid avoids the buildup of the diazo compound. This method enables the safe preparation of esters from simple precursors without isolation of diazo compounds.
Co-reporter:Edward I. Balmond, Brandon K. Tautges, Andrea L. Faulkner, Victor W. Or, Blanka M. Hodur, Jared T. Shaw, and Angelique Y. Louie
The Journal of Organic Chemistry 2016 Volume 81(Issue 19) pp:8744-8758
Publication Date(Web):August 16, 2016
DOI:10.1021/acs.joc.6b01193
Spiropyrans and spirooxazines represent an important class of photochromic compounds with a wide variety of applications. In order to effectively utilize and design these photoswitches it is desirable to understand how the substituents affect photochromic properties, and how the different structural motifs compare under identical conditions. In this work a small library of photoswitches was synthesized in order to comparatively evaluate the effect of substituent modifications and structure on photochromism. The library was designed to modify positions that were believed to have the greatest effect on C–O bond lability and therefore the photochromic properties. Herein we report a comparative analysis of the UV and visible light responses of 30 spiropyrans, spiroindolinonaphthopyrans, and spirooxazines. The influence of gadolinium(III) binding was also investigated on the library of compounds to determine its effect on photoswitching. Both assays demonstrated different trends in substituent and structural requirements for optimal photochromism.
Co-reporter:Gabriella M. Nepomuceno, Katie M. Chan, Valerie Huynh, Kevin S. Martin, Jared T. Moore, Terrence E. O’Brien, Luiz A. E. Pollo, Francisco J. Sarabia, Clarissa Tadeus, Zi Yao, David E. Anderson, James B. Ames, and Jared T. Shaw
ACS Medicinal Chemistry Letters 2015 Volume 6(Issue 3) pp:308
Publication Date(Web):January 7, 2015
DOI:10.1021/ml500497s
The bacterial cell division protein FtsZ is one of many potential targets for the development of novel antibiotics. Recently, zantrin Z3 was shown to be a cross-species inhibitor of FtsZ; however, its specific interactions with the protein are still unknown. Herein we report the synthesis of analogues that contain a more tractable core structure and an analogue with single-digit micromolar inhibition of FtsZ’s GTPase activity, which represents the most potent inhibitor of Escherichia coli FtsZ reported to date. In addition, the zantrin Z3 core has been converted to two potential photo-cross-linking reagents for proteomic studies that could shed light on the molecular interactions between FtsZ and molecules related to zantrin Z3.Keywords: bacterial cell division; FtsZ; SAR; zantrin Z3
Co-reporter:Cristian Soldi ; Kellan N. Lamb ; Richard A. Squitieri ; Marcos González-López ; Michael J. Di Maso
Journal of the American Chemical Society 2014 Volume 136(Issue 43) pp:15142-15145
Publication Date(Web):October 12, 2014
DOI:10.1021/ja508586t
The first asymmetric insertion reactions of donor–donor carbenoids, i.e., those with no pendant electron-withdrawing groups, are reported. This process enables the synthesis of densely substituted benzodihydrofurans with high levels of enantio- and diastereoselectivity. Preliminary results show similar efficiency in the preparation of indanes. This new method is used in the first enantioselective synthesis of an oligoresveratrol natural product (E-δ-viniferin).
Co-reporter:Nohemy A. Sorto, Michael J. Di Maso, Manuel A. Muñoz, Ryan J. Dougherty, James C. Fettinger, and Jared T. Shaw
The Journal of Organic Chemistry 2014 Volume 79(Issue 6) pp:2601-2610
Publication Date(Web):February 19, 2014
DOI:10.1021/jo500050n
Sulfone-substituted γ- and δ-lactams have been prepared in a single step with high diastereoselectivity. Sulfonylglutaric anhydrides produce intermediates that readily decarboxylate to provide δ-lactams with high diastereoselectivity. Substituents at the 3- or 4-position of the glutaric anhydride induce high levels of stereocontrol. Sulfonylsuccinic anhydrides produce intermediate carboxylic acids that can be trapped as methyl esters or allowed to decarboxylate under mild conditions. This method has been applied to a short synthesis of the pyrrolizidine alkaloid (±)-isoretronecanol.
Co-reporter:Gonzalo Jiménez-Osés ; Anthony J. Brockway ; Jared T. Shaw ;K. N. Houk
Journal of the American Chemical Society 2013 Volume 135(Issue 17) pp:6633-6642
Publication Date(Web):March 26, 2013
DOI:10.1021/ja4015937
The mechanism of direct displacement of alkoxy groups in vinylogous and aromatic esters by Grignard reagents, a reaction that is not observed with expectedly better tosyloxy leaving groups, is elucidated computationally. The mechanism of this reaction has been determined to proceed through the inner-sphere attack of nucleophilic alkyl groups from magnesium to the reacting carbons via a metalaoxetane transition state. The formation of a strong magnesium chelate with the reacting alkoxy and carbonyl groups dictates the observed reactivity and selectivity. The influence of ester, ketone, and aldehyde substituents was investigated. In some cases, the calculations predicted the formation of products different than those previously reported; these predictions were then verified experimentally. The importance of studying the actual system, and not simplified models as computational systems, is demonstrated.
Co-reporter:Jared T. Moore, Cristian Soldi, James C. Fettinger and Jared T. Shaw
Chemical Science 2013 vol. 4(Issue 1) pp:292-296
Publication Date(Web):25 Oct 2012
DOI:10.1039/C2SC21405A
A new catalytic synthesis of densely substituted tetrahydroquinolines is described. This reaction forms up to two rings, three bonds, and three stereogenic centers with excellent stereo- and regiocontrol in a single step. Although control experiments demonstrate that the active catalyst is protic acid, Sc(OTf)3 serves as an effective and practical pre-catalyst. The scope of this reaction is demonstrated with 21 monocyclizations and 14 bicyclization reactions.
Co-reporter:Jared T. Moore, Nadine V. Hanhan, Maximillian E. Mahoney, Stephen P. Cramer, and Jared T. Shaw
Organic Letters 2013 Volume 15(Issue 22) pp:5615-5617
Publication Date(Web):November 1, 2013
DOI:10.1021/ol402802g
A concise synthesis of homocitric acid lactone was developed to accommodate systematic placement of carbon isotopes (specifically 13C) for detailed studies of this cofactor. This new route uses a chiral allylic alcohol, available in multigram quantities from enzymatic resolution, as a starting material, which transposes asymmetry through an Ireland–Claisen rearrangement.
Co-reporter:Darlene Q. Tan, Ashkaan Younai, Ommidala Pattawong, James C. Fettinger, Paul Ha-Yeon Cheong, and Jared T. Shaw
Organic Letters 2013 Volume 15(Issue 19) pp:5126-5129
Publication Date(Web):September 26, 2013
DOI:10.1021/ol402554n
A reaction between imines and anhydrides has been developed with chiral disubstituted anhydrides and chiral imines. The synthesis of highly substituted γ-lactams with three stereogenic centers, including one quaternary center, proceeds at room temperature in high yield and with high diastereoselectivity in most cases. Enantiomerically pure alkyl-substituted anhydrides proceed with no epimerization, thus providing access to enantiomerically pure penta-substituted lactam products.
Co-reporter:Ommidala Pattawong, Darlene Q. Tan, James C. Fettinger, Jared T. Shaw, and Paul Ha-Yeon Cheong
Organic Letters 2013 Volume 15(Issue 19) pp:5130-5133
Publication Date(Web):September 26, 2013
DOI:10.1021/ol402561q
Computations (SCS-MP2//B3LYP) reveal that the asymmetric synthesis of highly substituted γ-lactams with three stereogenic centers, including one quaternary center, proceeds through a Mannich reaction between the enol form of the anhydride and the E-imine, followed by a transannular acylation. This new mechanistic picture accounts for both the observed reactivity and stereoselectivity. CH–O and hydrogen bonding interactions in the Mannich step and torsional steering effects in the acylation step are responsible for stereocontrol. It is demonstrated that this new mechanistic picture applies to the related reactions of homophthalic anhydrides with imines and presents new vistas for the design of a new reaction to access complex molecular architectures.
Co-reporter:Nohemy A. Sorto, Phillip P. Painter, James C. Fettinger, Dean J. Tantillo, and Jared T. Shaw
Organic Letters 2013 Volume 15(Issue 11) pp:2700-2703
Publication Date(Web):May 30, 2013
DOI:10.1021/ol4010068
Mimics of the T7-loop of the bacterial cell division protein FtsZ have been designed and synthesized. The design is based on the X-ray cocrystal structure of P. aeruginosa FtsZ:SulA. Fast Rigid Exhaustive Docking (FRED) was employed to select compounds that can mimic the key interacting residues. Bicyclic oxazolidinones 1a–d were selected for further study and synthesized from a key bicyclic aziridine intermediate, which is synthesized from a readily available unsaturated aldehyde and amides derived from α-amino acids.
Co-reporter:Kevin S. Martin, Michael J. Di Maso, James C. Fettinger, and Jared T. Shaw
ACS Combinatorial Science 2013 Volume 15(Issue 7) pp:356
Publication Date(Web):May 17, 2013
DOI:10.1021/co400049f
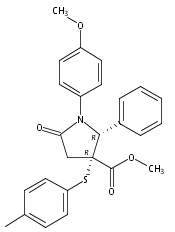
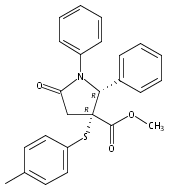
The synthesis of a pilot scale library of 116 structurally diverse γ-lactams is reported. The library core structure emanates from a γ-lactam forming one-pot, four-component reaction of ammonium acetate, p-methoxythiophenol, p-methoxybenzaldehyde, and maleic anhydride. Structural diversity then arises from amide coupling, thioaryl cleavage, N-functionalization, and heterocycle forming reactions on this core structure. Computational analysis reveals that the library contains molecular properties and shape diversity suitable for drug lead and biological probe discovery.Keywords: amide coupling; one-pot, four-component reaction; synthesis; thioaryl cleavage; γ-lactam
Co-reporter:Michael W. Lodewyk ; Cristian Soldi ; Paul B. Jones ; Marilyn M. Olmstead ; Juan Rita ; Jared T. Shaw ;Dean J. Tantillo
Journal of the American Chemical Society 2012 Volume 134(Issue 45) pp:18550-18553
Publication Date(Web):October 29, 2012
DOI:10.1021/ja3089394
Aquatolide has been reisolated from its natural source, and its structure has been revised on the basis of quantum-chemical NMR calculations, extensive experimental NMR analysis, and crystallography.
Co-reporter:Charles I. Grove, Michael J. Di Maso, Firoz A. Jaipuri, Michelle B. Kim, and Jared T. Shaw
Organic Letters 2012 Volume 14(Issue 17) pp:4338-4341
Publication Date(Web):August 13, 2012
DOI:10.1021/ol301743t
Efficient and stereoselective syntheses of pigmentosin A, talaroderxine A, and its diastereomer talaroderxine B are reported. The binaphthyl ring system is assembled by vanadium-catalyzed phenolic coupling of tricyclic precursors. These key intermediates were prepared by Michael–Dieckmann annulation of a protected orsellinate ester, with the requisite pyranones accessed by a new variant of Ghosez’s sulfone-epoxide annulation. Preliminary biological experiments are reported for pigmentosin.
Co-reporter:David E. Anderson, Michelle B. Kim, Jared T. Moore, Terrence E. O’Brien, Nohemy A. Sorto, Charles I. Grove, Laura L. Lackner, James B. Ames, and Jared T. Shaw
ACS Chemical Biology 2012 Volume 7(Issue 11) pp:1918
Publication Date(Web):September 8, 2012
DOI:10.1021/cb300340j
FtsZ is a guanosine triphosphatase (GTPase) that mediates cytokinesis in bacteria. FtsZ is homologous in structure to eukaryotic tubulin and polymerizes in a similar head-to-tail fashion. The study of tubulin’s function in eukaryotic cells has benefited greatly from specific and potent small molecule inhibitors, including colchicine and taxol. Although many small molecule inhibitors of FtsZ have been reported, none has emerged as a generally useful probe for modulating bacterial cell division. With the goal of establishing a useful and reliable small molecule inhibitor of FtsZ, a broad biochemical cross-comparison of reported FtsZ inhibitors was undertaken. Several of these molecules, including phenolic natural products, are unselective inhibitors that seem to derive their activity from the formation of microscopic colloids or aggregates. Other compounds, including the natural product viriditoxin and the drug development candidate PC190723, exhibit no inhibition of GTPase activity using protocols in this work or under published conditions. Of the compounds studied, only zantrin Z3 exhibits good levels of inhibition, maintains activity under conditions that disrupt small molecule aggregates, and provides a platform for exploration of structure–activity relationships (SAR). Preliminary SAR studies have identified slight modifications to the two side chains of this structure that modulate the inhibitory activity of zantrin Z3. Collectively, these studies will help focus future investigations toward the establishment of probes for FtsZ that fill the roles of colchicine and taxol in studies of tubulin.
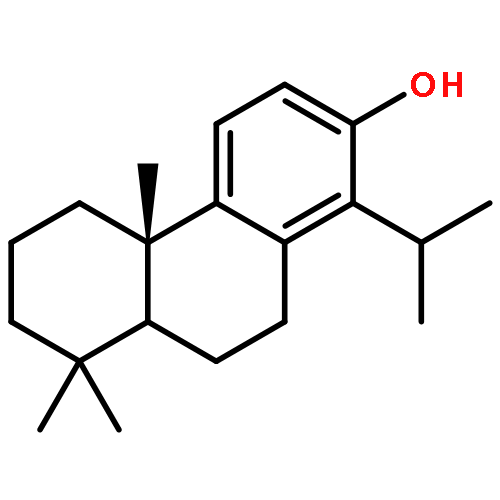
Co-reporter:Michelle B. Kim, Terrence E. O'Brien, Jared T. Moore, David E. Anderson, Marie H. Foss, Douglas B. Weibel, James B. Ames, and Jared T. Shaw
ACS Medicinal Chemistry Letters 2012 Volume 3(Issue 10) pp:818
Publication Date(Web):August 28, 2012
DOI:10.1021/ml3001775
The synthesis and antimicrobial activity of heterocyclic analogues of the diterpenoid totarol are described. An advanced synthetic intermediate with a ketone on the A-ring is used to attach fused heterocycles, and a carbon-to-nitrogen atom replacement is made on the B-ring by de novo synthesis. A-ring analogues with an indole attached exhibit, for the first time, enhanced antimicrobial activity relative to the parent natural product. Preliminary experiments demonstrate that the indole analogues do not target the bacterial cell division protein FtsZ as had been hypothesized for totarol.Keywords: antimicrobial activity; FtsZ; small molecule inhibitors; structure−activity relationships and optimization; totarol
Co-reporter:Darlene Q. Tan, Amy L. Atherton, Austin J. Smith, Cristian Soldi, Katherine A. Hurley, James C. Fettinger, and Jared T. Shaw
ACS Combinatorial Science 2012 Volume 14(Issue 3) pp:218
Publication Date(Web):January 6, 2012
DOI:10.1021/co2001873
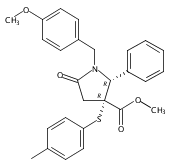
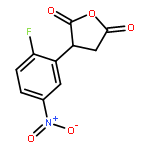
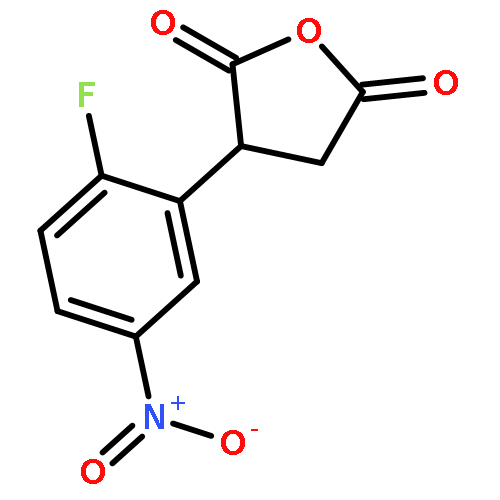
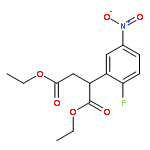
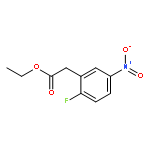
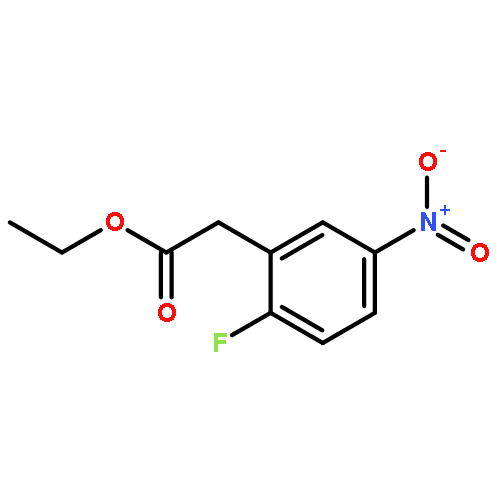
Formal cycloaddition reactions between imines and cyclic anhydrides serve as starting point for the synthesis of diverse libraries of small molecules. The synthesis of succinic anhydrides substituted with electron-withdrawing groups is facilitated by new mild conditions for alkylation of aryl-substituted acetyl esters with ethyl bromoacetate. These anhydrides are then used in formal cycloaddition reactions with imines to produce γ-lactams. 2-Fluoro-5-nitrophenylsuccinic anhydride reacts efficiently with imines to provide lactams that are further diversified by conversion of the nitro group to either an aniline and an azide for subsequent reactions with acylating agents and alkynes, respectively. The synthesis of cyanosuccinic anhydride is reported for the first time, and the use of this compound in reactions with imines and subsequent functionalization of the resultant lactams is demonstrated.Keywords: cyanosuccinic anhydrides; imine-anhydride formal cycloaddition; spirooxindole; γ-lactams
Co-reporter:Ashkaan Younai, James C. Fettinger, Jared T. Shaw
Tetrahedron 2012 68(22) pp: 4320-4327
Publication Date(Web):
DOI:10.1016/j.tet.2012.03.027
Co-reporter:James E. Biggs-Houck, Rebecca L. Davis, Jingqiang Wei, Brandon Q. Mercado, Marilyn M. Olmstead, Dean J. Tantillo, and Jared T. Shaw
The Journal of Organic Chemistry 2012 Volume 77(Issue 1) pp:160-172
Publication Date(Web):October 24, 2011
DOI:10.1021/jo201541e
Strategies for the formation of carbon–carbon bonds from the α-thioaryl carbonyl products of substituted lactams are described. Although direct functionalization is possible, a two step process of oxidation and magnesium-sulfoxide exchange has proven optimal. The oxidation step results in the formation of two diastereomers that exhibit markedly different levels of stability toward elimination, which is rationalized on the basis of quantum mechanical calculations and X-ray crystallography. Treatment of the sulfoxide with i-PrMgCl results in the formation of a magnesium enolate that will undergo an intramolecular Michael addition reaction to form two new stereogenic centers. The relationship between the substitution patterns of the sulfoxide substrate and the efficiency of the magnesium exchange reaction are also described.
Co-reporter:Marie H. Foss, Katherine A. Hurley, Nohemy A. Sorto, Laura L. Lackner, Kelsey M. Thornton, Jared T. Shaw, and Douglas B. Weibel
ACS Medicinal Chemistry Letters 2011 Volume 2(Issue 4) pp:289
Publication Date(Web):February 11, 2011
DOI:10.1021/ml1002822
This paper characterizes N-benzyl-3-sulfonamidopyrrolidines (gyramides) as DNA gyrase inhibitors. Gyramide A was previously shown to exhibit antimicrobial activity, which suggested it inhibited bacterial cell division. In this study, we conducted target identification studies and identified DNA gyrase as the primary target of gyramide A. The gyramide A resistance-determining region in DNA gyrase is adjacent to the DNA cleavage gate and is a new site for inhibitor design. We studied the antibiotic effects of gyramides A−C in combination with the Gram-negative efflux pump inhibitor MC-207,110 (60 μM). The gyramides had a minimum inhibitory concentration of 2.5−160 μM against Escherichia coli, Pseudomonas aeruginosa, Salmonella enterica, Staphylococcus aureus, and Streptococcus pneumoniae; the compounds were ineffective against Enterococcus faecalis. The IC50 of gyramides A−C against E. coli DNA gyrase was 0.7−3.3 μM. The N-benzyl-3-sulfonamidopyrrolidines described in this manuscript represent a starting point for development of antibiotics that bind a new site in DNA gyrase.Keywords: 534F6; antibiotics; DNA gyrase; gyramides; inhibitors
Co-reporter:Jared T. Moore, Cristian Soldi, James C. Fettinger and Jared T. Shaw
Chemical Science (2010-Present) 2013 - vol. 4(Issue 1) pp:NaN296-296
Publication Date(Web):2012/10/25
DOI:10.1039/C2SC21405A
A new catalytic synthesis of densely substituted tetrahydroquinolines is described. This reaction forms up to two rings, three bonds, and three stereogenic centers with excellent stereo- and regiocontrol in a single step. Although control experiments demonstrate that the active catalyst is protic acid, Sc(OTf)3 serves as an effective and practical pre-catalyst. The scope of this reaction is demonstrated with 21 monocyclizations and 14 bicyclization reactions.
.jpg)




![Quinazoline, 4-chloro-2-[(1E)-2-(4-chlorophenyl)ethenyl]-6,7-dimethoxy-](http://img.cochemist.com/ccimg/221100/221009-09-2.png)
![Quinazoline, 4-chloro-2-[(1E)-2-(4-chlorophenyl)ethenyl]-6,7-dimethoxy-](http://img.cochemist.com/ccimg/221100/221009-09-2_b.png)





![Spiro[2H-indole-2,3'-[3H]naphth[2,1-b][1,4]oxazine], 1,3-dihydro-1,3,3-trimethyl-5-nitro-](/data/chemimg/1076500/128171-73-3.png)
![Spiro[2H-indole-2,3'-[3H]naphth[2,1-b][1,4]oxazine], 1,3-dihydro-1,3,3-trimethyl-5-nitro-](/data/chemimg/1076500/128171-73-3_b.png)

![Spiro[2H-indole-2,3'-[3H]naphth[2,1-b][1,4]oxazine], 1,3-dihydro-1,3,3-trimethyl-8'-nitro-](/data/chemimg/1068900/121456-71-1.png)
![Spiro[2H-indole-2,3'-[3H]naphth[2,1-b][1,4]oxazine], 1,3-dihydro-1,3,3-trimethyl-8'-nitro-](/data/chemimg/1068900/121456-71-1_b.png)


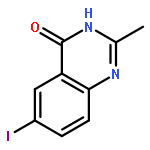
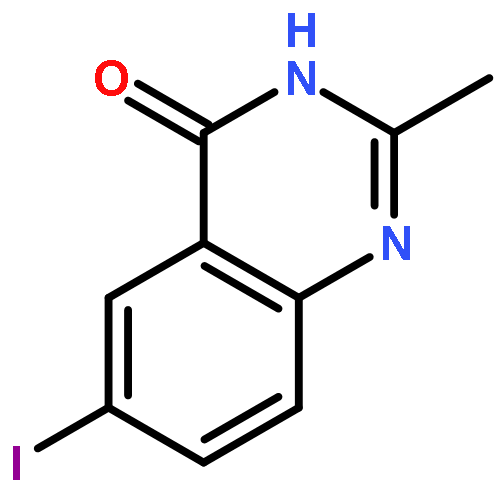
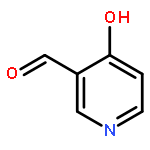
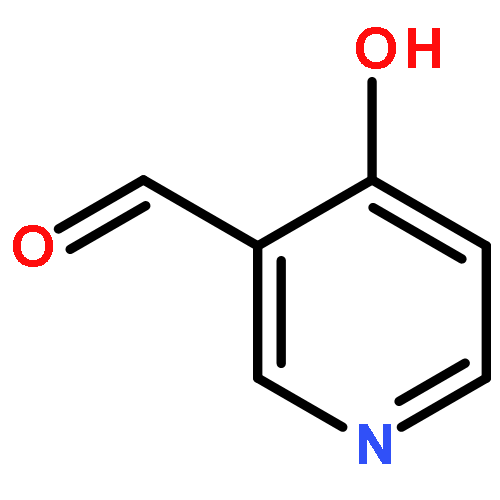







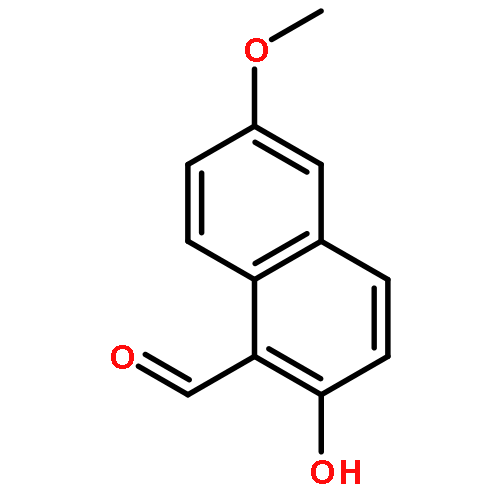
![Benzenamine, 4-methoxy-N-[(4-nitrophenyl)methylene]-, (E)-](http://img.cochemist.com/ccimg/40000/39963-93-4.png)
![Benzenamine, 4-methoxy-N-[(4-nitrophenyl)methylene]-, (E)-](http://img.cochemist.com/ccimg/40000/39963-93-4_b.png)
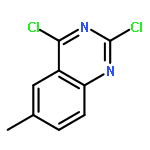
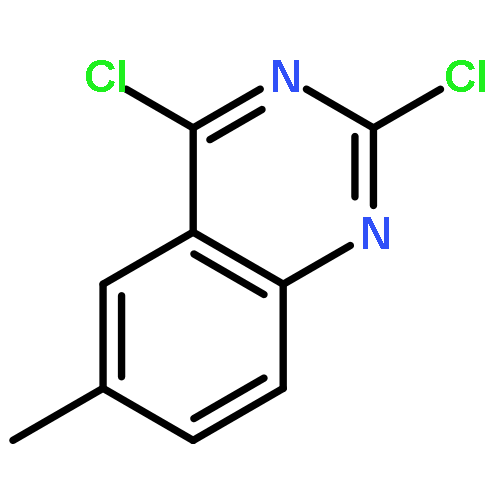
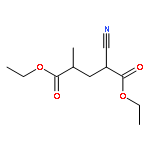



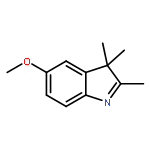
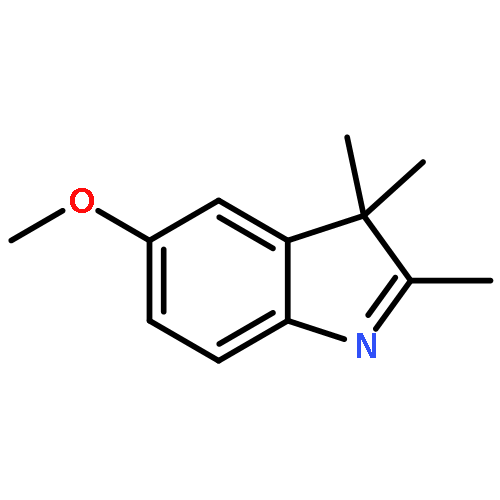
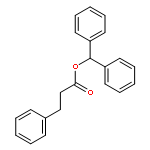
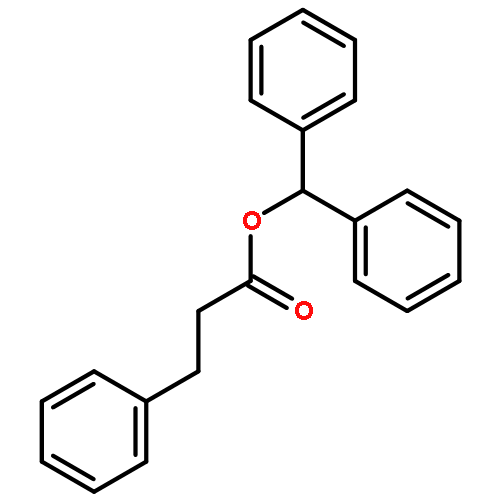






![Spiro[2H-1-benzopyran-2,2'-[2H]indole], 1',3'-dihydro-1',3',3'-trimethyl-6,8-dinitro-](/data/chemimg/16400/20200-64-0.png)
![Spiro[2H-1-benzopyran-2,2'-[2H]indole], 1',3'-dihydro-1',3',3'-trimethyl-6,8-dinitro-](/data/chemimg/16400/20200-64-0_b.png)
![Spiro[2H-indole-2,3'-[3H]naphtho[2,1-b]pyran], 1,3-dihydro-5-methoxy-1,3,3-trimethyl-](/data/chemimg/409500/20200-59-3.png)
![Spiro[2H-indole-2,3'-[3H]naphtho[2,1-b]pyran], 1,3-dihydro-5-methoxy-1,3,3-trimethyl-](/data/chemimg/409500/20200-59-3_b.png)


![Spiro[2H-1-benzopyran-2,2'-[2H]indole], 1',3'-dihydro-5'-methoxy-1',3',3'-trimethyl-6-nitro-](/data/chemimg/924500/16331-96-7.png)
![Spiro[2H-1-benzopyran-2,2'-[2H]indole], 1',3'-dihydro-5'-methoxy-1',3',3'-trimethyl-6-nitro-](/data/chemimg/924500/16331-96-7_b.png)




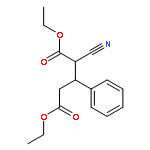



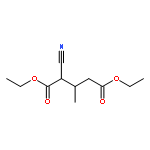
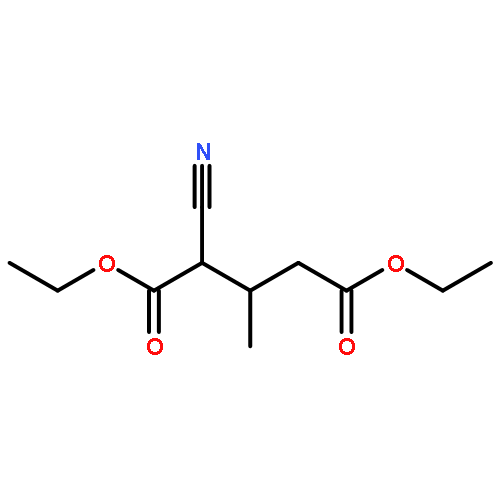
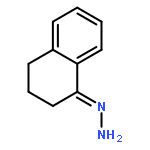
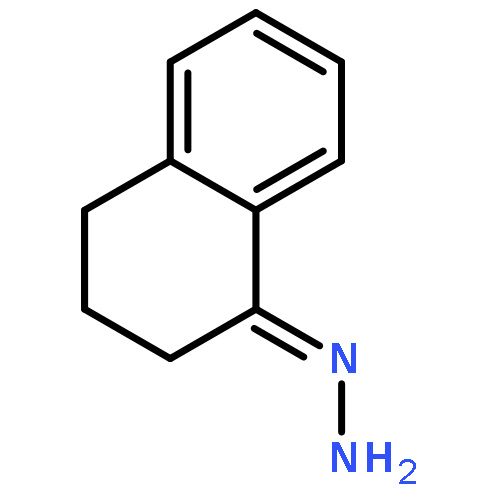


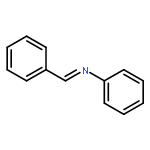
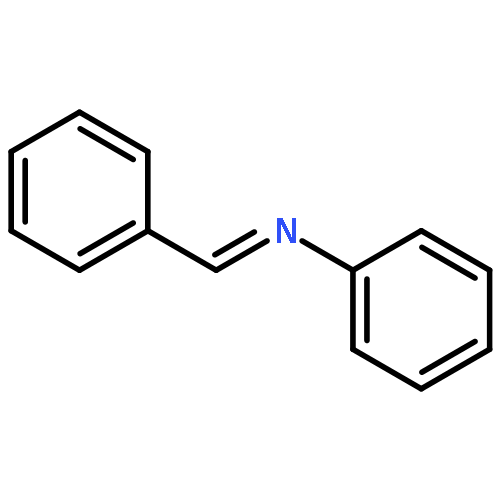
![Benzenamine, 4-methoxy-N-[(4-methoxyphenyl)methylene]-, (E)-](http://img.cochemist.com/ccimg/1700/1624-46-0.png)
![Benzenamine, 4-methoxy-N-[(4-methoxyphenyl)methylene]-, (E)-](http://img.cochemist.com/ccimg/1700/1624-46-0_b.png)
![Benzenamine, N-[(4-methoxyphenyl)methylene]-, (E)-](http://img.cochemist.com/ccimg/1700/1613-96-3.png)
![Benzenamine, N-[(4-methoxyphenyl)methylene]-, (E)-](http://img.cochemist.com/ccimg/1700/1613-96-3_b.png)
![2-[2-[2-(2,6-dichloroanilino)phenyl]acetyl]oxyacetic Acid](http://img.cochemist.com/ccimg/1000/954-67-6.png)
![2-[2-[2-(2,6-dichloroanilino)phenyl]acetyl]oxyacetic Acid](http://img.cochemist.com/ccimg/1000/954-67-6_b.png)
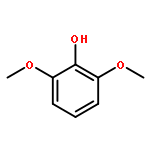
![6-Chloro-2-(iodomethyl)thiazolo[5,4-b]pyridine](http://img.cochemist.com/ccimg/1256500/1256478-42-8.png)
![6-Chloro-2-(iodomethyl)thiazolo[5,4-b]pyridine](http://img.cochemist.com/ccimg/1256500/1256478-42-8_b.png)
![6-Chloro-2-(chloromethyl)thiazolo[5,4-b]pyridine](http://img.cochemist.com/ccimg/1256500/1256478-41-7.png)
![6-Chloro-2-(chloromethyl)thiazolo[5,4-b]pyridine](http://img.cochemist.com/ccimg/1256500/1256478-41-7_b.png)
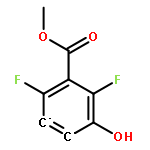
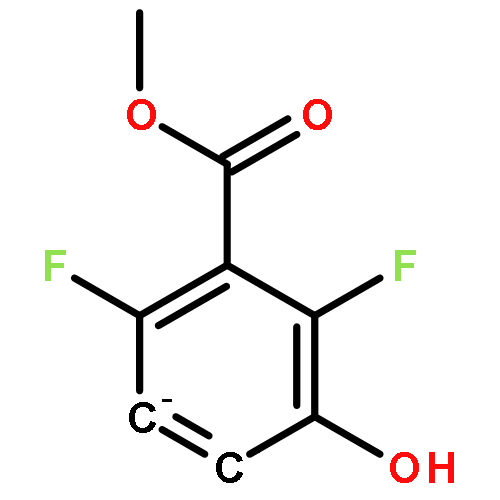







































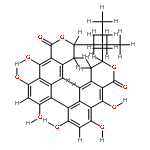
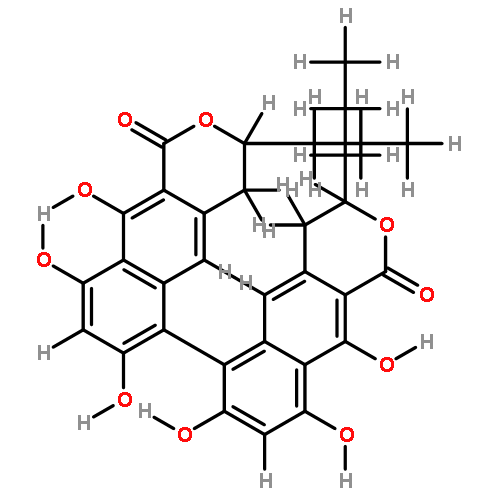
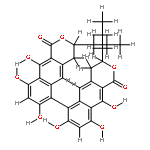
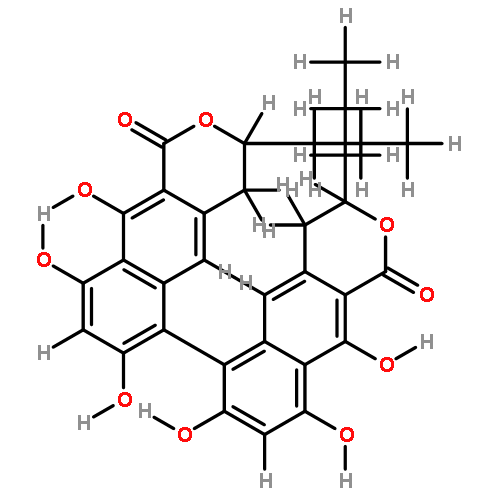
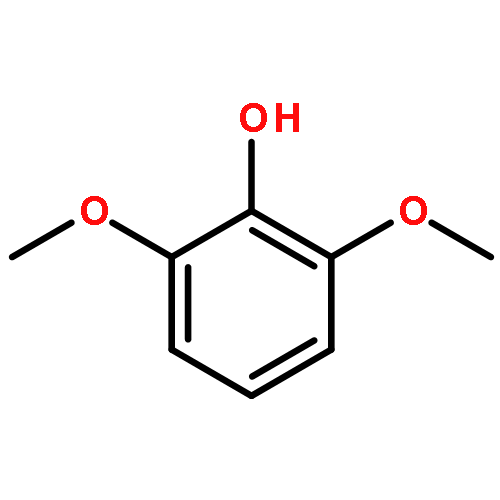
![[1,1'-Binaphthalene]-3,3'-dicarboxaldehyde, 2,2'-dihydroxy-, (1S)-](http://img.cochemist.com/data/images/141779-46-6.png)
![[1,1'-Binaphthalene]-3,3'-dicarboxaldehyde, 2,2'-dihydroxy-, (1S)-](http://img.cochemist.com/data/images/141779-46-6_b.png)
![Formamide, N-[(1R,2S)-2-hydroxy-1,2-diphenylethyl]-](/data/chemimg/122100/131350-15-7.png)
![Formamide, N-[(1R,2S)-2-hydroxy-1,2-diphenylethyl]-](/data/chemimg/122100/131350-15-7_b.png)
![Silane, (1,1-dimethylethyl)[2-(2R)-oxiranylethoxy]diphenyl-](http://img.cochemist.com/ccimg/126000/125975-53-3.png)
![Silane, (1,1-dimethylethyl)[2-(2R)-oxiranylethoxy]diphenyl-](http://img.cochemist.com/ccimg/126000/125975-53-3_b.png)










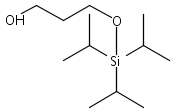
![Propanal, 3-[[tris(1-methylethyl)silyl]oxy]-](http://img.cochemist.com/ccimg/112000/111998-84-6.png)
![Propanal, 3-[[tris(1-methylethyl)silyl]oxy]-](http://img.cochemist.com/ccimg/112000/111998-84-6_b.png)
![5-Chloro-2-(chloromethyl)benzo[d]thiazole](http://img.cochemist.com/ccimg/110800/110704-19-3.png)
![5-Chloro-2-(chloromethyl)benzo[d]thiazole](http://img.cochemist.com/ccimg/110800/110704-19-3_b.png)

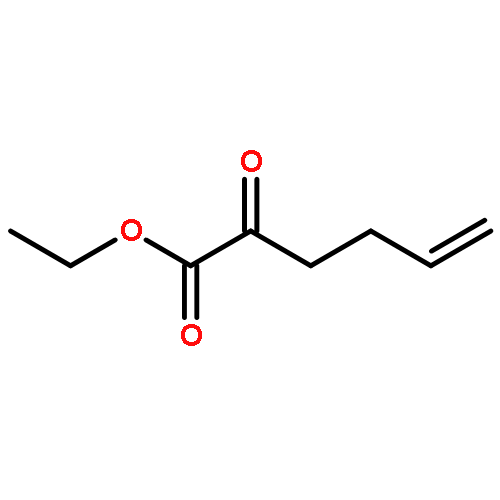

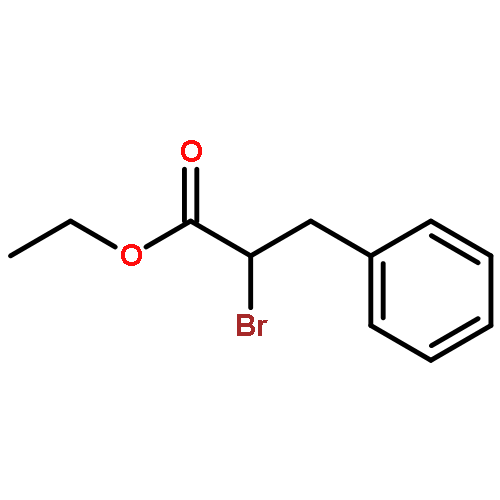
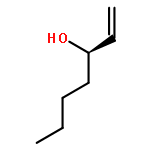
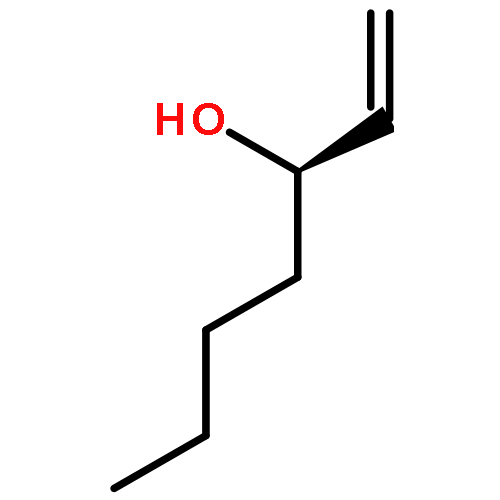


![Carbamic acid, N-[(1S)-2-oxo-1-(phenylmethyl)-2-[(phenylmethyl)amino]ethyl]-, 1,1-dimethylethyl ester](/data/chemimg/3758200/84235-32-5.png)
![Carbamic acid, N-[(1S)-2-oxo-1-(phenylmethyl)-2-[(phenylmethyl)amino]ethyl]-, 1,1-dimethylethyl ester](/data/chemimg/3758200/84235-32-5_b.png)




![Carbamicacid, N-[(1S)-2-methyl-1-[[(phenylmethyl)amino]carbonyl]propyl]-,1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/67200/67106-22-3.png)
![Carbamicacid, N-[(1S)-2-methyl-1-[[(phenylmethyl)amino]carbonyl]propyl]-,1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/67200/67106-22-3_b.png)











![DIMETHYL 2,2'-(9,9',10,10'-TETRAHYDROXY-7,7'-DIMETHOXY-1,1'-DIOXO<WBR />-3,3',4,4'-TETRAHYDRO-1H,1'H-8,8'-BIBENZO[G]ISOCHROMENE-3,3'-DIYL<WBR />)DIACETATE](http://img.cochemist.com/ccimg/35500/35483-50-2.png)
![DIMETHYL 2,2'-(9,9',10,10'-TETRAHYDROXY-7,7'-DIMETHOXY-1,1'-DIOXO<WBR />-3,3',4,4'-TETRAHYDRO-1H,1'H-8,8'-BIBENZO[G]ISOCHROMENE-3,3'-DIYL<WBR />)DIACETATE](http://img.cochemist.com/ccimg/35500/35483-50-2_b.png)



![Propanoic acid,2-[[(4-methylphenyl)sulfonyl]oxy]-, ethyl ester](http://img.cochemist.com/ccimg/33800/33798-77-5.png)
![Propanoic acid,2-[[(4-methylphenyl)sulfonyl]oxy]-, ethyl ester](http://img.cochemist.com/ccimg/33800/33798-77-5_b.png)






![2-Methylbenzo[g]quinazolin-4(3H)-one](http://img.cochemist.com/ccimg/16700/16673-88-4.png)
![2-Methylbenzo[g]quinazolin-4(3H)-one](http://img.cochemist.com/ccimg/16700/16673-88-4_b.png)







![Phenol,4-chloro-2,6-bis[(5-chloro-2-hydroxyphenyl)methyl]-](http://img.cochemist.com/ccimg/6700/6642-07-5.png)
![Phenol,4-chloro-2,6-bis[(5-chloro-2-hydroxyphenyl)methyl]-](http://img.cochemist.com/ccimg/6700/6642-07-5_b.png)








![5,9-DIHYDROXY-7-(3-HYDROXY-3-METHYLBUTYL)-8-METHOXY-2,2-DIMETHYL-<WBR />3,4-DIHYDRO-2H,6H-PYRANO[3,2-B]XANTHEN-6-ONE](http://img.cochemist.com/ccimg/3800/3772-56-3.png)
![5,9-DIHYDROXY-7-(3-HYDROXY-3-METHYLBUTYL)-8-METHOXY-2,2-DIMETHYL-<WBR />3,4-DIHYDRO-2H,6H-PYRANO[3,2-B]XANTHEN-6-ONE](http://img.cochemist.com/ccimg/3800/3772-56-3_b.png)






![3-[(6-Chlorothiazolo[5,4-b]pyridin-2-yl)methoxy]-2,6-difluorobenzamide](http://img.cochemist.com/ccimg/951200/951120-33-5.png)
![3-[(6-Chlorothiazolo[5,4-b]pyridin-2-yl)methoxy]-2,6-difluorobenzamide](http://img.cochemist.com/ccimg/951200/951120-33-5_b.png)