Co-reporter:Yang Gao;Yue-Jian Lin;Guo-Xin Jin
Dalton Transactions 2017 vol. 46(Issue 5) pp:1585-1592
Publication Date(Web):2017/01/31
DOI:10.1039/C6DT04550B
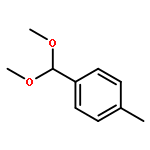
An iridium(III)-catalyzed C–N-bond-forming direct synthesis of carborane azo derivatives was developed. The reaction of o-carborane monocarboxylate with aryldiazonium salt in the presence of a catalytic amount of [Cp*IrCl2]2 and NaOAc in CH3CN at 80 °C gave the corresponding carborane azo derivative in good yield. A series of cyclometalated iridium/rhodium complexes that contain Cp*M–B (M = Ir, Rh) bonds, and are stabilised by the intramolecular coordination of nitrogen donors, were successfully isolated and structurally characterised.
Co-reporter:Wen-Ying Zhang, Yue-Jian Lin, Ying-Feng Han, and Guo-Xin Jin
Journal of the American Chemical Society 2016 Volume 138(Issue 33) pp:10700-10707
Publication Date(Web):July 27, 2016
DOI:10.1021/jacs.6b06622
Owing to the often-similar physical and chemical properties of structural isomers of organic molecules, large efforts have been made to develop efficient strategies to isolate specific isomers. However, facile separation of regioisomeric compounds remains difficult. Here we demonstrate a universal organometallic capsule in which two silver centers are rigidly separated from each other by two tetranuclear [Rh4] pyramidal frustums, which selectively encapsulate a specific isomer from mixtures. Not only is the present heterometallic capsule suitable as a host for the encapsulation of a series of aromatic compounds, but also the receptor shows widely differing specificity for the various isomers. Direct experimental evidence is provided for the selective encapsulation of a series of para (p)-disubstituted benzene derivatives, such as p-xylene, p-dichlorobenzene, p-dibromobenzene, and p-diiodobenzene. The size and shape matching, as well as the Ag−π interactions, are the main forces governing the extent of molecular recognition. The encapsulated guest p-xylene can be released by using the solid–liquid solvent washing strategy, and the other guest molecules are easily liberated by using light stimulus.
Co-reporter:Long Zhang, Tao Yan, Ying-Feng Han, F. Ekkehardt Hahn and Guo-Xin Jin
Dalton Transactions 2015 vol. 44(Issue 19) pp:8797-8800
Publication Date(Web):15 Apr 2015
DOI:10.1039/C5DT01311A
A series of complexes of novel chelating thione-alkene S(η2-CC)S tridentate ligands bound to late transition metals (Ir, Rh, Pd) were isolated and characterised. Counter-anions play an important role in the binding of the alkene moiety to the metals. Different solvents were observed to affect the stability of the rhodium complexes.
Co-reporter:Tao Yan;Li-Ying Sun;Yu-Xin Deng; Ying-Feng Han; Guo-Xin Jin
Chemistry - A European Journal 2015 Volume 21( Issue 49) pp:17610-17613
Publication Date(Web):
DOI:10.1002/chem.201503709
Abstract
A synthetic strategy for the preparation of a series of polyimidazolium macrocycles from the corresponding dicarbene-derived metallacycles is described. Photodimerization of terminal cinnamic esters (UV-irradiation, λ=365 nm) produces the closed metallacycles with perfect stereoselectivity and high yields. Subsequent removal of the template from the photodimerization product results in polyimidazolium macrocycles. The size and shape of the receptor can be tuned easily by changing the length and breadth of the internal bridging groups of the ligands. Preliminary investigation shows the potential of the macrocycle as iodide sensor.
Co-reporter: Ying-Feng Han; Guo-Xin Jin;Constantin G. Daniliuc; F. Ekkehardt Hahn
Angewandte Chemie International Edition 2015 Volume 54( Issue 16) pp:4958-4962
Publication Date(Web):
DOI:10.1002/anie.201411006
Abstract
Molecular rectangles were obtained from two bis(NHC) ligands, each featuring two terminal coumarin groups and two Ag+, Au+, or Cu+ ions. Upon UV irradiation (λ=365 nm), the dinuclear complexes undergo photochemical modification through a [2+2] cycloaddition reaction of two adjacent coumarin moieties to give a macrocyclic tetra(NHC) ligand. The photodimerization of the coumarin pendants proceeds stereoselectively to give the syn-head-head isomers in all cases. Subsequent irradiation at λ=254 nm initiates a photocleavage reaction with reconstitution of the initial dinuclear complexes with coumarin pendants.
Co-reporter:Chun-Xiang Wang, Yang Gao, Yu-Xin Deng, Yue-Jian Lin, Ying-Feng Han, and Guo-Xin Jin
Organometallics 2015 Volume 34(Issue 24) pp:5801-5806
Publication Date(Web):December 15, 2015
DOI:10.1021/acs.organomet.5b00977
The modification of dinuclear silver metallcycles via photochemical [2 + 2] cycloaddition is described. Reaction of benzimidazole with trans-4,4′-dibromostilbene gives trans-4,4′-bis(1H-benzo[d]imidazolyl)stilbene (L), which after subsequent dialkylation with alkyl bromides and anion exchange yields the corresponding dibenzimidazolium salts H2-1(PF6)2 and H2-2(PF6)2. The dibenzimidazolium salts react with Ag2O to give the dinuclear silver(I) tetracarbene metallacycles Ag2(1)2(PF6)2 and Ag2(2)2(PF6)2 in high yield. Irradiation (UV light, λ 365 nm) of Ag2(1)2(PF6)2 and Ag2(2)2(PF6)2 in [D6]DMSO resulted in rapid conversion into the corresponding dinuclear cyclobutane-carbene complexes Ag2(3)(PF6)2 and Ag2(4)(PF6)2 quantitatively. The cyclobutane-bridged polycarbene precursors were isolated in good yields as their tetrabenzimidazolium salts.
Co-reporter: Ying-Feng Han; Guo-Xin Jin;Constantin G. Daniliuc; F. Ekkehardt Hahn
Angewandte Chemie 2015 Volume 127( Issue 16) pp:5042-5046
Publication Date(Web):
DOI:10.1002/ange.201411006
Abstract
Molekulare Rechtecke werden aus zwei Bis(NHC)-Liganden, die jeweils zwei endständige Cumaringruppen tragen, sowie zwei Ag+-, Au+- oder Cu+-Ionen erhalten. Bei UV-Bestrahlung (λ=365 nm) erfolgt eine photochemische Modifikation der zweikernigen Komplexe durch eine [2+2]-Cycloaddition von zwei gegenüberliegenden Cumaringruppen unter Bildung eines makrocyclischen Tetra(NHC)-Liganden. Die Photodimerisierung der Cumarinsubstituenten verläuft stereoselektiv und ergibt ausschließlich das syn-Kopf-Kopf-Isomer. Eine nachfolgende Bestrahlung bei λ=254 nm initiiert die Photospaltung unter Rückbildung des zweikernigen Ausgangskomplexes mit vier Cumarinsubstituenten.
Co-reporter:Ying-Feng Han and Guo-Xin Jin
Chemical Society Reviews 2014 vol. 43(Issue 8) pp:2799-2823
Publication Date(Web):05 Feb 2014
DOI:10.1039/C3CS60343A
Half-sandwich Cp*Ir and Cp*Rh metalacycles have been successfully applied in traditional domains encompassing organic transformations and catalysis in recent years, especially the catalytic activation of C–H bonds. Cyclometalation has proven to be a highly attractive and versatile synthetic method for the formation of organometallic metalacycles. This review intends to describe isolated and well-defined cyclometalated iridium/rhodium complexes that contain a Cp*M–C (M = Ir, Rh) bond stabilised by the intramolecular coordination of neutral donor atoms (N, C, O or P). The formation of metalamacrocycles and cages employing cyclometalated approaches is discussed. In focusing on selected mechanistic insights garnered from iridium/rhodium-catalysed functionalisation of C–H bonds involving cyclometalated complexes, a limited number of substrates will be discussed, but a broad range of mechanistic features is highlighted.
Co-reporter:Ying-Feng Han ; Long Zhang ; Lin-Hong Weng ;Guo-Xin Jin
Journal of the American Chemical Society 2014 Volume 136(Issue 41) pp:14608-14615
Publication Date(Web):September 12, 2014
DOI:10.1021/ja508543y
Although reversible covalent activation of molecular hydrogen (H2) by transition-metal complexes is a common reaction, H2-mediated sophisticated reversible arrangements of organometallic frameworks have not yet been described. Herein, we report unusual organometallic transformations in solution that can be accomplished by uptake or release of H2. An efficient route for synthesizing air- and moisture-stable 16-electron M2L2-type metallacycles under very mild conditions has been developed. The new organometallic metallacycles favor the binding of small ligands such as MeCN, Cl–, CO, and pyridine. The reaction of a coordinatively unsaturated 16-electron M2L2-type macrocyclic complex featuring thione ligands with 1 atm of H2 leads to the isolation of a 18-electron M2L3-type cylinder, along with hydride species. Remarkably, the obtained mixture underwent loss of H2 in a facile manner upon heating to re-form the starting M2L2-type complex. A possible mechanism is proposed for the reversible transformations.
Co-reporter:Wen-Ying Zhang, Ying-Feng Han, Lin-Hong Weng, and Guo-Xin Jin
Organometallics 2014 Volume 33(Issue 12) pp:3091-3095
Publication Date(Web):June 6, 2014
DOI:10.1021/om500338w
Tetranuclear half-sandwich iridium or rhodium complexes were obtained in good yields from the reactions of the binuclear half-sandwich metal precursors [Cp*2M2(μ-CA)]Cl2 (1a, M = Ir; 1b, M = Rh; CA = chloranilate) or [Cp*2M2(μ-DHNA)]Cl2 (2a, M = Ir; 2b, M = Rh; H2DHNA = 6,11-dihydroxynaphthacene-5,12-dione) with diPyNI (diPyNI = N,N-bis(4-pyridyl)-1,4,5,8-naphthalenetetracarboxydiimide) in the presence of AgOTf (OTf = CF3SO3) in CH3OH, respectively. The new metallarectangles have been characterized by elemental analysis, FT-IR, 1H NMR, electrospray mass spectrometry (ESI-MS), and UV/vis absorption spectroscopy. The interactions of these metallarectangles with aromatic molecules, especially pyrene, in solution have been studied by various NMR techniques (1D, DOSY, and ROESY) and UV–vis absorption. DOSY measurements suggest that the interactions between metallarectangle 4a and pyrene are outside of the cavity. The strong π···π interactions between pyrene and the naphthalenetetracarboxydiimide ring of metallarectangle 4a were further supported by single-crystal X-ray diffraction data; pyrene molecules are found outside the cavity of the metallarectangle.
Co-reporter:Long Zhang, Tao Yan, Ying-Feng Han, F. Ekkehardt Hahn and Guo-Xin Jin
Dalton Transactions 2015 - vol. 44(Issue 19) pp:NaN8800-8800
Publication Date(Web):2015/04/15
DOI:10.1039/C5DT01311A
A series of complexes of novel chelating thione-alkene S(η2-CC)S tridentate ligands bound to late transition metals (Ir, Rh, Pd) were isolated and characterised. Counter-anions play an important role in the binding of the alkene moiety to the metals. Different solvents were observed to affect the stability of the rhodium complexes.
Co-reporter:Yang Gao, Yue-Jian Lin, Ying-Feng Han and Guo-Xin Jin
Dalton Transactions 2017 - vol. 46(Issue 5) pp:NaN1592-1592
Publication Date(Web):2016/12/22
DOI:10.1039/C6DT04550B
An iridium(III)-catalyzed C–N-bond-forming direct synthesis of carborane azo derivatives was developed. The reaction of o-carborane monocarboxylate with aryldiazonium salt in the presence of a catalytic amount of [Cp*IrCl2]2 and NaOAc in CH3CN at 80 °C gave the corresponding carborane azo derivative in good yield. A series of cyclometalated iridium/rhodium complexes that contain Cp*M–B (M = Ir, Rh) bonds, and are stabilised by the intramolecular coordination of nitrogen donors, were successfully isolated and structurally characterised.
Co-reporter:Ying-Feng Han and Guo-Xin Jin
Chemical Society Reviews 2014 - vol. 43(Issue 8) pp:NaN2823-2823
Publication Date(Web):2014/02/05
DOI:10.1039/C3CS60343A
Half-sandwich Cp*Ir and Cp*Rh metalacycles have been successfully applied in traditional domains encompassing organic transformations and catalysis in recent years, especially the catalytic activation of C–H bonds. Cyclometalation has proven to be a highly attractive and versatile synthetic method for the formation of organometallic metalacycles. This review intends to describe isolated and well-defined cyclometalated iridium/rhodium complexes that contain a Cp*M–C (M = Ir, Rh) bond stabilised by the intramolecular coordination of neutral donor atoms (N, C, O or P). The formation of metalamacrocycles and cages employing cyclometalated approaches is discussed. In focusing on selected mechanistic insights garnered from iridium/rhodium-catalysed functionalisation of C–H bonds involving cyclometalated complexes, a limited number of substrates will be discussed, but a broad range of mechanistic features is highlighted.










![Benzo[lmn][3,8]phenanthroline-1,3,6,8(2H,7H)-tetrone, 2,7-di-4-pyridinyl-](/data/chemimg/102500/34151-49-0.png)
![Benzo[lmn][3,8]phenanthroline-1,3,6,8(2H,7H)-tetrone, 2,7-di-4-pyridinyl-](/data/chemimg/102500/34151-49-0_b.png)


![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)

