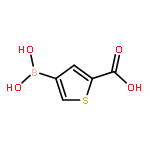
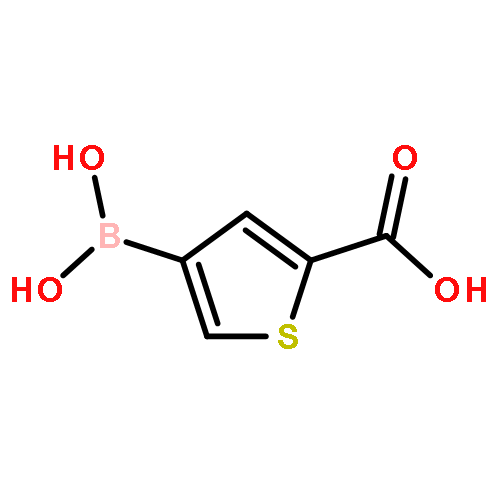
Co-reporter:Mahesh P. Paudyal, Li Wu, Zhong-Yin Zhang, Christopher D. Spilling, Chung F. Wong
Bioorganic & Medicinal Chemistry 2014 22(24) pp: 6781-6788
Publication Date(Web):
DOI:10.1016/j.bmc.2014.10.042
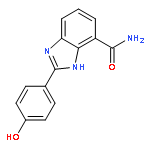
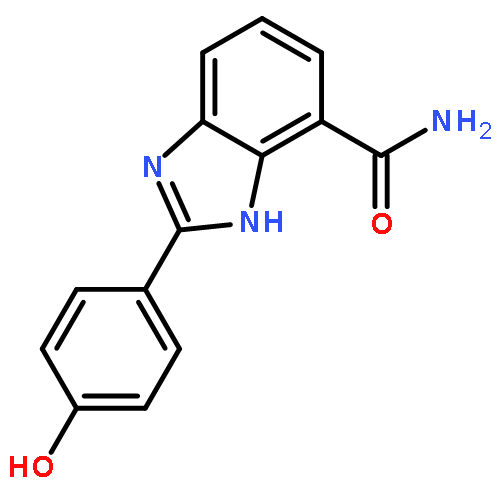
Co-reporter:Zunnan Huang;Yantao He;Xian Zhang;Andrea Gunawan;Li Wu;Zhong-Yin Zhang;Chung F. Wong
Chemical Biology & Drug Design 2010 Volume 76( Issue 2) pp:85-99
Publication Date(Web):
DOI:10.1111/j.1747-0285.2010.00996.x
Yersinia pestis causes diseases ranging from gastrointestinal syndromes to bubonic plague and could be misused as a biological weapon. As its protein tyrosine phosphatase YopH has already been demonstrated as a potential drug target, we have developed two series of forty salicylic acid derivatives and found sixteen to have micromolar inhibitory activity. We designed these ligands to have two chemical moieties connected by a flexible hydrocarbon linker to target two pockets in the active site of the protein to achieve binding affinity and selectivity. One moiety possessed the salicylic acid core intending to target the phosphotyrosine-binding pocket. The other moiety contained different chemical fragments meant to target a nearby secondary pocket. The two series of compounds differed by having hydrocarbon linkers with different lengths. Before experimental co-crystal structures are available, we have performed molecular docking to predict how these compounds might bind to the protein and to generate structural models for performing binding affinity calculation to aid future optimization of these series of compounds.
Co-reporter:Mayank Goyal ; Michael Rizzo ; Frank Schumacher ;Chung F. Wong
Journal of Medicinal Chemistry 2009 Volume 52(Issue 18) pp:5582-5585
Publication Date(Web):August 24, 2009
DOI:10.1021/jm900974p
We modeled the kinetics of drug binding to protein kinases in the EGF signaling pathway relevant to non-small-cell lung cancer and found that binding kinetics could influence therapeutic potential, that fast binding kinetics was advantageous for most targets with a couple of exceptions, that targeting some protein kinases could enhance rather than attenuate the pathway, and that IC50 could be sensitive to the kinetic parameters of drug binding.
Co-reporter:Zunnan Huang and Chung F. Wong
The Journal of Physical Chemistry B 2009 Volume 113(Issue 43) pp:14343-14354
Publication Date(Web):October 7, 2009
DOI:10.1021/jp907375b
Reliable prediction of protein−ligand docking pose requires proper account of induced fit effects. Treating both the ligand and the protein as flexible molecules is still challenging because many degrees of freedom are involved. Peptides are one type of ligand that are particularly difficult to study because of their extreme flexibility. In this study, we tested a molecular dynamics-based simulated-annealing cycling protocol in docking peptides to four protein kinases and two phosphatases using two implicit-solvent models: a distance-dependent dielectric model (ε(r) = 4r) and a version of the Generalized Born model termed GBMV. We found that the simpler ε(r) = 4r model identified docking pose better than the more expensive GBMV model. In addition, rescoring structures obtained from one implicit-solvent model with the other identified good docking poses for all six systems studied. Including protein energy in scoring also improved results.
Co-reporter:Baojing Zhou and Chung F. Wong
The Journal of Physical Chemistry A 2009 Volume 113(Issue 17) pp:5144-5150
Publication Date(Web):March 31, 2009
DOI:10.1021/jp810827w
Although various groups have studied the phosphorylation mechanism of the insulin receptor tyrosine kinase (IRK), an unanimous picture has not yet emerged. In this work, we performed a computational study to gain further insights. We first built a structural model of the reactant complex with the guide of several crystal structures and previous computational studies of the cyclic AMP-dependent protein kinase. We then optimized the structure by performing geometry optimization using a quantum mechanical model containing nearly 300 atoms. A reaction path was then traced between the reactant and the product by using a multiple coordinate-driven method. The calculations mapped out a sequence of structural changes depicting the conversion of the reactant to the product. Analysis of the structural changes revealed the formation of a dissociative transition state and the involvement of a proton transfer from the hydroxyl group of the tyrosyl residue of the peptide substrate to a conserved aspartate in the active site of the enzyme. The proton transfer began well before the transition state was reached and finished only shortly before the product was completely formed. In addition, the formation of a hydrogen bonding network among Arg1136, Asp1132, the γ-phosphate of ATP, and the tyrosine residue of the substrate appeared to hold the latter two in a near-attack position for reaction. The model estimated a reaction barrier of 14 kcal/mol, semiquantitatively in accord with experiment.

![N-[3-[5-CHLORO-2-[2-METHOXY-4-(4-METHYLPIPERAZIN-1-YL)ANILINO]PYRIMIDIN-4-YL]OXYPHENYL]PROP-2-ENAMIDE](http://img.cochemist.com/ccimg/1213300/1213269-23-8.png)
![N-[3-[5-CHLORO-2-[2-METHOXY-4-(4-METHYLPIPERAZIN-1-YL)ANILINO]PYRIMIDIN-4-YL]OXYPHENYL]PROP-2-ENAMIDE](http://img.cochemist.com/ccimg/1213300/1213269-23-8_b.png)
![(S)-4-(2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-7-(piperidin-3-ylmethoxy)-1H-imidazo[4,5-c]pyridin-4-yl)-2-methylbut-3-yn-2-ol](http://img.cochemist.com/ccimg/937200/937174-76-0.png)
![(S)-4-(2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-7-(piperidin-3-ylmethoxy)-1H-imidazo[4,5-c]pyridin-4-yl)-2-methylbut-3-yn-2-ol](http://img.cochemist.com/ccimg/937200/937174-76-0_b.png)
![Pyrido[2,3-d]pyrimidin-7(8H)-one,6-(2,6-dichlorophenyl)-2-[(4-fluoro-3-methylphenyl)amino]-8-methyl-](http://img.cochemist.com/ccimg/287300/287204-45-9.png)
![Pyrido[2,3-d]pyrimidin-7(8H)-one,6-(2,6-dichlorophenyl)-2-[(4-fluoro-3-methylphenyl)amino]-8-methyl-](http://img.cochemist.com/ccimg/287300/287204-45-9_b.png)