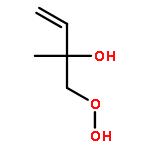
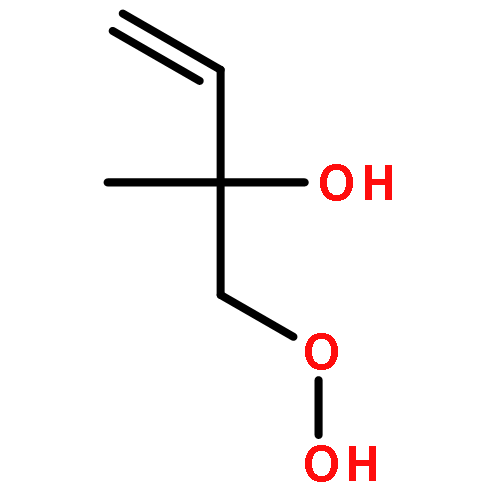
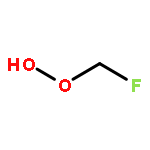
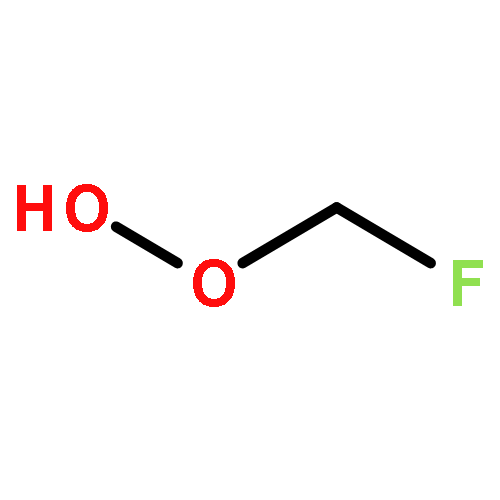
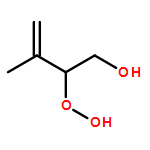
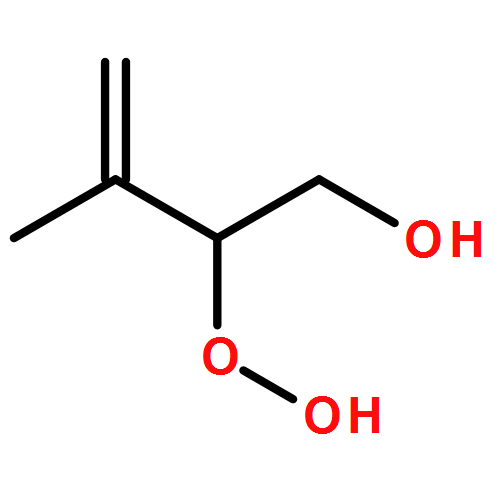
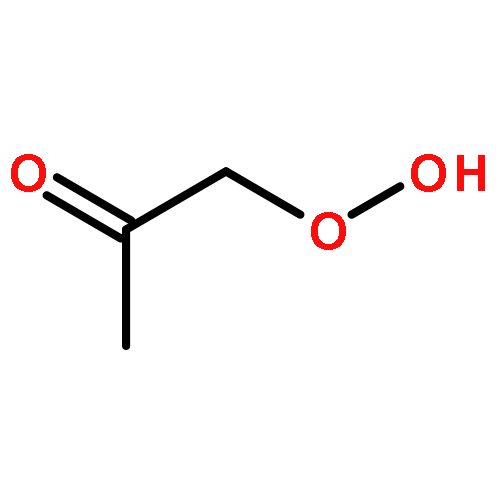
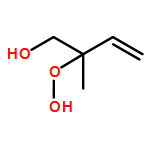
Co-reporter:Paul M. Cropper, Devon K. Overson, Robert A. Cary, Delbert J. Eatough, Judith C. Chow, Jaron C. Hansen
Atmospheric Environment 2017 Volume 169(Volume 169) pp:
Publication Date(Web):1 November 2017
DOI:10.1016/j.atmosenv.2017.09.019
•A field instrument capable of measuring particle phase organic compounds on an hourly averaged basis has been developed.•Measurement of atmospherically relevant organic compounds such as levoglucosan, and PAHs has been demonstrated.•The integration of a miniature GC-MS provides a cost efficient method for the measurement of organic compounds in PM2.5.Particulate matter (PM) is among the most harmful air pollutants to human health, but due to its complex chemical composition is poorly characterized. A large fraction of PM is composed of organic compounds, but these compounds are not regularly monitored due to limitations in current sampling and analysis techniques. The Organic Aerosol Monitor (GC-MS OAM) combines a collection device with thermal desorption, gas chromatography and mass spectrometry to quantitatively measure the carbonaceous components of PM on an hourly averaged basis. The GC-MS OAM is fully automated and has been successfully deployed in the field. It uses a chemically deactivated filter for collection followed by thermal desorption and GC-MS analysis. Laboratory tests show that detection limits range from 0.2 to 3 ng for 16 atmospherically relevant compounds, with the possibility for hundreds more. The GC-MS OAM was deployed in the field for semi-continuous measurement of the organic markers, levoglucosan, dehydroabietic acid, and polycyclic aromatic hydrocarbons (PAHs) from January to March 2015. Results illustrate the significance of this monitoring technique to characterize the organic components of PM and identify sources of pollution.Download high-res image (154KB)Download full-size image
Co-reporter:Jaron C. Hansen, Randall R. Friedl and Stanley P. Sander
The Journal of Physical Chemistry A 2008 Volume 112(Issue 39) pp:9229-9237
Publication Date(Web):August 15, 2008
DOI:10.1021/jp8007706
The rate coefficients for the reactions OH + ClOOCl → HOCl + ClOO (eq 5) and OH + Cl2O → HOCl + ClO (eq 6) were measured using a fast flow reactor coupled with molecular beam quadrupole mass spectrometry. OH was detected using resonance fluorescence at 309 nm. The measured Arrhenius expressions for these reactions are k5 = (6.0 ± 3.5) × 10−13 exp((670 ± 230)/T) cm3 molecule−1 s−1 and k6 = (5.1 ± 1.5) × 10−12 exp((100 ± 92)/T) cm3 molecule−1 s−1, respectively, where the uncertainties are reported at the 2σ level. Investigation of the OH + ClOOCl potential energy surface using high level ab initio calculations indicates that the reaction occurs via a chlorine abstraction from ClOOCl by the OH radical. The lowest energy pathway is calculated to proceed through a weak ClOOCl−OH prereactive complex that is bound by 2.6 kcal mol−1 and leads to ClOO and HOCl products. The transition state to product formation is calculated to be 0.59 kcal mol−1 above the reactant energy level. Inclusion of the OH + ClOOCl rate data into an atmospheric model indicates that this reaction contributes more than 15% to ClOOCl loss during twilight conditions in the Arctic stratosphere. Reducing the rate of ClOOCl photolysis, as indicated by a recent re-examination of the ClOOCl UV absorption spectrum, increases the contribution of the OH + ClOOCl reaction to polar stratospheric loss of ClOOCl.
Co-reporter:Alecia M. English and Jaron C. Hansen, Joseph J. Szente and M. Matti Maricq
The Journal of Physical Chemistry A 2008 Volume 112(Issue 39) pp:9220-9228
Publication Date(Web):August 16, 2008
DOI:10.1021/jp800727a
The gas phase reactions of CH3O2 + CH3O2, HO2 + HO2, and CH3O2 + HO2 in the presence of water vapor have been studied at temperatures between 263 and 303 K using laser flash photolysis coupled with UV time-resolved absorption detection at 220 and 260 nm. Water vapor concentrations were quantified using tunable diode laser spectroscopy operating in the mid-IR. The HO2 self-reaction rate constant is significantly enhanced by water vapor, consistent with what others have reported, whereas the CH3O2 self-reaction and the cross-reaction (CH3O2 + HO2) rate constants are nearly unaffected. The enhancement in the HO2 self-reaction rate coefficient occurs because of the formation of a strongly bound (6.9 kcal mol−1) HO2·H2O complex during the reaction mechanism where the H2O acts as an energy chaperone. The nominal impact of water vapor on the CH3O2 self-reaction rate coefficient is consistent with recent high level ab initio calculations that predict a weakly bound CH3O2·H2O complex (2.3 kcal mol−1). The smaller binding energy of the CH3O2·H2O complex does not favor its formation and consequent participation in the methyl peroxy self-reaction mechanism.
Co-reporter:Jaron C. Hansen, Bradley A. Flowers, John F. Stanton
Journal of Molecular Structure: THEOCHEM 2006 Volume 768(1–3) pp:111-118
Publication Date(Web):31 August 2006
DOI:10.1016/j.theochem.2006.05.045
Perturbations of the vibrational and electronic energy levels of isolated HO2 by formation of a doubly hydrogen bound association complex with H2O2 have been investigated using ab initio calculations. Minimum potential energy surfaces for a six-membered and five-membered ring structures are found. The binding energy, vibrational frequencies and low lying electronic excited states are predicted for the lowest energy structure of the complex and are calibrated by calculations of the separated monomers. The lowest energy conformation of the complex is found to be bound by 7.9 kcal mol−1. We find that complexation with H2O2 blue shifts HO2 electronic excited states by ∼0.1 eV and redshifts H2O2 states by ∼0.1 eV. Two additional, though exceedingly weak, electronic transitions are found to borrow oscillator strength from the ground state of the hydrogen bonded complex. The enthalpy and entropy of formation of the complex is estimated to be ΔH0 (270 K)=-35.8 kJ mol-1 and ΔS0 (270 K)=-138.3 J mol-1 K-1. The existence of a HO2–H2O2 complex is discussed in terms of its potential role in atmospheric photochemistry and laboratory kinetic studies.
.jpg)