Co-reporter:Gummadi Durgaprasad, Javier A. Luna, Kyle D. Spielvogel, Christian Haas, Scott K. Shaw, and Scott R. Daly
Organometallics October 23, 2017 Volume 36(Issue 20) pp:4020-4020
Publication Date(Web):October 11, 2017
DOI:10.1021/acs.organomet.7b00623
Here we describe the synthesis, structures, and reactivity of Ru complexes containing a triaryl, redox-active S2N2 ligand derived from o-phenylenediamine and thioanisole subunits. The coordination chemistry of N,N′-bis[2-(methylthio)phenyl]-1,2-diaminobenzene [H2(MeSNNSMe)] was established by treating RuCl2(PPh3)3 with H2(MeSNNSMe) to yield {Ru[H2(MeSNNSMe)]Cl(PPh3)}Cl (1). Coordinated H2(MeSNNSMe) was sequentially deprotonated to form Ru[H(MeSNNSMe)]Cl(PPh3) (2) followed by the five-coordinate, square pyramidal complex Ru(MeSNNSMe)(PPh3) (3). Single-crystal X-ray diffraction (XRD) studies revealed that the ligand structurally rearranged around the metal at each deprotonation step to conjugate the adjacent aryl groups with the o-phenylenediamine backbone. Deprotonation of 2 with NaBH4 or treatment of 3 with BH3·tetrahydrofuran (THF) yielded Ru[(μ-H)BH2](MeSNNSMe)(PPh3) (5) with BH3 bound across a Ru–N bond in a metal–ligand cooperative fashion. The cyclic voltammogram of 3 in THF revealed three redox events consistent with one-electron oxidations and reductions of the o-phenylenediamine backbone and the metal (Ru3+/Ru2+). Reactions of 3 with CO, HBF4, and benzoic acid yielded the new complexes Ru(MeSNNSMe)(CO)(PPh3), {Ru[H(MeSNNSMe)](PPh3)(THF)}BF4, and Ru[H(MeSNNSMe)](PPh3)(PhCO2), indicating broader suitability for small molecule binding and reactivity studies. Subsequent nuclear magnetic resonance spectroscopy, infrared spectroscopy, and mass spectrometry data are reported in addition to molecular structures obtained from single-crystal XRD studies.
Co-reporter:Kyounghoon Lee;Courtney M. Donahue
Dalton Transactions 2017 vol. 46(Issue 29) pp:9394-9406
Publication Date(Web):2017/07/25
DOI:10.1039/C7DT02144E
The synthesis, coordination chemistry, and reactivity of two diphosphines containing the cyclic triaminoborane 1,8,10,9-triazaboradecalin (TBD) are described. To evaluate the ligand-centered reactivity of PhTBDPhos and iPrTBDPhos, the complexes (PhTBDPhos)MCl2 and (iPrTBDPhos)MCl2, where M = Ni and Pd, were prepared and characterized by elemental analysis, multinuclear NMR spectroscopy (1H, 13C, 31P, and 11B), and single-crystal X-ray diffraction (XRD). Despite very low boron Lewis acidity in the TBD backbone, (PhTBDPhos)NiCl2 (1) and (PhTBDPhos)PdCl2 (3) react with H2O, alcohols, and hydrated fluoride reagents in the presence of NEt3 to yield trans H–O or H–F addition across the bridgehead N–B bond. In contrast, iPrTBDPhos shows no appreciable reactivity when bound to NiCl2 (2) and PdCl2 (4), which is attributed to the sterically-bulky isopropyl substituents blocking substrate access to boron in the TBD backbone. The new complexes {[(PhTBDPhos-H2O)Ni]2(μ-OH)2}Cl2 (5), {[(PhTBDPhos-H2O)Pd]2(μ-OH)2}Cl2 (6), (PhTBDPhos-MeOH)NiCl2 (7), (PhTBDPhos-MeOH)PdCl2 (8), (PhTBDPhos-C3H5OH)PdCl2 (9), and {[(PhTBDPhos-HF)Ni]2(μ-OH)2}Cl2 (10) were isolated, and all but 6 were structurally characterized by single-crystal XRD. Multinuclear NMR studies revealed that isolated, crystallographically-authenticated samples of 5–9 lose ligand-bound water or alcohol with reappearance of starting materials 1 and 3 when dissolved in NMR solvents. Addition of NEt3 attenuated the water and alcohol loss from 5–9 to allow 1H, 13C, 31P, and 11B NMR data to be collected for all the compounds, confirming the determined structures. Additional reactivity experiments with NaOMe and fluoride reagents suggested that participation of the bridgehead nitrogen in the TBD backbone is important for promoting reactivity at boron when PhTBDPhos is bound to Ni and Pd. The term “cooperative ligand-centered reactivity” (CLR) is proposed to define chemical reactions that appear to require participation of more than one atom on the ligand, such as those reported here.
Co-reporter:Kyounghoon Lee, Haochuan Wei, Anastasia V. Blake, Courtney M. Donahue, Jason M. Keith and Scott R. Daly
Dalton Transactions 2016 vol. 45(Issue 24) pp:9774-9785
Publication Date(Web):24 May 2016
DOI:10.1039/C6DT00200E
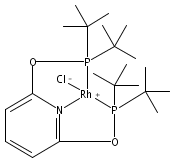
Here we report P K-edge, Cl K-edge, and Rh L3-edge X-ray absorption spectroscopy (XAS) data for Rh[C5H3N-2,6-(XPtBu2)2]Cl, where X = O (tBuPONOP; 1) or CH2 (tBuPNP; 2). Solid-state XAS data for 1 and 2 were compared to density functional theory (DFT) and time-dependent density functional theory (TDDFT) calculations to identify how changing the PNP pincer linker from O to CH2 affected electronic structure and bonding at Rh(I). Pronounced differences in XAS peak intensities and energies were observed. The P K-edge XAS data revealed a large increase in Rh 4dx2−y2 and P 3p orbital-mixing (Rh–P σ*) in 1 compared to 2, and pronounced transition energy variations reflected marked differences in orbital energies and compositions. By comparison, the Cl K-edge XAS data revealed only subtle differences in Rh–Cl covalency, although larger splitting between the Rh–Cl π* and σ* transitions was observed in 2. Analysis of the occupied MOs from DFT (HOMO, HOMO−1, HOMO−2, and HOMO−3) and comparison to the unoccupied MOs involved in XAS revealed a relatively uniform energy increase (ca. 0.3–0.5 eV) for all five 4d-derived molecular orbitals in Rh(tBuPNP)Cl (2) compared to Rh(tBuPONOP)Cl (1). The energy shift was relatively invariant with respect to differences in orbital symmetry, bonding type (σ or π), and orbital mixing, which suggested that the increase could be attributed to electrostatic effects. The change in d-orbital energies are consistent with known reactivity differences of Rh(tBuPONOP)+ and Rh(tBuPNP)+ towards CO, H2, and CH2Cl2, and are explained here by considering how d-orbital energies affect covalent L → M σ bonding and M → L π backbonding.
Co-reporter:Kyounghoon Lee, Haochuan Wei, Anastasia V. Blake, Courtney M. Donahue, Hyuk Yong Kwon, Jason M. Keith and Scott R. Daly
Dalton Transactions 2016 vol. 45(Issue 27) pp:11198-11198
Publication Date(Web):22 Jun 2016
DOI:10.1039/C6DT90109C
Correction for ‘Ligand K-edge XAS, DFT, and TDDFT analysis of pincer linker variations in Rh(I) PNP complexes: reactivity insights from electronic structure’ by Jason M. Keith, Scott R. Daly, et al., Dalton Trans., 2016, 45, 9774–9785.
Co-reporter:Courtney M. Donahue, Samuel P. McCollom, Chelsie M. Forrest, Anastasia V. Blake, Brian J. Bellott, Jason M. Keith, and Scott R. Daly
Inorganic Chemistry 2015 Volume 54(Issue 12) pp:5646-5659
Publication Date(Web):May 21, 2015
DOI:10.1021/ic503125b
Despite the long-standing use of phosphine and diphosphine ligands in coordination chemistry and catalysis, questions remain as to their effects on metal–ligand bonding in transition metal complexes. Here we report ligand K-edge XAS, DFT, and TDDFT studies aimed at quantifying the impact of coordination geometry, diphosphine bite angle, and phosphine trans influence on covalency in M–P and M–Cl bonds. A series of four-coordinate NiCl2 and PdCl2 complexes containing PPh3 or Ph2P(CH2)nPPh2, where n = 1 (dppm), 2 (dppe), 3 (dppp), and 4 (dppb), was analyzed. The XAS data revealed that changing the coordination geometry from tetrahedral in Ni(PPh3)2Cl2 (1) to square planar in Ni(dppe)Cl2 (2) more than doubles the intensity of pre-edge features assigned to Ni–P and Ni–Cl 1s → σ* transitions. By way of comparison, varying the diphosphine in Pd(dppm)Cl2 (4), Pd(dppp)Cl2 (6), and Pd(dppb)Cl2 (7) yielded Pd–P 1s → σ* transitions with identical intensities, but a 10% increase was observed in the P K-edge XAS spectrum of Pd(dppe)Cl2 (5). A similar observation was made when comparing Ni(dppe)Cl2 (2) to Ni(dppp)Cl2 (3), and DFT and TDDFT calculations corroborated XAS results obtained for both series. Comparison of the spectroscopic and theoretical results to the diphosphine structures revealed that changes in M–P covalency were not correlated to changes in bite angles or coordination geometry. As a final measure, P and Cl K-edge XAS data were collected on trans-Pd(PPh3)2Cl2 (8) for comparison to the cis diphosphine complex Pd(dppe)Cl2 (5). Consistent with phosphine’s stronger trans influence compared to chloride, a 35% decrease in the intensity of the Pd–P 1s → σ* pre-edge feature and a complementary 34% increase in Pd–Cl 1s → σ* feature was observed for 8 (trans) compared to 5 (cis). Overall, the results reveal how coordination geometry, ligand arrangement, and diphosphine structure affect covalent metal–phosphorus and metal–chloride bonding in these late transition metal complexes.
Co-reporter:Kyounghoon Lee, Haochuan Wei, Anastasia V. Blake, Courtney M. Donahue, Hyuk Yong Kwon, Jason M. Keith and Scott R. Daly
Dalton Transactions 2016 - vol. 45(Issue 27) pp:NaN11198-11198
Publication Date(Web):2016/06/22
DOI:10.1039/C6DT90109C
Correction for ‘Ligand K-edge XAS, DFT, and TDDFT analysis of pincer linker variations in Rh(I) PNP complexes: reactivity insights from electronic structure’ by Jason M. Keith, Scott R. Daly, et al., Dalton Trans., 2016, 45, 9774–9785.
Co-reporter:Kyounghoon Lee, Courtney M. Donahue and Scott R. Daly
Dalton Transactions 2017 - vol. 46(Issue 29) pp:NaN9406-9406
Publication Date(Web):2017/07/11
DOI:10.1039/C7DT02144E
The synthesis, coordination chemistry, and reactivity of two diphosphines containing the cyclic triaminoborane 1,8,10,9-triazaboradecalin (TBD) are described. To evaluate the ligand-centered reactivity of PhTBDPhos and iPrTBDPhos, the complexes (PhTBDPhos)MCl2 and (iPrTBDPhos)MCl2, where M = Ni and Pd, were prepared and characterized by elemental analysis, multinuclear NMR spectroscopy (1H, 13C, 31P, and 11B), and single-crystal X-ray diffraction (XRD). Despite very low boron Lewis acidity in the TBD backbone, (PhTBDPhos)NiCl2 (1) and (PhTBDPhos)PdCl2 (3) react with H2O, alcohols, and hydrated fluoride reagents in the presence of NEt3 to yield trans H–O or H–F addition across the bridgehead N–B bond. In contrast, iPrTBDPhos shows no appreciable reactivity when bound to NiCl2 (2) and PdCl2 (4), which is attributed to the sterically-bulky isopropyl substituents blocking substrate access to boron in the TBD backbone. The new complexes {[(PhTBDPhos-H2O)Ni]2(μ-OH)2}Cl2 (5), {[(PhTBDPhos-H2O)Pd]2(μ-OH)2}Cl2 (6), (PhTBDPhos-MeOH)NiCl2 (7), (PhTBDPhos-MeOH)PdCl2 (8), (PhTBDPhos-C3H5OH)PdCl2 (9), and {[(PhTBDPhos-HF)Ni]2(μ-OH)2}Cl2 (10) were isolated, and all but 6 were structurally characterized by single-crystal XRD. Multinuclear NMR studies revealed that isolated, crystallographically-authenticated samples of 5–9 lose ligand-bound water or alcohol with reappearance of starting materials 1 and 3 when dissolved in NMR solvents. Addition of NEt3 attenuated the water and alcohol loss from 5–9 to allow 1H, 13C, 31P, and 11B NMR data to be collected for all the compounds, confirming the determined structures. Additional reactivity experiments with NaOMe and fluoride reagents suggested that participation of the bridgehead nitrogen in the TBD backbone is important for promoting reactivity at boron when PhTBDPhos is bound to Ni and Pd. The term “cooperative ligand-centered reactivity” (CLR) is proposed to define chemical reactions that appear to require participation of more than one atom on the ligand, such as those reported here.
Co-reporter:Kyounghoon Lee, Haochuan Wei, Anastasia V. Blake, Courtney M. Donahue, Jason M. Keith and Scott R. Daly
Dalton Transactions 2016 - vol. 45(Issue 24) pp:NaN9785-9785
Publication Date(Web):2016/05/24
DOI:10.1039/C6DT00200E
Here we report P K-edge, Cl K-edge, and Rh L3-edge X-ray absorption spectroscopy (XAS) data for Rh[C5H3N-2,6-(XPtBu2)2]Cl, where X = O (tBuPONOP; 1) or CH2 (tBuPNP; 2). Solid-state XAS data for 1 and 2 were compared to density functional theory (DFT) and time-dependent density functional theory (TDDFT) calculations to identify how changing the PNP pincer linker from O to CH2 affected electronic structure and bonding at Rh(I). Pronounced differences in XAS peak intensities and energies were observed. The P K-edge XAS data revealed a large increase in Rh 4dx2−y2 and P 3p orbital-mixing (Rh–P σ*) in 1 compared to 2, and pronounced transition energy variations reflected marked differences in orbital energies and compositions. By comparison, the Cl K-edge XAS data revealed only subtle differences in Rh–Cl covalency, although larger splitting between the Rh–Cl π* and σ* transitions was observed in 2. Analysis of the occupied MOs from DFT (HOMO, HOMO−1, HOMO−2, and HOMO−3) and comparison to the unoccupied MOs involved in XAS revealed a relatively uniform energy increase (ca. 0.3–0.5 eV) for all five 4d-derived molecular orbitals in Rh(tBuPNP)Cl (2) compared to Rh(tBuPONOP)Cl (1). The energy shift was relatively invariant with respect to differences in orbital symmetry, bonding type (σ or π), and orbital mixing, which suggested that the increase could be attributed to electrostatic effects. The change in d-orbital energies are consistent with known reactivity differences of Rh(tBuPONOP)+ and Rh(tBuPNP)+ towards CO, H2, and CH2Cl2, and are explained here by considering how d-orbital energies affect covalent L → M σ bonding and M → L π backbonding.
.jpg)



![Dichloro[bis(1,3-diphenylphosphino)propane]palladium(II)](http://img.cochemist.com/ccimg/59900/59831-02-6.png)
![Dichloro[bis(1,3-diphenylphosphino)propane]palladium(II)](http://img.cochemist.com/ccimg/59900/59831-02-6_b.png)