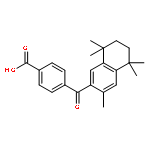
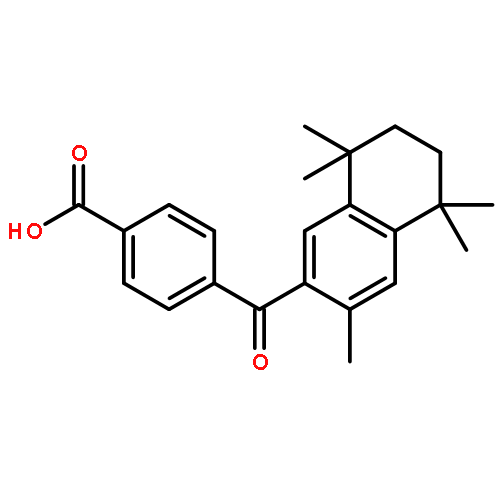
Co-reporter:Bo-Liang Dong;Qing-Hua Liao
Journal of Molecular Modeling 2011 Volume 17( Issue 7) pp:1727-1734
Publication Date(Web):2011 July
DOI:10.1007/s00894-010-0881-7
Extensive studies suggest direct links between cholesteryl ester transfer protein (CETP), high-density lipoproteins-cholesterol level and cardiovascular diseases. Many therapeutic approaches are aimed at the CETP. A series of N, N-disubstituted-trifluoro-3-amino-2-propanol analogues are among the most highly potent and selective inhibitors of CETP described to date. For in-depth investigation into the structural and chemical features responsible for exploring the binding pocket of these compounds, as well as for the binding recognition mechanism concerned, we performed a series of automated molecular docking operations. Moreover, the docking results were quite robust as further validated by molecular dynamics. The docking results reveal that the binding site mainly consists of two hydrophobic regions (P1 and P2 site) which are able to accommodate the lipophilic arms of the compounds investigated. Val421 in P1 site and Met194 in P2 site could be considered to be two important residues in forming the two hydrophobic regions. The presence of residues Phe197 and Phe463 in P2 site may be responsible for the binding recognition through π-π stacking interactions. The hydrophobic 3-phenoxy substituent may be important in creating the preferable inhibitive capability for increasing the binding potency. The hydrophobic character of the tetrafluoroethoxybenzyl group at position 3 displays better hydrophobicity than a shorter hydrophobic substituent. An interaction model of CETP-inhibitors is derived that can be successfully used to explain the different biologic activities of these inhibitors. It is anticipated that the findings reported here may provide very useful information or clues for designing effective drugs for the therapeutic treatment of CETP-related cardiovascular diseases.
Co-reporter:Peng Zhao;Qing-hua Liao;Cheng-Feng Ren
Journal of Molecular Modeling 2011 Volume 17( Issue 6) pp:1259-1265
Publication Date(Web):2011 June
DOI:10.1007/s00894-010-0822-5
Retinoid X receptors (RXRα, β and γ) are recently known to be cancer chemotherapies targets. The ligand binding domains of RXRs have been crystallized, but the information of RXRγ ligand binding site is not yet available due to the lack of liganded complex. A thorough understanding of the ligand binding sites is essential to study RXRs and may result in cancer therapeutic breakthrough. Thus we aimed to study the RXRγ ligand binding site and find out the differences between the three subtypes. Alignment and molecular simulation were carried out for identifying the RXRγ ligand binding site, characterizing the RXRγ ligand binding mode and comparing the three RXRs. The result has indicated that the RXRγ ligand binding site is defined by helices H5, H10, β-sheet s1 and the end loop. Besides hydrophobic interactions, the ligand 9-cis retinoic acid interacts with RXRγ through a hydrogen bond with Ala106, a salt bridge with Arg95 and the π-π interactions with Phe217 and Phe218. The binding modes exhibit some similarities among RXRs, such as the interactions with Arg95 and Ala106. Nonetheless, owing to the absence of Ile47, Cys48, Ala50, Ala51 and residues 225∼237 in the active site, the binding pocket in RXRγ is two times larger than those of RXRα and RXRβ. Meanwhile, spatial effects of Trp84, Arg95, Ala106, Phe217 and Phe218 help to create a differently shaped binding pocket as compared to those of RXRα and RXRβ. Consequently, the ligand in RXRγ undergoes a “standing” posing which is distinct from the other two RXRs.
Co-reporter:Yingda Ye, Qinghua Liao, Jing Wei, Qingzhi Gao
Neurochemistry International (January 2010) Volume 56(Issue 1) pp:107-117
Publication Date(Web):1 January 2010
DOI:10.1016/j.neuint.2009.09.008
Corticotropin-releasing factor (CRF) is a neuropeptide that falls into the broad spectrum of having neurotransmitter/neurohormonal/neuromodulator activities. The design and synthesis of low molecular weight non-peptide antagonists for the CRF receptors is a very important area of research as they can be employed in the treatment of a wide variety of disorders. To investigate the ligand–receptor binding mode and design novel CRF1 antagonists, both quantitative and qualitative 3D-QSAR analysis have been performed on a data set of CRF1 antagonists by using HypoGen and HipHopRefine programs of Catalyst software. The training set of HypoGen study included twenty-five structurally diverse CRF1 antagonists with Ki values ranging from 0.5 nM to 10 μM. The common feature-based 3D-QSAR study used eight highly potent CRF1 antagonists and four poor antagonistic ligands to generate 3D-pharmacophore models with excluded volumes. The obtained 3D-pharmacophore models from each study served as queries for virtual screening with a ‘focused compound library’ for novel CRF1 antagonist development. Pharmacophore models obtained for antagonist binding are useful for CRF related chemical biology and drug design. Strategies and methods employed in this paper are simple and practical for medicinal chemists in drug R&D.
Co-reporter:Yacong Zhao, Xiafei Hao, Jiannan Feng, Beifen Shen, Jing Wei, Jian Sun
International Immunopharmacology (February 2015) Volume 24(Issue 2) pp:219-223
Publication Date(Web):1 February 2015
DOI:10.1016/j.intimp.2014.12.013
•Peptide 814 from library and peptide TA by CADD inhibited BLyS–TACI interaction.•814-Fc and TA-Fc fusion proteins inhibited BLyS activity as the peptides did.•814 and 814-Fc protein had two-fold higher affinity than TA and TA-Fc protein.•Both BLyS affinity maturation library and CADD can produce BLyS-binding peptides.BLyS antagonists have become the therapeutic reagents in the treatment of autoimmune disorders. BLyS binding peptides and their Fc fusion proteins may be alternative BLyS antagonists in such application. In this study, the activity of BLyS binding peptide 814 obtained from phage display library and peptide TA designed by computer-aided modeling on the interaction of BLyS–TACI was compared. In addition, to maintain the spatial conformation and stability of the peptides, human IgG1 Fc fragment was fused to peptides 814 and TA to form peptide-Fc fusion proteins, steady and innovative peptibodies. The prokaryotic expression plasmids pET30a-814-Fc and pET30a-TA-Fc for these peptibodies were acquired by genetic engineering, and confirmed by DNA sequencing. After the right plasmids were transformed into Escherichia coli BL21 (DE3), the fusion proteins were expressed and purified by protein A affinity column. As a result of competitive ELISA, peptides 814 and TA at 100 μg/ml displayed 52.2% and 28.6% inhibition on the interaction of TACI-Fc with BLyS respectively. Moreover, 814-Fc and TA-Fc fusion proteins could bind to BLyS in a dosage-dependent manner as TACI-Fc did, and displayed 54.7% and 26.1% inhibition on the interaction of TACI-Fc-Myc with BLyS at 100 μg/ml respectively. So 814-Fc and TA-Fc proteins had the similar bioactivity as the peptides did. Furthermore, compared with TA-Fc, 814-Fc showed two-fold inhibition effect on BLyS binding to TACI, suggesting that 814-Fc could inhibit BLyS bioactivity significantly and might serve as a potential antagonist to treat autoimmune diseases associated with BLyS overexpression.

![2-[2-CHLORO-5-[(2S)-3-(5-CHLOROSPIRO[3H-1-BENZOFURAN-2,4'-PIPERIDINE]-1'-YL)-2-HYDROXYPROPOXY]-4-(METHYLCARBAMOYL)PHENOXY]-2-METHYLPROPANOIC ACID](/data/chemimg/321800/1003566-93-5.png)
![2-[2-CHLORO-5-[(2S)-3-(5-CHLOROSPIRO[3H-1-BENZOFURAN-2,4'-PIPERIDINE]-1'-YL)-2-HYDROXYPROPOXY]-4-(METHYLCARBAMOYL)PHENOXY]-2-METHYLPROPANOIC ACID](/data/chemimg/321800/1003566-93-5_b.png)
![3-[4-[4-[2-[3-[(dimethylamino)methyl]phenyl]-1H-pyrrolo[2,3-b]pyridin-4-yl]-1-ethylpyrazol-3-yl]phenyl]-1,1-dimethylurea](http://img.cochemist.com/ccimg/943000/942918-07-2.png)
![3-[4-[4-[2-[3-[(dimethylamino)methyl]phenyl]-1H-pyrrolo[2,3-b]pyridin-4-yl]-1-ethylpyrazol-3-yl]phenyl]-1,1-dimethylurea](http://img.cochemist.com/ccimg/943000/942918-07-2_b.png)
![4H-Pyrazolo[4,3-c]quinolin-4-one, 2,5-dihydro-2-(4-methylphenyl)-](http://img.cochemist.com/ccimg/863700/863641-72-9.png)
![4H-Pyrazolo[4,3-c]quinolin-4-one, 2,5-dihydro-2-(4-methylphenyl)-](http://img.cochemist.com/ccimg/863700/863641-72-9_b.png)
![5-Isoquinolinamine, N-[1-(phenylmethyl)-3-piperidinyl]-](http://img.cochemist.com/ccimg/675200/675133-21-8.png)
![5-Isoquinolinamine, N-[1-(phenylmethyl)-3-piperidinyl]-](http://img.cochemist.com/ccimg/675200/675133-21-8_b.png)
![5-(1-METHYL-1H-PYRAZOL-4-YL)-1-(PHENYLSULFONYL)-1H-PYRROLO[2,3-B]PYRIDINE](http://img.cochemist.com/ccimg/574800/574753-99-4.png)
![5-(1-METHYL-1H-PYRAZOL-4-YL)-1-(PHENYLSULFONYL)-1H-PYRROLO[2,3-B]PYRIDINE](http://img.cochemist.com/ccimg/574800/574753-99-4_b.png)
![(2E)-3-[4-(5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalen-2-yl)-4,5,6,7-tetrahydro-1-benzofuran-2-yl]prop-2-enoic acid](http://img.cochemist.com/ccimg/503700/503620-39-1.png)
![(2E)-3-[4-(5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalen-2-yl)-4,5,6,7-tetrahydro-1-benzofuran-2-yl]prop-2-enoic acid](http://img.cochemist.com/ccimg/503700/503620-39-1_b.png)
![5-Isoquinolinamine, N-[1-(2-methylpropyl)-4-piperidinyl]-](http://img.cochemist.com/ccimg/353600/353555-32-5.png)
![5-Isoquinolinamine, N-[1-(2-methylpropyl)-4-piperidinyl]-](http://img.cochemist.com/ccimg/353600/353555-32-5_b.png)
![1H-Indazol-5-amine, N-[1-[(2-chlorophenyl)methyl]-3-piperidinyl]-](http://img.cochemist.com/ccimg/353600/353546-84-6.png)
![1H-Indazol-5-amine, N-[1-[(2-chlorophenyl)methyl]-3-piperidinyl]-](http://img.cochemist.com/ccimg/353600/353546-84-6_b.png)
![1H-Indazol-5-amine, N-[1-(phenylmethyl)-3-pyrrolidinyl]-](http://img.cochemist.com/ccimg/353600/353545-89-8.png)
![1H-Indazol-5-amine, N-[1-(phenylmethyl)-3-pyrrolidinyl]-](http://img.cochemist.com/ccimg/353600/353545-89-8_b.png)
![1H-Indazol-5-amine, N-[1-(phenylmethyl)-4-piperidinyl]-](http://img.cochemist.com/ccimg/353600/353544-77-1.png)
![1H-Indazol-5-amine, N-[1-(phenylmethyl)-4-piperidinyl]-](http://img.cochemist.com/ccimg/353600/353544-77-1_b.png)


![Urea, N-[[2-fluoro-6-(trifluoromethyl)phenyl]methyl]-N'-4-pyridinyl-](http://img.cochemist.com/ccimg/353600/353541-23-8.png)
![Urea, N-[[2-fluoro-6-(trifluoromethyl)phenyl]methyl]-N'-4-pyridinyl-](http://img.cochemist.com/ccimg/353600/353541-23-8_b.png)
![Urea, N-[2-fluoro-6-(trifluoromethyl)phenyl]-N'-4-pyridinyl-](http://img.cochemist.com/ccimg/353600/353540-83-7.png)
![Urea, N-[2-fluoro-6-(trifluoromethyl)phenyl]-N'-4-pyridinyl-](http://img.cochemist.com/ccimg/353600/353540-83-7_b.png)
![1H-Indazol-5-amine, N-[1-(phenylmethyl)-3-piperidinyl]-](http://img.cochemist.com/ccimg/353600/353538-63-3.png)
![1H-Indazol-5-amine, N-[1-(phenylmethyl)-3-piperidinyl]-](http://img.cochemist.com/ccimg/353600/353538-63-3_b.png)
![Acetamide,N-(4-cyanophenyl)-2-[4-(2,3,6,9-tetrahydro-2,6-dioxo-1,3-dipropyl-1H-purin-8-yl)phenoxy]-](http://img.cochemist.com/ccimg/264700/264622-58-4.png)
![Acetamide,N-(4-cyanophenyl)-2-[4-(2,3,6,9-tetrahydro-2,6-dioxo-1,3-dipropyl-1H-purin-8-yl)phenoxy]-](http://img.cochemist.com/ccimg/264700/264622-58-4_b.png)
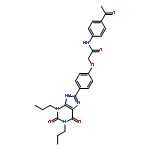
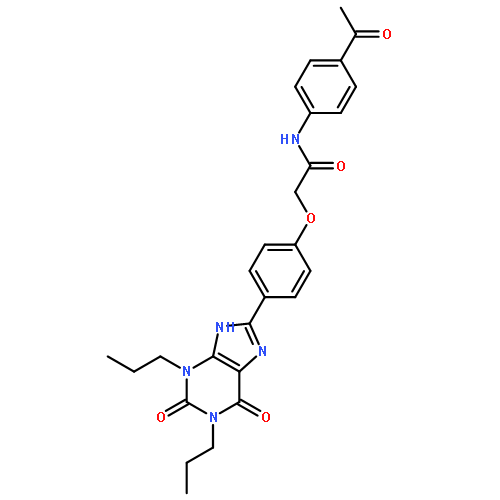
![5-Butyl-8-[4-(trifluoromethyl)phenyl]-1H-[1,2,4]triazolo[5,1-i]purine](http://img.cochemist.com/ccimg/246100/246031-23-2.png)
![5-Butyl-8-[4-(trifluoromethyl)phenyl]-1H-[1,2,4]triazolo[5,1-i]purine](http://img.cochemist.com/ccimg/246100/246031-23-2_b.png)
![2,4,6-Heptatrienoic acid,6-[6-(1,1-dimethylethyl)-2,3-dihydro-1,1-dimethyl-1H-inden-4-yl]-,(2E,4E)-](http://img.cochemist.com/ccimg/221700/221652-58-0.png)
![2,4,6-Heptatrienoic acid,6-[6-(1,1-dimethylethyl)-2,3-dihydro-1,1-dimethyl-1H-inden-4-yl]-,(2E,4E)-](http://img.cochemist.com/ccimg/221700/221652-58-0_b.png)
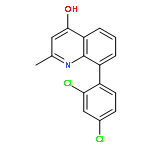
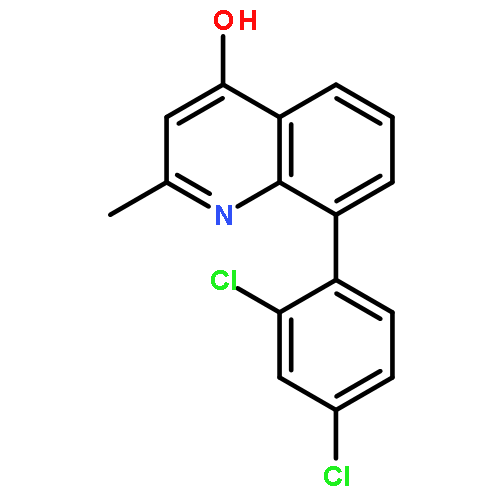
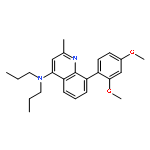
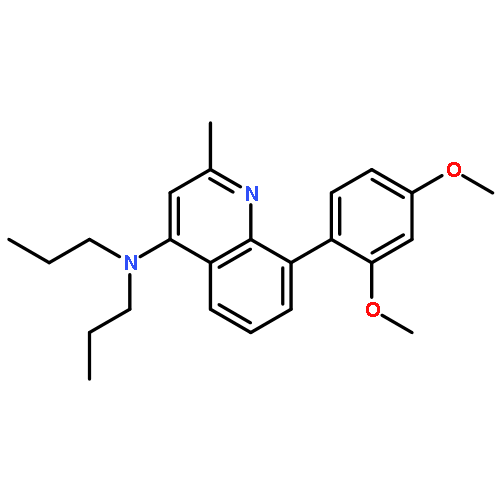
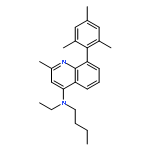
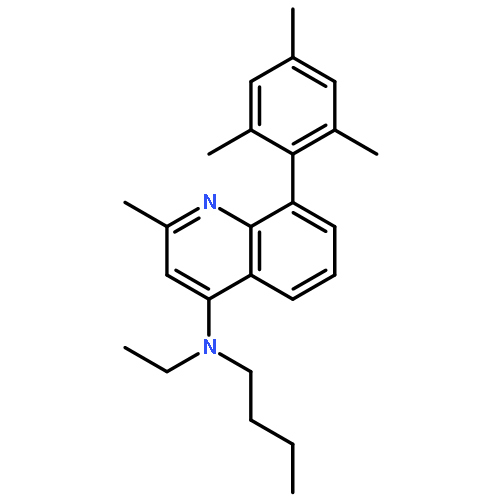
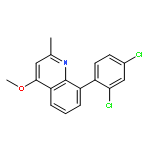
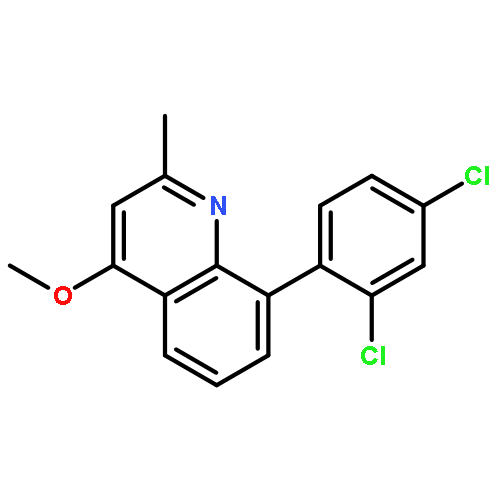
![5-AMINO-1H-PYRROLO[2,3-B]PYRIDINE-3-CARBOXYLIC ACID](http://img.cochemist.com/ccimg/212800/212790-31-3.png)
![5-AMINO-1H-PYRROLO[2,3-B]PYRIDINE-3-CARBOXYLIC ACID](http://img.cochemist.com/ccimg/212800/212790-31-3_b.png)
![N-Butyl-N-[3-(2,4-dichlorophenyl)-2,5-dimethylpyrazolo[1,5-a]pyrimidin-7-yl]-N-ethylamine](http://img.cochemist.com/ccimg/202600/202579-59-7.png)
![N-Butyl-N-[3-(2,4-dichlorophenyl)-2,5-dimethylpyrazolo[1,5-a]pyrimidin-7-yl]-N-ethylamine](http://img.cochemist.com/ccimg/202600/202579-59-7_b.png)


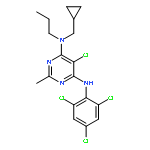
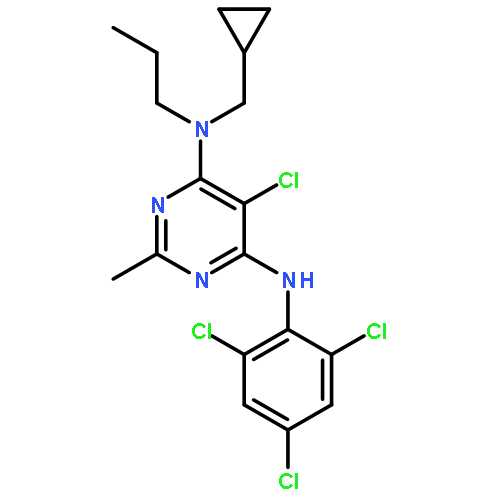


![4-[(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)sulfonyl]benzoic acid](http://img.cochemist.com/ccimg/179800/179762-66-4.png)
![4-[(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)sulfonyl]benzoic acid](http://img.cochemist.com/ccimg/179800/179762-66-4_b.png)
![4-[(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)sulfinyl]benzoic acid](http://img.cochemist.com/ccimg/179800/179762-65-3.png)
![4-[(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)sulfinyl]benzoic acid](http://img.cochemist.com/ccimg/179800/179762-65-3_b.png)
![4-[(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)sulfanyl]benzoic acid](http://img.cochemist.com/ccimg/173200/173156-91-7.png)
![4-[(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)sulfanyl]benzoic acid](http://img.cochemist.com/ccimg/173200/173156-91-7_b.png)
![2-(Furan-2-yl)-7-phenethyl-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amine](http://img.cochemist.com/ccimg/160100/160098-96-4.png)
![2-(Furan-2-yl)-7-phenethyl-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amine](http://img.cochemist.com/ccimg/160100/160098-96-4_b.png)
![Benzoic acid,4-[1-(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)cyclopropyl]-](http://img.cochemist.com/ccimg/157000/156910-31-5.png)
![Benzoic acid,4-[1-(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)cyclopropyl]-](http://img.cochemist.com/ccimg/157000/156910-31-5_b.png)
![6-[1-(3,5,5,8,8-PENTAMETHYL-5,6,7,8-TETRAHYDRO-NAPHTHALEN-2-YL)-CYCLOPROPYL]-NICOTINIC ACID](http://img.cochemist.com/ccimg/153600/153559-76-3.png)
![6-[1-(3,5,5,8,8-PENTAMETHYL-5,6,7,8-TETRAHYDRO-NAPHTHALEN-2-YL)-CYCLOPROPYL]-NICOTINIC ACID](http://img.cochemist.com/ccimg/153600/153559-76-3_b.png)
![Benzoic acid,4-[1-(3-bromo-5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)ethenyl]-](http://img.cochemist.com/ccimg/153600/153559-60-5.png)
![Benzoic acid,4-[1-(3-bromo-5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)ethenyl]-](http://img.cochemist.com/ccimg/153600/153559-60-5_b.png)






![4-[2-(5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalen-2-yl)-1,3-dithiolan-2-yl]benzoic acid](http://img.cochemist.com/ccimg/146700/146670-37-3.png)
![4-[2-(5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalen-2-yl)-1,3-dithiolan-2-yl]benzoic acid](http://img.cochemist.com/ccimg/146700/146670-37-3_b.png)
![Benzoic acid,4-[2-methyl-1-(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)-1-propen-1-yl]-](http://img.cochemist.com/ccimg/146700/146670-35-1.png)
![Benzoic acid,4-[2-methyl-1-(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)-1-propen-1-yl]-](http://img.cochemist.com/ccimg/146700/146670-35-1_b.png)
![Dibenz[b,e]oxepin-2-carboxylicacid, 6,11-dihydro-11-[[2-[(2-thienylsulfonyl)amino]ethyl]thio]-](http://img.cochemist.com/ccimg/123300/123226-56-2.png)
![Dibenz[b,e]oxepin-2-carboxylicacid, 6,11-dihydro-11-[[2-[(2-thienylsulfonyl)amino]ethyl]thio]-](http://img.cochemist.com/ccimg/123300/123226-56-2_b.png)
![9-bromo-11-({2-[(phenylsulfonyl)amino]ethyl}sulfanyl)-6,11-dihydrodibenzo[b,e]oxepine-2-carboxylic acid](http://img.cochemist.com/ccimg/123300/123226-48-2.png)
![9-bromo-11-({2-[(phenylsulfonyl)amino]ethyl}sulfanyl)-6,11-dihydrodibenzo[b,e]oxepine-2-carboxylic acid](http://img.cochemist.com/ccimg/123300/123226-48-2_b.png)



![(1r,3s,5z)-5-[(2e)-2-[(1r,3as,7ar)-1-[(2r)-6-hydroxy-6-methylheptan-2-yl]-7a-methyl-2,3,3a,5,6,7-hexahydro-1h-inden-4-ylidene]ethylidene]-4-methylidenecyclohexane-1,3-diol](http://img.cochemist.com/ccimg/32300/32222-06-3.png)
![(1r,3s,5z)-5-[(2e)-2-[(1r,3as,7ar)-1-[(2r)-6-hydroxy-6-methylheptan-2-yl]-7a-methyl-2,3,3a,5,6,7-hexahydro-1h-inden-4-ylidene]ethylidene]-4-methylidenecyclohexane-1,3-diol](http://img.cochemist.com/ccimg/32300/32222-06-3_b.png)








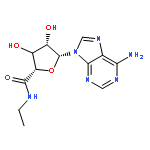
![N-[5-Chloro-2-[2-[4-(4-fluorobenzyl)-2(R)-methylpiperazin-1-yl]-2-oxoethoxy]phenyl]urea hydrochloride](http://img.cochemist.com/ccimg/288300/288262-96-4.png)
![N-[5-Chloro-2-[2-[4-(4-fluorobenzyl)-2(R)-methylpiperazin-1-yl]-2-oxoethoxy]phenyl]urea hydrochloride](http://img.cochemist.com/ccimg/288300/288262-96-4_b.png)
![1H-[1,2,4]Triazolo[5,1-i]purine, 5-butyl-8-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/246100/246030-67-1.png)
![1H-[1,2,4]Triazolo[5,1-i]purine, 5-butyl-8-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/246100/246030-67-1_b.png)
![(Z)-4-[(Hydroxyimino)(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)methyl]benzoic acid](http://img.cochemist.com/ccimg/221400/221343-42-6.png)
![(Z)-4-[(Hydroxyimino)(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)methyl]benzoic acid](http://img.cochemist.com/ccimg/221400/221343-42-6_b.png)
![2-[(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)sulfanyl]-1,3-thiazole-5-carboxylic acid](http://img.cochemist.com/ccimg/179800/179762-67-5.png)
![2-[(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)sulfanyl]-1,3-thiazole-5-carboxylic acid](http://img.cochemist.com/ccimg/179800/179762-67-5_b.png)
![6-[(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)sulfanyl]pyridine-3-carboxylic acid](http://img.cochemist.com/ccimg/173200/173156-98-4.png)
![6-[(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)sulfanyl]pyridine-3-carboxylic acid](http://img.cochemist.com/ccimg/173200/173156-98-4_b.png)