Co-reporter:Sourav Ghorai, Daesung Lee
Tetrahedron 2017 Volume 73, Issue 29(Issue 29) pp:
Publication Date(Web):20 July 2017
DOI:10.1016/j.tet.2017.02.008
Despite their electrophilic nature, arynes are not known to react with nitriles under typical conditions for their formation. We demonstrated, however, that structurally diverse arynes could be generated via the hexadehydro Diels-Alder reaction and trapped with nitriles to induce the Ritter-type reaction when catalyzed by a cationic silver species. Presumably, under these conditions, the transiently formed aryne-silver complexes react with nitriles initially to form nitrilium ion intermediates, which then subsequently react with either water or carboxylic acid to yield the observed arylamides or arylimides.Download high-res image (123KB)Download full-size image
Co-reporter:Rajdip Karmakar and Daesung Lee
Chemical Society Reviews 2016 vol. 45(Issue 16) pp:4459-4470
Publication Date(Web):26 Jan 2016
DOI:10.1039/C5CS00835B
Arynes are unique aromatic species that display versatile reactivity in a variety of transformations. It has been demonstrated that the reaction profiles of arynes can be altered by transition metal additives, and one of the earliest examples of the metal additive effect was observed with silver ions (Ag+). Especially, in the presence of Ag+, benzyne showed distinctive reactivity and selectivity toward π-nucleophiles compared to the corresponding reaction in its absence. Although more experimental data need to be accumulated to accurately define the Ag+ additive effect, based on its role displayed in various transformations, we can infer that Ag+ interacts with arynes to form a reactive intermediate, which behaves like a silver-bound aryl cation or 1,2-carbene-silver carbenoid. In this tutorial review, various aryne-mediated reactions involving Ag+ or other organosilver species are discussed, which show a bird's-eye view on the Ag+ effect in aryne chemistry such that further explorations of the unique reactivity of arynes under the influence Ag+ will be inspired.
Co-reporter:Rajdip Karmakar, Phani Mamidipalli, Ryan M. Salzman, Seongwon Hong, Sang Young Yun, Wei Guo, Yuanzhi Xia, and Daesung Lee
Organic Letters 2016 Volume 18(Issue 15) pp:3530-3533
Publication Date(Web):July 22, 2016
DOI:10.1021/acs.orglett.6b01443
A new benzannulation reaction with accompanied trifluoromethylthiolation is described. This benzannulation can generate a range of trifluoromethylthiolated benzolactams and benzolactones from 1,3,8-triynes and a stoichiometric amount of AgSCF3 at 90 °C through an initial Alder-ene reaction, 1,4-addition of AgSCF3, and a series of bond-reorganization processes that include double bond migration, 6π-electrocyclization, and a [1,3]-H shift. For certain substrates containing a triisopropylsilyl (TIPS) group, the final [1,3]-H shift-interrupted products, were obtained.
Co-reporter:Rajdip Karmakar and Daesung Lee
Organic Letters 2016 Volume 18(Issue 23) pp:6105-6107
Publication Date(Web):November 18, 2016
DOI:10.1021/acs.orglett.6b03241
The total syntheses of selaginpulvilins C and D is described. The key strategy for the construction of the core fluorene moiety involves in situ formation of an aryne intermediate followed by its formal hydrogenation. The precursor tetraynes that undergo aromatization via hexadehydro Diels–Alder reaction were prepared from readily available building blocks through typical alkyne-coupling reactions.
Co-reporter:Rajdip Karmakar, Kung-Pern Wang, Sang Young Yun, Phani Mamidipalli and Daesung Lee
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 21) pp:4782-4788
Publication Date(Web):26 Apr 2016
DOI:10.1039/C6OB00524A
A new functionalization method of arynes promoted by a novel catalytic role of the Grubbs-type ruthenium alkylidene complex is described. Through a sequence of aryne formation followed by their halo-functionalization, various bis-1,3-diynes were directly converted to functionalized aryl chlorides, bromides and iodides in good yields in the presence of a catalytic amount of a ruthenium alkylidene complex and halogenated hydrocarbons such as CH2Cl2, CHCl3, CH2Br2, and CH2I2. The utility of this novel transformation is demonstrated by a formal synthesis of herbindole B.
Co-reporter:Matthew J. O'Connor;Chunrui Sun;Xinyu Guan;Venkata R. Sabbasani ; Daesung Lee
Angewandte Chemie International Edition 2016 Volume 55( Issue 6) pp:2222-2225
Publication Date(Web):
DOI:10.1002/anie.201510152
Abstract
α,β-Unsaturated ketones generally undergo addition reactions with nucleophiles with a preference for either 1,2- or 1,4-addition, but rarely both. However, the right combination of reagents allows for consecutive 1,4- and 1,2-additions to occur: Cyclic α,β-unsaturated ketones undergo double additions with lithium(trimethylsilyl)diazomethane, effectively generating various molecular frameworks with complexity and diversity. Owing to the sequential generation of several intermediates of multifaceted reactivity, including diazoalkane derivatives and alkylidene carbenes, it is possible to induce novel Grob-type C−C fragmentations, alkylidene carbene mediated Li−N insertions, and dipolar cycloadditions by controlling the reaction parameters.
Co-reporter:Matthew J. O'Connor;Chunrui Sun;Xinyu Guan;Venkata R. Sabbasani ; Daesung Lee
Angewandte Chemie International Edition 2016 Volume 55( Issue 6) pp:
Publication Date(Web):
DOI:10.1002/anie.201511326
Co-reporter:Matthew J. O'Connor;Chunrui Sun;Xinyu Guan;Venkata R. Sabbasani ; Daesung Lee
Angewandte Chemie 2016 Volume 128( Issue 6) pp:2262-2265
Publication Date(Web):
DOI:10.1002/ange.201510152
Abstract
α,β-Unsaturated ketones generally undergo addition reactions with nucleophiles with a preference for either 1,2- or 1,4-addition, but rarely both. However, the right combination of reagents allows for consecutive 1,4- and 1,2-additions to occur: Cyclic α,β-unsaturated ketones undergo double additions with lithium(trimethylsilyl)diazomethane, effectively generating various molecular frameworks with complexity and diversity. Owing to the sequential generation of several intermediates of multifaceted reactivity, including diazoalkane derivatives and alkylidene carbenes, it is possible to induce novel Grob-type C−C fragmentations, alkylidene carbene mediated Li−N insertions, and dipolar cycloadditions by controlling the reaction parameters.
Co-reporter:Matthew J. O'Connor;Chunrui Sun;Xinyu Guan;Venkata R. Sabbasani ; Daesung Lee
Angewandte Chemie 2016 Volume 128( Issue 6) pp:
Publication Date(Web):
DOI:10.1002/ange.201511326
Co-reporter:Chunrui Sun, Hyunjin Lee, and Daesung Lee
Organic Letters 2015 Volume 17(Issue 21) pp:5348-5351
Publication Date(Web):October 26, 2015
DOI:10.1021/acs.orglett.5b02709
The carbocyclic core of massadine has been synthesized relying on a stereoselective formal [3 + 2] cycloaddition of lithiumtrimethylsilyldiazomethane with α,β-unsaturated esters to form a Δ2-pyrazoline moiety followed by facile N–N bond cleavage. A unique feature of the current approach is the direct installation of the tertiary α-amino center and a β-cyano group in a cis arrangement on the resulting cyclopentane framework via a previously developed formal aminocyanation protocol.
Co-reporter:Venkata R. Sabbasani and Daesung Lee
Organic Letters 2015 Volume 17(Issue 19) pp:4878-4881
Publication Date(Web):September 24, 2015
DOI:10.1021/acs.orglett.5b02433
Novel oxidative dimerization of silylallenes is described. Treatment of silylallenes with a catalytic amount of copper(I) chloride, a substoichiometric amount of N-hydroxyphthalamide, and a stoichiometric amount of a terminal oxidant diacetoxyiodobenzene afforded head-to-head dimers as the main products. Silyallenes containing a small ring afforded only dimers, whereas as the ring size increased 1,3-enynes became more favorable products. For silylallenes containing an acyclic substituent, dimer formation is a norm with exceptions where N-hydroxyphthalimide reacts at the propargylic center to generate the corresponding aminoxy ethers.
Co-reporter:Matthew O'Connor;Chunrui Sun; Daesung Lee
Angewandte Chemie International Edition 2015 Volume 54( Issue 34) pp:9963-9966
Publication Date(Web):
DOI:10.1002/anie.201503982
Abstract
Concise routes for the total and formal syntheses of the amathaspiramides were developed through a formal [3+2] cycloaddition between lithium(trimethylsilyl)diazomethane and α,β-unsaturated esters. The effectiveness of this new cycloaddition for the construction of Δ2-pyrazolines containing a α-tert-alkylamino carbon center and subsequent facile protonolytic NN bond cleavage allows the synthesis of a key intermediate of the amathaspiramides and other α,α-disubstituted amino acid derivatives.
Co-reporter:Yanhua Mi;Tao Zhou;Kung-Pern Wang; Daesung Lee; Yuanzhi Xia
Chemistry - A European Journal 2015 Volume 21( Issue 48) pp:17256-17268
Publication Date(Web):
DOI:10.1002/chem.201502469
Abstract
The mechanisms of regiodivergent cyclizations of o-alkynylbenzaldehyde acetals and thioacetals catalyzed by Pd and Pt halides are studied. DFT calculations found that both reactions are initiated by electrophilic activation of the acetylenic moiety instead of the previously proposed metal-triggered CX (X=O, S) cleavage. Both the regioselective cyclization of the π-alkyne complex and the chemoselective [1,2]-migration in the carbenoid intermediate were determined as key steps to achieving the observed divergence. For acetal derivatives containing an internal alkyne, the 6-endo-dig cyclization is more favorable and leads to the carbenoid intermediate easily through further steps of CX fragmentation and carbocation cyclization. Then, from the carbenoid intermediate, the [1,2]-migration of sulfur is easier than that of H, Me, and Ph; whereas, a reversed aptitude was predicted for the oxygen analogue, which is consistent with the greater ability of sulfur atoms to stabilize β-carbocations. However, for precursors containing a terminal alkyne, the 5-exo-dig pathway is preferred and only the 1,2-disubstituted indene product is seen, irrespective of the nature of the acetal; thus, a different product from that reported in the literature is predicted for benzaldehyde acetal with a terminal alkyne at the ortho position. This prediction led us to reconsider some of the reported results and hidden realities were uncovered with solid new experimental evidence.
Co-reporter:Venkata R. Sabbasani;Dr. Genping Huang; Yuanzhi Xia; Daesung Lee
Chemistry - A European Journal 2015 Volume 21( Issue 48) pp:17210-17214
Publication Date(Web):
DOI:10.1002/chem.201503243
Abstract
Facile and selective Alder-ene reactions of silylallenes involving the activation of an allenic C(sp2)H over an allylic C(sp3)H bond is described. In this ene reaction, the presence of a silyl substituent was found to be critical for the observed reactivity and selectivity since the corresponding alkyl-substituted allenes show different reaction profiles. Computational studies show that the origin of this unusual reactivity is the lower bond dissociation energy of the α-C(sp2)H bond in silylallenes compared to the corresponding nonsilylated allenes.
Co-reporter:Yanhua Mi;Tao Zhou;Kung-Pern Wang; Daesung Lee; Yuanzhi Xia
Chemistry - A European Journal 2015 Volume 21( Issue 48) pp:
Publication Date(Web):
DOI:10.1002/chem.201503938
Abstract
Invited for the cover of this issue are the groups of Yuanzhi Xia at Wenzhou University (P.R. China) and Daesung Lee at University of Illinois at Chicago (USA). The image depicts the mechanistic understanding of the regiodivergent cyclizations of o-alkynylbenzaldehyde acetals and thioacetals catalyzed by metal halides. Read the full text of the article at 10.1002/chem.201502469.
Co-reporter:Yanhua Mi;Tao Zhou;Kung-Pern Wang; Daesung Lee; Yuanzhi Xia
Chemistry - A European Journal 2015 Volume 21( Issue 48) pp:
Publication Date(Web):
DOI:10.1002/chem.201584801
Co-reporter:Matthew O'Connor;Chunrui Sun; Daesung Lee
Angewandte Chemie 2015 Volume 127( Issue 34) pp:10101-10104
Publication Date(Web):
DOI:10.1002/ange.201503982
Abstract
Concise routes for the total and formal syntheses of the amathaspiramides were developed through a formal [3+2] cycloaddition between lithium(trimethylsilyl)diazomethane and α,β-unsaturated esters. The effectiveness of this new cycloaddition for the construction of Δ2-pyrazolines containing a α-tert-alkylamino carbon center and subsequent facile protonolytic NN bond cleavage allows the synthesis of a key intermediate of the amathaspiramides and other α,α-disubstituted amino acid derivatives.
Co-reporter:Ivan Volchkov and Daesung Lee
Chemical Society Reviews 2014 vol. 43(Issue 13) pp:4381-4394
Publication Date(Web):24 Apr 2014
DOI:10.1039/C4CS00036F
The direct metal-catalyzed [1,3]-transposition of allylic alcohols and allylic silyl ethers is a synthetically useful isomerization process that occurs via [3,3]-sigmatropic rearrangement induced by high oxidation state oxometal complexes. The isomerization requires only a catalytic amount of promoter, and high chirality transfer can be achieved. Thus, it bears a significant potential to become a powerful tool in multistep synthesis. Although [1,3]-transposition of allylic alcohols has been known since the late 1960s, the development of synthetically useful protocols that allow for a high level of regio- and stereoselectivity control and their synthetic applications have emerged only recently. This tutorial review summarizes recently developed regioselective [1,3]-transpositions of allylic alcohols and silyl ethers and their applications to natural product synthesis.
Co-reporter:Nam-Kyu Lee ; Sang Young Yun ; Phani Mamidipalli ; Ryan M. Salzman ; Daesung Lee ; Tao Zhou ;Yuanzhi Xia
Journal of the American Chemical Society 2014 Volume 136(Issue 11) pp:4363-4368
Publication Date(Web):March 2, 2014
DOI:10.1021/ja500292x
A new biaryl synthesis via silver-catalyzed hydroarylation of arynes from acyclic building blocks with unactivated arenes in intra- and intermolecular manners has been developed. The previously observed Diels–Alder reactions of arynes with arene were not observed under the current silver-catalyzed conditions. Deuterium scrambling and DFT calculations suggest a stepwise electrophilic aromatic substitution mechanism through the formation of a Wheland-type intermediate followed by a water-catalyzed proton transfer in the final step of the hydroarylation.
Co-reporter:Phani Mamidipalli, Sang Young Yun, Kung-Pern Wang, Tao Zhou, Yuanzhi Xia and Daesung Lee
Chemical Science 2014 vol. 5(Issue 6) pp:2362-2367
Publication Date(Web):19 Feb 2014
DOI:10.1039/C3SC53478B
Formal hydrogenation of arynes was realized by using trialkylsilyl groups tethered to the arynes as the hydride source. In stark contrast to the effective 1°, 2°, and 3° C–H bond insertion of alkyl groups tethered to arynes, the 2° and 3° C–H bonds on the β-carbon of silyl groups show high tendency for hydride transfer rather than C–H insertion, whereas the corresponding 1° C–H bonds exclusively undergo C–H insertion. The exclusive hydride transfer and C–H insertion behavior of different C–H bonds in these two reaction pathways were rationalized by the stability of the incipient carbocationic intermediates which could be further bolstered by DFT-calculations.
Co-reporter:Rajdip Karmakar, Sang Young Yun, Kung-Pern Wang, and Daesung Lee
Organic Letters 2014 Volume 16(Issue 1) pp:6-9
Publication Date(Web):December 6, 2013
DOI:10.1021/ol403237z
The regioselectivity in nucleophile trapping is investigated with arynes generated directly from bis-1,3-diynes. The regioselectivity is profoundly influenced by not only the nature of nucleophiles but also the substituents on the arynes, which is the consequence of both the unfavorable steric interaction between the incoming nucleophile and the nearby substituent and the inherent electronic bias induced by different substituents on the arynes.
Co-reporter:Jun-Cheng Zheng;Huaqing Liu;Nam-Kyu Lee
European Journal of Organic Chemistry 2014 Volume 2014( Issue 3) pp:506-510
Publication Date(Web):
DOI:10.1002/ejoc.201301575
Abstract
The different reaction profiles of lithium trimethylsilyldiazomethane with various 4-alkenyl ketones are described. It was found that the individual steps along the reaction pathway including a Brook rearrangement, elimination of lithium trimethylsilanolate to form alkylidene carbenes, and their addition to an alkene to form strained bicyclo[3.1.0]hex-1-ene derivatives were significantly affected by the substituents near or on the alkene moiety. Especially, the dimerization of bicyclo[3.1.0]hex-1-enes depends critically on the substituent pattern of the alkene, which controls the reaction to proceed in a concerted or a stepwise manner. On the basis of X-ray diffraction analysis, the dimers, which are of unprecedented structural complexity, were unambiguously elucidated.
Co-reporter:Chunrui Sun;Matthew J. O'Connor; Daesung Lee; Donald J. Wink ;Robert D. Milligan
Angewandte Chemie International Edition 2014 Volume 53( Issue 12) pp:3197-3200
Publication Date(Web):
DOI:10.1002/anie.201309435
Abstract
Installation of amino functionality on organic molecules through direct CN bond formation is an important research objective. To achieve this goal, a 1,2-aminocyanation reaction was developed. The reaction occurs through the formation of pyrazolines by means of a formal dipolar cycloaddition of cyclic α,β-unsaturated ketones with lithium trimethylsilyldiazomethane followed by novel protonolytic NN bond cleavage under mild conditions. This two-step process provides a diverse array of structurally complex free and mono-alkylated α-amino ketones in excellent yields.
Co-reporter:Dr. Shu-Lin Liu;Dr. Ren Sheng;Matthew J. O'Connor;Yang Cui;Dr. Youngdae Yoon;Svetlana Kurilova;Dr. Daesung Lee;Dr. Wonhwa Cho
Angewandte Chemie International Edition 2014 Volume 53( Issue 52) pp:14387-14391
Publication Date(Web):
DOI:10.1002/anie.201408153
Abstract
Lipids regulate a wide range of biological activities. Since their local concentrations are tightly controlled in a spatiotemporally specific manner, the simultaneous quantification of multiple lipids is essential for elucidation of the complex mechanisms of biological regulation. Here, we report a new method for the simultaneous in situ quantification of two lipid pools in mammalian cells using orthogonal fluorescent sensors. The sensors were prepared by incorporating two environmentally sensitive fluorophores with minimal spectral overlap separately into engineered lipid-binding proteins. Dual ratiometric analysis of imaging data allowed accurate, spatiotemporally resolved quantification of two different lipids on the same leaflet of the plasma membrane or a single lipid on two opposite leaflets of the plasma membrane of live mammalian cells. This new imaging technology should serve as a powerful tool for systems-level investigation of lipid-mediated cell signaling and regulation.
Co-reporter:Chunrui Sun;Matthew J. O'Connor; Daesung Lee; Donald J. Wink ;Robert D. Milligan
Angewandte Chemie 2014 Volume 126( Issue 12) pp:3261-3264
Publication Date(Web):
DOI:10.1002/ange.201309435
Abstract
Installation of amino functionality on organic molecules through direct CN bond formation is an important research objective. To achieve this goal, a 1,2-aminocyanation reaction was developed. The reaction occurs through the formation of pyrazolines by means of a formal dipolar cycloaddition of cyclic α,β-unsaturated ketones with lithium trimethylsilyldiazomethane followed by novel protonolytic NN bond cleavage under mild conditions. This two-step process provides a diverse array of structurally complex free and mono-alkylated α-amino ketones in excellent yields.
Co-reporter:Dr. Shu-Lin Liu;Dr. Ren Sheng;Matthew J. O'Connor;Yang Cui;Dr. Youngdae Yoon;Svetlana Kurilova;Dr. Daesung Lee;Dr. Wonhwa Cho
Angewandte Chemie 2014 Volume 126( Issue 52) pp:14615-14619
Publication Date(Web):
DOI:10.1002/ange.201408153
Abstract
Lipids regulate a wide range of biological activities. Since their local concentrations are tightly controlled in a spatiotemporally specific manner, the simultaneous quantification of multiple lipids is essential for elucidation of the complex mechanisms of biological regulation. Here, we report a new method for the simultaneous in situ quantification of two lipid pools in mammalian cells using orthogonal fluorescent sensors. The sensors were prepared by incorporating two environmentally sensitive fluorophores with minimal spectral overlap separately into engineered lipid-binding proteins. Dual ratiometric analysis of imaging data allowed accurate, spatiotemporally resolved quantification of two different lipids on the same leaflet of the plasma membrane or a single lipid on two opposite leaflets of the plasma membrane of live mammalian cells. This new imaging technology should serve as a powerful tool for systems-level investigation of lipid-mediated cell signaling and regulation.
Co-reporter:Sang Young Yun ; Kung-Pern Wang ; Nam-Kyu Lee ; Phani Mamidipalli
Journal of the American Chemical Society 2013 Volume 135(Issue 12) pp:4668-4671
Publication Date(Web):March 11, 2013
DOI:10.1021/ja400477r
Arynes generated directly from alkyne building blocks in the presence of silver catalysts effectively activate primary, secondary, and tertiary alkane C–H bonds. This C–H insertion requires only a catalytic amount of silver complex and modest heating compared to harsh conditions and extra promoters including directing groups, oxidants, and bases in typical transition-metal-based C–H bond functionalizations. Preliminary mechanistic studies suggest that the C–H bond-breaking and new bond-forming events take place in a concerted manner, rendering a formal 1,2-addition of C–H bond across the π-bond of arynes.
Co-reporter:Ivan Volchkov and Daesung Lee
Journal of the American Chemical Society 2013 Volume 135(Issue 14) pp:5324-5327
Publication Date(Web):March 21, 2013
DOI:10.1021/ja401717b
An asymmetric total synthesis of (−)-amphidinolide V was accomplished. The synthesis features a base-catalyzed alkynyl silane alcoholysis/ring-closing enyne metathesis sequence for facile construction of a 1,3-diene motif. A diene RCM followed by a ring-contractive allylic transposition of cyclic silyl ethers was incorporated for the stereoselective installation of a functionalized 1,5-diene subunit. An efficient proline-mediated direct cross-aldol condensation of two advanced aldehyde intermediates was utilized for the construction of a key α,β-unsaturated epoxyaldehyde. This total synthesis demonstrates the prowess of metal-catalyzed transformations in complex molecule synthesis.
Co-reporter:Venkata R. Sabbasani, Phani Mamidipalli, Huijie Lu, Yuanzhi Xia, and Daesung Lee
Organic Letters 2013 Volume 15(Issue 7) pp:1552-1555
Publication Date(Web):March 21, 2013
DOI:10.1021/ol400331r
A general and stereoselective rearrangement of allenic alcohols to (E,E)-1,3-dien-2-yl triflates and chlorides was developed under metal-free conditions. Subtle electronic effects of the alkyl and aryl substituents on the carbon bearing the hydroxyl group has a profound impact on the reaction rate and efficiency such that vinyl triflates were obtained from electron-deficient substrates and trimethylsilyl triflate whereas vinyl chlorides were generated with an electron-rich substrate and trimethylsilyl chloride.
Co-reporter:Rajdip Karmakar, Phani Mamidipalli, Sang Young Yun, and Daesung Lee
Organic Letters 2013 Volume 15(Issue 8) pp:1938-1941
Publication Date(Web):March 29, 2013
DOI:10.1021/ol4005905
Efficient Alder-ene reactions of various arynes generated directly from bis-1,3-diynes are described. The reactivity of ene donors with different tethers was examined under thermal and metal-catalyzed conditions, which indicates that both the formation of aryne intermediates and their ene reactions are less sensitive to the catalyst than to the structural features of the substrates.
Co-reporter:Venkata R. Sabbasani and Daesung Lee
Organic Letters 2013 Volume 15(Issue 15) pp:3954-3957
Publication Date(Web):July 22, 2013
DOI:10.1021/ol401735v
An efficient nitration of silyl allenes with nitrogen dioxide radical, generated from NaNO2 and AcOH, to form α-nitro-α,β-unsaturated silyl oximes has been developed. A similar nitration could be achieved by using Fe(NO3)3·9H2O and FeCl3·6H2O, but different from the regioselective oxime formation, two regioisomeric chloride-trapped products were isolated with varying ratios depending on the steric bulk of the silyl group. A novel ring-closure reaction of α-nitro-α,β-unsaturated silyl oximes upon treating with TBAF to form isooxazolidinone derivatives was also developed.
Co-reporter:Huaqing Liu, Matthew J. O’Connor, Chunrui Sun, Donald J. Wink, and Daesung Lee
Organic Letters 2013 Volume 15(Issue 12) pp:2974-2977
Publication Date(Web):May 30, 2013
DOI:10.1021/ol401124t
A sequence of a carbonyl addition between 4-alkenyl ketones and aldehydes with trimethylsilyldiazomethane followed by a 1,3-Brook rearrangement and an intramolecular 1,3-dipolar cycloaddition was promoted by Lewis bases tetrabutylammonium triphenyldifluorosilicate (TBAT) or potassium tert-butoxide (KO-t-Bu). Through these concatenated bond-forming processes, a variety of novel bi- and tricyclic Δ1-pyrazolines were synthesized.
Co-reporter:Kung-Pern Wang, Eun Jin Cho, Sang Young Yun, Jee Young Rhee, Daesung Lee
Tetrahedron 2013 69(43) pp: 9105-9110
Publication Date(Web):
DOI:10.1016/j.tet.2013.08.024
Co-reporter:Genping Huang, Yuanzhi Xia, Chunrui Sun, Jingwei Li, and Daesung Lee
The Journal of Organic Chemistry 2013 Volume 78(Issue 3) pp:988-995
Publication Date(Web):January 8, 2013
DOI:10.1021/jo302466z
DFT/M06 calculations were carried out to better understand the mechanism and regio- and chemoselectivities of our previously discovered formal C–H amination of silyl cyclopropenes by azodicarboxylates ( Chem. Commun. 2012, 48, 10990). The results revealed that the initial Alder-ene reaction between the two reactants follows a stepwise mechanism and the subsequent allylic transposition proceeds via a concerted [1,3]-migration of hydrazodicarboxylate. For the Alder-ene process of 1-silyl-2-methylcyclopropene, different electronic effects of the substituents make the C1 much more negatively charged and thus more reactive than C2 in the regiochemistry-determining electrophilic azodicarboxylate addition step. In addition, the poor regioselectivity caused by a C2 ether linkage and the lower reactivity of ene donors other than cyclopropenes with azodicarboxylate were well explained by the computational results. Furthermore, the divergent allylic transposition of the Alder-ene intermediates was rationalized, and the steric repulsion between the silyl group and the hydrazodicarboxylate moiety was suggested as the driving force in promoting the allylic transposition. The barrier for the rate-controlling [1,3]-migration of hydrazodicarboxylate from an intermediate containing the C1–N bond is 21.1 kcal/mol, whereas a higher barrier of 27.5 kcal/mol is required for the similar rearrangement of the thermodynamically more stable intermediate containing the C2–N bond.
Co-reporter:Sang Young Yun ; Kung-Pern Wang ; Mansuk Kim
Journal of the American Chemical Society 2012 Volume 134(Issue 26) pp:10783-10786
Publication Date(Web):June 18, 2012
DOI:10.1021/ja304255h
An efficient 1,4-hydrovinylative cyclization reaction of triynes and tetraynes catalyzed by ruthenium alkylidene complexes under ethylene is described. The regioselectivity of vinyl group incorporation can be controlled by the nature of the substituent on the alkyne, and the Grubbs second-generation catalyst is the most effective among typical ruthenium alkylidene complexes.
Co-reporter:Jingwei Li and Daesung Lee
Chemical Science 2012 vol. 3(Issue 11) pp:3296-3301
Publication Date(Web):17 Aug 2012
DOI:10.1039/C2SC20812A
A highly diastereoselective ring-rearrangement metathesis of cyclopentene derivatives containing all-carbon quaternary centres is reported. This tandem metathesis process provides cyclohexene derivatives with two contiguous stereogenic centres, one of which is an all-carbon quaternary stereogenic centre, in high yields and excellent diastereoselecitivity. The efficacy of this methodology is showcased in the total synthesis of spiropiperidine alkaloid nitramine. In the meantime, it has been found that metathesis of the corresponding acyclic triene derivatives affords the same products with the same level of efficiency and diastereoselectivity.
Co-reporter:Chunrui Sun, Jingwei Li, Daesung Lee, Genping Huang and Yuanzhi Xia
Chemical Communications 2012 vol. 48(Issue 89) pp:10990-10992
Publication Date(Web):24 Sep 2012
DOI:10.1039/C2CC35329F
A novel C(sp3)–H amination of trimethylsilyl-substituted cyclopropenes is described. This C–H amination proceeds via a tandem regioselective ene reaction between cyclopropenes and azodicarboxylate to generate a hydrazodicarboxylate intermediate followed by its site-selective allylic transposition.
Co-reporter:Jingwei Li, Xiaoguang Liu, and Daesung Lee
Organic Letters 2012 Volume 14(Issue 1) pp:410-413
Publication Date(Web):December 21, 2011
DOI:10.1021/ol203206k
Gold-catalyzed ring closure of 1,5-enyne containing a silyl ether at the allylic position induces a skeletal rearrangement to form an oxonium intermediate, which then undergoes a smooth allylation in both an intra- and intermolecular manner. In the intramolecular allyl transfer, the additive alcohol becomes positioned on the silicon of the silyl ether by forming a new Si–OR bond, whereas, in the intermolecular allylation, the alcohol is incorporated in the homoallylic carbon by forming a C–OR bond.
Co-reporter:Huaqing Liu;Chunrui Sun;Nam-Kyu Lee;Dr. Roger F. Henry;Dr. Daesung Lee
Chemistry - A European Journal 2012 Volume 18( Issue 38) pp:11889-11893
Publication Date(Web):
DOI:10.1002/chem.201201346
Co-reporter:Jingwei Li ; Chunrui Sun ; Silviya Demerzhan
Journal of the American Chemical Society 2011 Volume 133(Issue 33) pp:12964-12967
Publication Date(Web):July 26, 2011
DOI:10.1021/ja204979r
A novel transition-metal-catalyzed rearrangement of silylated cyclopropenes to the corresponding allenes is described. The presence of both the trimethylsilyl group on the cyclopropene and the platinum catalyst are crucial for this rearrangement.
Co-reporter:Jingwei Li
European Journal of Organic Chemistry 2011 Volume 2011( Issue 23) pp:
Publication Date(Web):
DOI:10.1002/ejoc.201190060
Abstract
The cover picture shows a chain of dominoes in motion where one domino topples the one next to it, and so on. The individual dominoes symbolize elementary bond-forming events by which simple alkene and alkyne building blocks are processed in tandem to form products of extended conjugation and polycyclic systems. Many enyne-metathesis-based tandem reactions developed and applied to the synthesis of natural and unnatural products are detailed in the Microreview by J. Li and D. Lee on p. 4269 ff.
Co-reporter:Jingwei Li
European Journal of Organic Chemistry 2011 Volume 2011( Issue 23) pp:4269-4287
Publication Date(Web):
DOI:10.1002/ejoc.201100438
Abstract
Enyne metathesis is a powerful synthetic tool for generating 1,3-dienes by redistributing unsaturated functionalities between an alkene and an alkyne via vinylalkylidene intermediates. Although less explored, enyne metathesis has a unique capacity for forming multiple C=C bonds and polycyclic systems in a tandem fashion. By exploiting these characteristics, various tandem reactions have been developed and applied to the synthesis of natural and unnatural products. This microreview recapitulates the recent advances in enyne-metathesis-based tandem processes under two main categories: Enyne metathesis/metathesis and enyne metathesis/nonmetathesis.
Co-reporter:Ivan Volchkov;Dr. Kamlesh Sharma;Dr. Eun Jin Cho;Dr. Daesung Lee
Chemistry – An Asian Journal 2011 Volume 6( Issue 8) pp:1961-1966
Publication Date(Web):
DOI:10.1002/asia.201100131
Co-reporter:Jingwei Li ; Chunrui Sun
Journal of the American Chemical Society 2010 Volume 132(Issue 19) pp:6640-6641
Publication Date(Web):April 22, 2010
DOI:10.1021/ja101998w
A new cyclopropanation reaction involving Cα−Si bond insertion of alkylidene carbenes derived from α-silyl ketones has been developed. This unprecedented alkylidene carbene reactivity features excellent selectivity for insertion into Cα−Si bonds rather than insertion into Cγ−H bonds or addition to γ,δ-double or -triple bonds. The selectivity trend clearly indicates that the α-oxygen in the tether significantly promotes Cγ−H insertion, although the Cα−Si bond insertion still competes effectively.
Co-reporter:Sang Young Yun ; Kung-Pern Wang ; Mansuk Kim
Journal of the American Chemical Society 2010 Volume 132(Issue 26) pp:8840-8841
Publication Date(Web):June 15, 2010
DOI:10.1021/ja1020525
The cross-metathesis of terminal alkyne and alkene using Ru-based Grubbs catalyst generally undergoes α-insertion. In this study, excellent control over α- and β-insertion of ruthenium alkylidene into terminal alkynes has been achieved by using a substituent at a remote site from the reaction center. While the origin of this regioselectivity of insertion and metallotropic shift remains to be established, the trend indicates that the α-insertion with a concomitant metallotropic shift is favored with small substituents whereas the β-insertion without a metallotropic shift is favored with bulky substituents such as tert-butyl and trialkyl silyl groups.
Co-reporter:SangYoung Yun Dr.;EricC. Hansen Dr.;Ivan Volchkov;EunJin Cho Dr.;WaiYip Lo
Angewandte Chemie International Edition 2010 Volume 49( Issue 25) pp:4261-4263
Publication Date(Web):
DOI:10.1002/anie.201001681
Co-reporter:SangYoung Yun Dr.;EricC. Hansen Dr.;Ivan Volchkov;EunJin Cho Dr.;WaiYip Lo
Angewandte Chemie 2010 Volume 122( Issue 25) pp:4357-4359
Publication Date(Web):
DOI:10.1002/ange.201001681
Co-reporter:Jingwei Li Dr.
Chemistry – An Asian Journal 2010 Volume 5( Issue 6) pp:1298-1302
Publication Date(Web):
DOI:10.1002/asia.200900724
Co-reporter:Sang Young Yun ; Jun-Cheng Zheng
Journal of the American Chemical Society 2009 Volume 131(Issue 24) pp:8413-8415
Publication Date(Web):May 27, 2009
DOI:10.1021/ja903526g
A systematic study of C−H insertion reactions with variously substituted and conformationally constrained substrates was carried out. High selectivity in the insertion between two competing C−H bonds caused by a strong stereoelectronic effect of an oxygen substituent was achieved. This regioselective C−H insertion reaction was employed as a platform to develop a concise asymmetric synthesis of platensimycin.
Co-reporter:Kung-Pern Wang ; Sang Young Yun ; Daesung Lee ;Donald J. Wink
Journal of the American Chemical Society 2009 Volume 131(Issue 42) pp:15114-15115
Publication Date(Web):October 2, 2009
DOI:10.1021/ja907155r
The isolation and structural characterization of alkyne-bound Ru alkylidene complexes has been elusive. However, a sequential enyne ring-closing metathesis of a diyne moiety and metallotropic [1,3]-shift followed by a second enyne ring-closing metathesis allowed the formation of a highly stable alkyne-chelated ruthenium complex. The full characterization of this complex was realized by NMR spectroscopy and X-ray crystallography.
Co-reporter:Sumit Mukherjee and Daesung Lee
Organic Letters 2009 Volume 11(Issue 13) pp:2916-2919
Publication Date(Web):June 9, 2009
DOI:10.1021/ol900923c
A tandem ring-closing metathesis of a silaketal-based dienyne substrate proceeded efficiently to provide a bicyclic siloxane, which upon removal of the silicon tether afforded an (E,Z)-1,3-dienediol. Further manipulation of this key functional motif rendered synthesis of the entire C1−C19 linear skeleton of (−)-cochleamycin A, a late-stage intermediate employed in the previous total synthesis of (+)-cochleamycin A by Roush and co-workers.
Co-reporter:Eun Jin Cho
Advanced Synthesis & Catalysis 2008 Volume 350( Issue 17) pp:2719-2723
Publication Date(Web):
DOI:10.1002/adsc.200800544
Abstract
A propargylic ester containing a propargylic alkoxy group has been observed to preferentially undergo [1,2]-acyl shift over [1,3]-shift. In addition, the complementary and contrasting reactivity of vinyl vs. alkynyl platinum carbenoids has been discovered. Vinyl platinum carbenoids are more prone to undergo [1,2]-H shift over addition to π-bonds whereas alkynyl platinum carbenoids preferentially add to π-bonds.
Co-reporter:SangYoung Yun Dr.;Jun-Cheng Zheng Dr.
Angewandte Chemie 2008 Volume 120( Issue 33) pp:6297-6299
Publication Date(Web):
DOI:10.1002/ange.200801587
Co-reporter:SangYoung Yun Dr.;Jun-Cheng Zheng Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 33) pp:6201-6203
Publication Date(Web):
DOI:10.1002/anie.200801587
Co-reporter:Daesung Lee and Mansuk Kim
Organic & Biomolecular Chemistry 2007 vol. 5(Issue 21) pp:3418-3427
Publication Date(Web):06 Sep 2007
DOI:10.1039/B710379D
Metal complexes of alkynyl carbenes undergo a [1,3]-bond shift known as a metallotropic shift. Since the discovery of the metallotropic [1,3]-shift of Rh-carbenoid, many more alkynyl carbene complexes with Ti, Cr, Mn, Mo, Ru, W, Re, Pt, and Au have been discovered to show metallotropic shift behavior. This article briefly summarizes the [1,3]-bond shift of alkynyl carbenes and their metal complexes.
Co-reporter:Chunrui Sun, Jingwei Li, Daesung Lee, Genping Huang and Yuanzhi Xia
Chemical Communications 2012 - vol. 48(Issue 89) pp:NaN10992-10992
Publication Date(Web):2012/09/24
DOI:10.1039/C2CC35329F
A novel C(sp3)–H amination of trimethylsilyl-substituted cyclopropenes is described. This C–H amination proceeds via a tandem regioselective ene reaction between cyclopropenes and azodicarboxylate to generate a hydrazodicarboxylate intermediate followed by its site-selective allylic transposition.
Co-reporter:Venkata R. Sabbasani, Sang Young Yun and Daesung Lee
Inorganic Chemistry Frontiers 2016 - vol. 3(Issue 8) pp:NaN943-943
Publication Date(Web):2016/05/23
DOI:10.1039/C6QO00148C
Novel ruthenium alkylidene complexes are prepared via enyne ring-closing metathesis, and the structural features of acetate- and sulfonamide-chelates are explored by single crystal X-ray diffraction analyses. All 5-membered sulfonamide chelates and 6-membered acetate chelates have the chelated oxygen and the NHC ligand in cis relationship, which force the two chloride ligands into cis orientation. Upon exposure to carbon monoxide, while acetate chelates decomposed to generate vinyl acetates, the sulfonamide chelate formed an unprecedented CO-bound octahedral ruthenium alkylidene complex. In this complex, the alkylidene carbene moiety and the NHC ligand have a trans relationship, which is the first example of Grubbs-type ruthenium alkylidene to have this unique trans geometry. This complex is devoid of metathesis activity because of the coordinatively saturated nature of the ruthenium center with a completely locked ligand environment, which does not allow either associative or dissociative ligand–substrate exchange with alkene substrates.
Co-reporter:Rajdip Karmakar and Daesung Lee
Chemical Society Reviews 2016 - vol. 45(Issue 16) pp:NaN4470-4470
Publication Date(Web):2016/01/26
DOI:10.1039/C5CS00835B
Arynes are unique aromatic species that display versatile reactivity in a variety of transformations. It has been demonstrated that the reaction profiles of arynes can be altered by transition metal additives, and one of the earliest examples of the metal additive effect was observed with silver ions (Ag+). Especially, in the presence of Ag+, benzyne showed distinctive reactivity and selectivity toward π-nucleophiles compared to the corresponding reaction in its absence. Although more experimental data need to be accumulated to accurately define the Ag+ additive effect, based on its role displayed in various transformations, we can infer that Ag+ interacts with arynes to form a reactive intermediate, which behaves like a silver-bound aryl cation or 1,2-carbene-silver carbenoid. In this tutorial review, various aryne-mediated reactions involving Ag+ or other organosilver species are discussed, which show a bird's-eye view on the Ag+ effect in aryne chemistry such that further explorations of the unique reactivity of arynes under the influence Ag+ will be inspired.
Co-reporter:Phani Mamidipalli, Sang Young Yun, Kung-Pern Wang, Tao Zhou, Yuanzhi Xia and Daesung Lee
Chemical Science (2010-Present) 2014 - vol. 5(Issue 6) pp:NaN2367-2367
Publication Date(Web):2014/02/19
DOI:10.1039/C3SC53478B
Formal hydrogenation of arynes was realized by using trialkylsilyl groups tethered to the arynes as the hydride source. In stark contrast to the effective 1°, 2°, and 3° C–H bond insertion of alkyl groups tethered to arynes, the 2° and 3° C–H bonds on the β-carbon of silyl groups show high tendency for hydride transfer rather than C–H insertion, whereas the corresponding 1° C–H bonds exclusively undergo C–H insertion. The exclusive hydride transfer and C–H insertion behavior of different C–H bonds in these two reaction pathways were rationalized by the stability of the incipient carbocationic intermediates which could be further bolstered by DFT-calculations.
Co-reporter:Jingwei Li and Daesung Lee
Chemical Science (2010-Present) 2012 - vol. 3(Issue 11) pp:NaN3301-3301
Publication Date(Web):2012/08/17
DOI:10.1039/C2SC20812A
A highly diastereoselective ring-rearrangement metathesis of cyclopentene derivatives containing all-carbon quaternary centres is reported. This tandem metathesis process provides cyclohexene derivatives with two contiguous stereogenic centres, one of which is an all-carbon quaternary stereogenic centre, in high yields and excellent diastereoselecitivity. The efficacy of this methodology is showcased in the total synthesis of spiropiperidine alkaloid nitramine. In the meantime, it has been found that metathesis of the corresponding acyclic triene derivatives affords the same products with the same level of efficiency and diastereoselectivity.
Co-reporter:Ivan Volchkov and Daesung Lee
Chemical Society Reviews 2014 - vol. 43(Issue 13) pp:NaN4394-4394
Publication Date(Web):2014/04/24
DOI:10.1039/C4CS00036F
The direct metal-catalyzed [1,3]-transposition of allylic alcohols and allylic silyl ethers is a synthetically useful isomerization process that occurs via [3,3]-sigmatropic rearrangement induced by high oxidation state oxometal complexes. The isomerization requires only a catalytic amount of promoter, and high chirality transfer can be achieved. Thus, it bears a significant potential to become a powerful tool in multistep synthesis. Although [1,3]-transposition of allylic alcohols has been known since the late 1960s, the development of synthetically useful protocols that allow for a high level of regio- and stereoselectivity control and their synthetic applications have emerged only recently. This tutorial review summarizes recently developed regioselective [1,3]-transpositions of allylic alcohols and silyl ethers and their applications to natural product synthesis.
Co-reporter:Daesung Lee and Mansuk Kim
Organic & Biomolecular Chemistry 2007 - vol. 5(Issue 21) pp:NaN3427-3427
Publication Date(Web):2007/09/06
DOI:10.1039/B710379D
Metal complexes of alkynyl carbenes undergo a [1,3]-bond shift known as a metallotropic shift. Since the discovery of the metallotropic [1,3]-shift of Rh-carbenoid, many more alkynyl carbene complexes with Ti, Cr, Mn, Mo, Ru, W, Re, Pt, and Au have been discovered to show metallotropic shift behavior. This article briefly summarizes the [1,3]-bond shift of alkynyl carbenes and their metal complexes.
Co-reporter:Rajdip Karmakar, Kung-Pern Wang, Sang Young Yun, Phani Mamidipalli and Daesung Lee
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 21) pp:NaN4788-4788
Publication Date(Web):2016/04/26
DOI:10.1039/C6OB00524A
A new functionalization method of arynes promoted by a novel catalytic role of the Grubbs-type ruthenium alkylidene complex is described. Through a sequence of aryne formation followed by their halo-functionalization, various bis-1,3-diynes were directly converted to functionalized aryl chlorides, bromides and iodides in good yields in the presence of a catalytic amount of a ruthenium alkylidene complex and halogenated hydrocarbons such as CH2Cl2, CHCl3, CH2Br2, and CH2I2. The utility of this novel transformation is demonstrated by a formal synthesis of herbindole B.

![Benzene, [1-[3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]propyl]-4,4-dimethyl-1,2-pentadien-1-yl]-](/data/chemimg/3722300/1838597-99-1.png)
![Benzene, [1-[3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]propyl]-4,4-dimethyl-1,2-pentadien-1-yl]-](/data/chemimg/3722300/1838597-99-1_b.png)
![Benzene, [1-(3,3-dimethyl-1-buten-1-ylidene)heptyl]-](/data/chemimg/3722100/1838597-98-0.png)
![Benzene, [1-(3,3-dimethyl-1-buten-1-ylidene)heptyl]-](/data/chemimg/3722100/1838597-98-0_b.png)
![Benzene, [1-[2-[(1,1-dimethylethyl)dimethylsilyl]ethenylidene]heptyl]-](/data/chemimg/3722200/1448195-59-2.png)
![Benzene, [1-[2-[(1,1-dimethylethyl)dimethylsilyl]ethenylidene]heptyl]-](/data/chemimg/3722200/1448195-59-2_b.png)


![1-Propene, 3-[(3-bromo-2-propynyl)oxy]-](http://img.cochemist.com/ccimg/874900/874894-24-3.png)
![1-Propene, 3-[(3-bromo-2-propynyl)oxy]-](http://img.cochemist.com/ccimg/874900/874894-24-3_b.png)

![BENZALDEHYDE, 5-METHOXY-2-[(TRIMETHYLSILYL)ETHYNYL]-](http://img.cochemist.com/ccimg/681500/681427-53-2.png)
![BENZALDEHYDE, 5-METHOXY-2-[(TRIMETHYLSILYL)ETHYNYL]-](http://img.cochemist.com/ccimg/681500/681427-53-2_b.png)




![1,7-Diazaspiro[4.4]non-1-en-6-one,9-(2,4-dibromo-5-methoxyphenyl)-8-hydroxy-7-methyl-, (5S,8R,9R)-](http://img.cochemist.com/ccimg/227000/226993-88-0.png)
![1,7-Diazaspiro[4.4]non-1-en-6-one,9-(2,4-dibromo-5-methoxyphenyl)-8-hydroxy-7-methyl-, (5S,8R,9R)-](http://img.cochemist.com/ccimg/227000/226993-88-0_b.png)
![1,7-Diazaspiro[4.4]nonan-6-one,9-(2,4-dibromo-5-methoxyphenyl)-8-hydroxy-7-methyl-, (5S,8R,9R)-](http://img.cochemist.com/ccimg/227000/226993-86-8.png)
![1,7-Diazaspiro[4.4]nonan-6-one,9-(2,4-dibromo-5-methoxyphenyl)-8-hydroxy-7-methyl-, (5S,8R,9R)-](http://img.cochemist.com/ccimg/227000/226993-86-8_b.png)
![1,7-Diazaspiro[4.4]nonane-2,6-dione,9-(2,4-dibromo-5-methoxyphenyl)-8-hydroxy-1,7-dimethyl-, (5S,8R,9R)-](http://img.cochemist.com/ccimg/227000/226993-85-7.png)
![1,7-Diazaspiro[4.4]nonane-2,6-dione,9-(2,4-dibromo-5-methoxyphenyl)-8-hydroxy-1,7-dimethyl-, (5S,8R,9R)-](http://img.cochemist.com/ccimg/227000/226993-85-7_b.png)
![1,7-Diazaspiro[4.4]nonan-6-one,9-(2,4-dibromo-5-methoxyphenyl)-8-hydroxy-1,7-dimethyl-, (5S,8R,9R)-](http://img.cochemist.com/ccimg/227000/226993-84-6.png)
![1,7-Diazaspiro[4.4]nonan-6-one,9-(2,4-dibromo-5-methoxyphenyl)-8-hydroxy-1,7-dimethyl-, (5S,8R,9R)-](http://img.cochemist.com/ccimg/227000/226993-84-6_b.png)




![Benzene, [[(3-bromo-1-methyl-2-propynyl)oxy]methyl]-](http://img.cochemist.com/ccimg/184400/184370-62-5.png)
![Benzene, [[(3-bromo-1-methyl-2-propynyl)oxy]methyl]-](http://img.cochemist.com/ccimg/184400/184370-62-5_b.png)
![1-Butanone, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-1-phenyl-](http://img.cochemist.com/ccimg/143900/143878-47-1.png)
![1-Butanone, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-1-phenyl-](http://img.cochemist.com/ccimg/143900/143878-47-1_b.png)
![1H-Inden-1-one, 2,3-dihydro-2-[(3,4,5-trimethoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/141300/141247-28-1.png)
![1H-Inden-1-one, 2,3-dihydro-2-[(3,4,5-trimethoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/141300/141247-28-1_b.png)
![Benzenepropanol, α-[2-(trimethylsilyl)ethynyl]-](/data/chemimg/1719800/140149-77-5.png)
![Benzenepropanol, α-[2-(trimethylsilyl)ethynyl]-](/data/chemimg/1719800/140149-77-5_b.png)



![Silane, [[4-(1,1-dimethylethyl)-1-cyclohexen-1-yl]ethynyl]trimethyl-](http://img.cochemist.com/ccimg/91200/91158-83-7.png)
![Silane, [[4-(1,1-dimethylethyl)-1-cyclohexen-1-yl]ethynyl]trimethyl-](http://img.cochemist.com/ccimg/91200/91158-83-7_b.png)
![Pentanoic acid, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, methyl ester](http://img.cochemist.com/ccimg/87800/87729-38-2.png)
![Pentanoic acid, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, methyl ester](http://img.cochemist.com/ccimg/87800/87729-38-2_b.png)


![Benzene, 1,1'-[1-(3,3-dimethyl-1-butenylidene)-1,2-ethanediyl]bis- (9CI)](/data/chemimg/3722200/74762-22-4.png)
![Benzene, 1,1'-[1-(3,3-dimethyl-1-butenylidene)-1,2-ethanediyl]bis- (9CI)](/data/chemimg/3722200/74762-22-4_b.png)
![TRICYCLO[3.3.1.13,7]DECAN-2-OL, 2-ETHYNYL-](http://img.cochemist.com/ccimg/70900/70887-49-9.png)
![TRICYCLO[3.3.1.13,7]DECAN-2-OL, 2-ETHYNYL-](http://img.cochemist.com/ccimg/70900/70887-49-9_b.png)














![1(2H)-Naphthalenone, 3,4-dihydro-2-[(4-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/49700/49629-37-0.png)
![1(2H)-Naphthalenone, 3,4-dihydro-2-[(4-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/49700/49629-37-0_b.png)





![Bicyclo[3.1.1]hept-2-ene-2-carboxylic acid, 6,6-dimethyl-, methyl ester, (1S,5R)-](/data/chemimg/1197000/33741-30-9.png)
![Bicyclo[3.1.1]hept-2-ene-2-carboxylic acid, 6,6-dimethyl-, methyl ester, (1S,5R)-](/data/chemimg/1197000/33741-30-9_b.png)


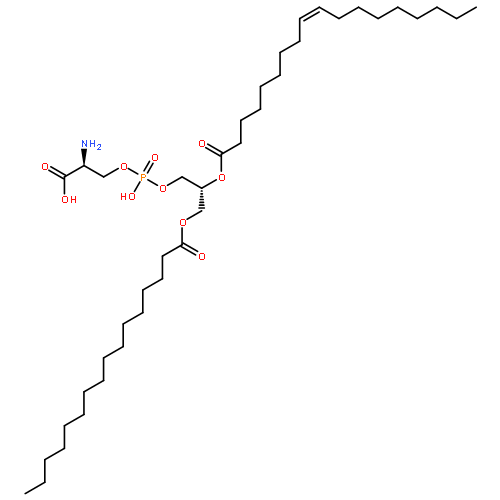
![3,5,8-Trioxa-4-phosphahexacos-17-en-1-aminium,4-hydroxy-N,N,N-trimethyl-9-oxo-7-[[(1-oxohexadecyl)oxy]methyl]-, inner salt,4-oxide, (7R,17Z)-](http://img.cochemist.com/ccimg/26900/26853-31-6.png)
![3,5,8-Trioxa-4-phosphahexacos-17-en-1-aminium,4-hydroxy-N,N,N-trimethyl-9-oxo-7-[[(1-oxohexadecyl)oxy]methyl]-, inner salt,4-oxide, (7R,17Z)-](http://img.cochemist.com/ccimg/26900/26853-31-6_b.png)











![Cyclohexanone, 2-[(4-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/5800/5765-29-7.png)
![Cyclohexanone, 2-[(4-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/5800/5765-29-7_b.png)

![2-phenylethyl 4-[4-(benzyloxy)-3-methoxyphenyl]-7-(4-chlorophenyl)-2-methyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate](http://img.cochemist.com/ccimg/5800/5706-14-9.png)
![2-phenylethyl 4-[4-(benzyloxy)-3-methoxyphenyl]-7-(4-chlorophenyl)-2-methyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate](http://img.cochemist.com/ccimg/5800/5706-14-9_b.png)










![Bicyclo[3.1.1]hept-3-en-2-one,4,6,6-trimethyl-](http://img.cochemist.com/ccimg/100/80-57-9.png)
![Bicyclo[3.1.1]hept-3-en-2-one,4,6,6-trimethyl-](http://img.cochemist.com/ccimg/100/80-57-9_b.png)


![LITHIUM, [DIAZO(TRIMETHYLSILYL)METHYL]-](http://img.cochemist.com/ccimg/84700/84645-45-4.png)
![LITHIUM, [DIAZO(TRIMETHYLSILYL)METHYL]-](http://img.cochemist.com/ccimg/84700/84645-45-4_b.png)

![Benzene, [3-methyl-5-(trimethylsilyl)-3,4-pentadien-1-yl]-](/data/chemimg/125700/110211-33-1.png)
![Benzene, [3-methyl-5-(trimethylsilyl)-3,4-pentadien-1-yl]-](/data/chemimg/125700/110211-33-1_b.png)