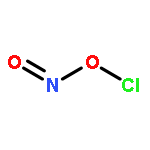
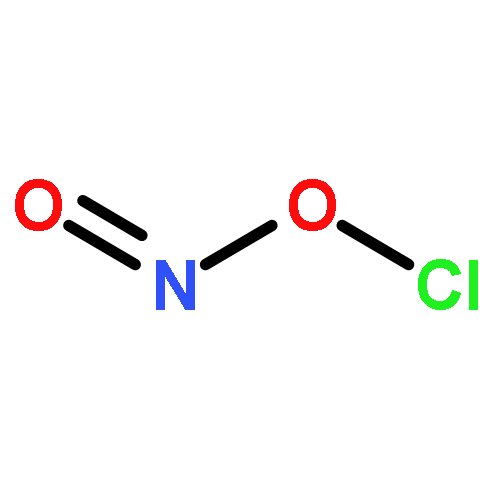
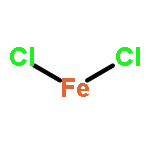
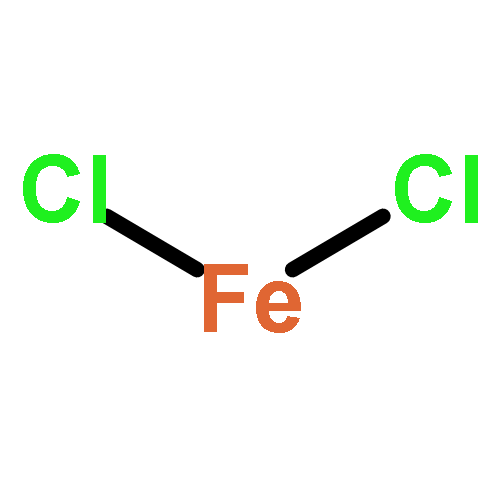
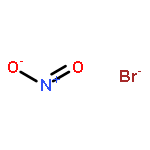
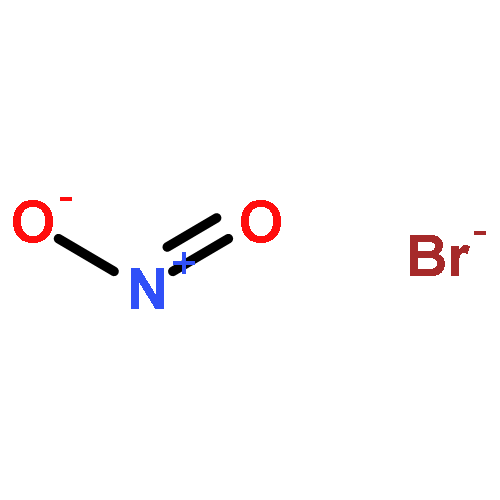
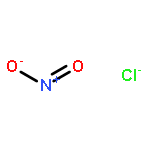
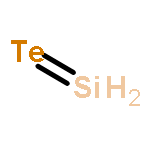
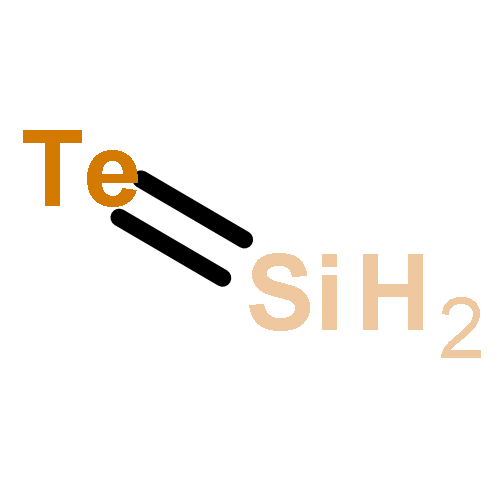
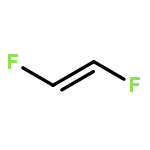
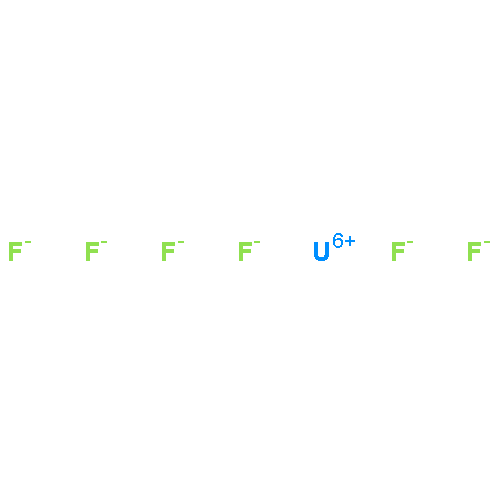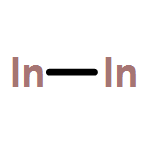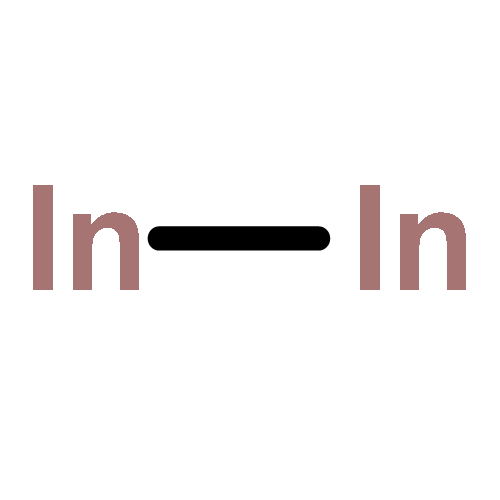Co-reporter:Michael H. Palmer, Malgorzata Biczysko, Kirk A. Peterson, Christopher S. Stapleton, and Simon P. Wells
The Journal of Physical Chemistry A October 19, 2017 Volume 121(Issue 41) pp:7917-7917
Publication Date(Web):September 25, 2017
DOI:10.1021/acs.jpca.7b08399
A combined study of the vibrational spectroscopy of iodopentafluorobenzene by new Raman and Fourier transform infrared (FTIR) spectroscopies, over the spectral range 300 to 3200 cm–1 (Raman) and 50 to 3400 cm–1 (FTIR), with a state-of-the-art theoretical investigation is reported. This has enabled reliable identification of numerous fundamental, overtone, and combination band transitions in unprecedented detail. The theoretical analysis, beyond the double-harmonic approximation, is based on generalized second-order vibrational perturbation theory (GVPT2) with a hybrid coupled cluster/density functional theory (CC/DFT) approach. Anharmonic contributions to structural parameters, rotational constants, vibrational frequencies, and spectral intensities are incorporated. The procedures, of general applicability, enable rigorous comparison of theoretical methods with experimental results in vibrational spectroscopy.
Co-reporter:Rulin Feng;Monica Vasiliu;David A. Dixon
The Journal of Physical Chemistry A February 9, 2017 Volume 121(Issue 5) pp:1041-1050
Publication Date(Web):January 11, 2017
DOI:10.1021/acs.jpca.6b11889
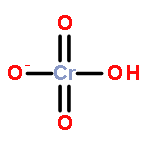
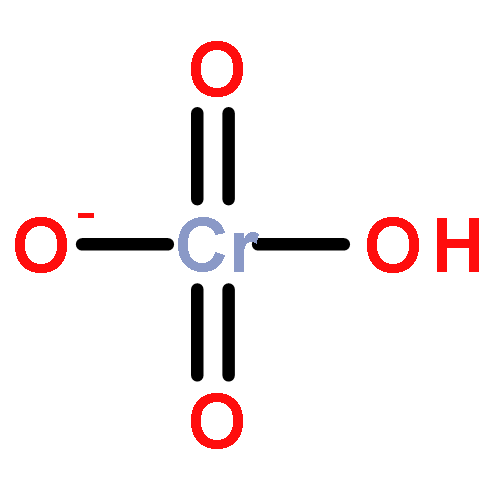
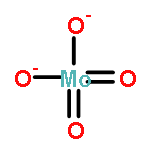
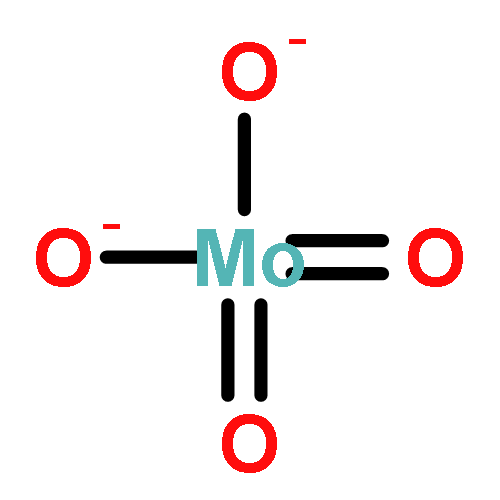
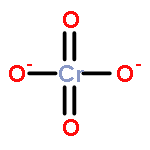
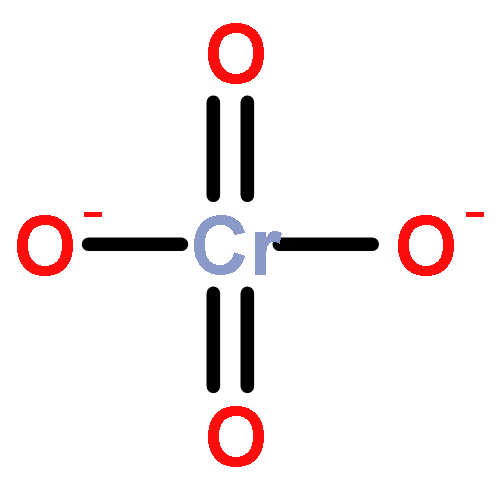
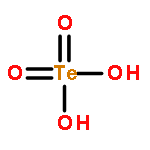
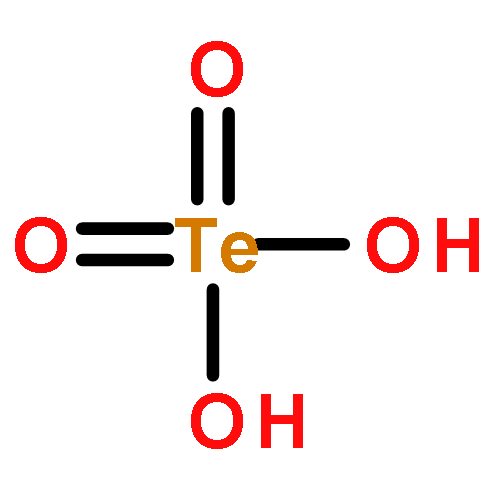
Gas-phase acidities and aqueous solution pKa’s are predicted for MO2(OH)2, where the center atom M is a main Group 6, 16, and U atom using the Feller–Peterson–Dixon approach based on coupled cluster CCSD(T) calculations with additional corrections. The gas-phase acidities of the MO2(OH)2 compounds are essentially the same for elements (M) of the same group, 304–310 kcal/mol at 298 K. All of the Group 6 compounds are 5–6 kcal/mol less acidic in the gas phase than H2SO4. The gas-phase acidity of UO2(OH)2 is calculated to be up to 338.0 kcal/mol, ∼10% less acidic in the gas phase than the other MO2(OH)2 acids. The most acidic molecule in aqueous solution is predicted to be H2SO4. Overall, for the Group 16 compounds, the pKa’s increase going down the group, with H2PoO4 predicted to be slightly more acidic than nitric acid. H2CrO4 is the most acidic of the Group 6 transition metal compounds. The aqueous acidities of H2MoO4 and H2WO4 are comparable and about 3 pKa units less acidic than H2CrO4 and comparable in acidity to HNO3. H2UO4 is not acidic at all in aqueous solution with a pKa near 20 pKa units and is also not predicted to readily undergo hydrolysis reactions.
Co-reporter:Payal Parmar, Kirk A. Peterson, and Aurora E. Clark
The Journal of Physical Chemistry A 2013 Volume 117(Issue 46) pp:11874-11880
Publication Date(Web):May 16, 2013
DOI:10.1021/jp403078j
High-quality static electric dipole polarizabilities have been determined for the ground states of the hard-sphere cations of U, Np, and Pu in the III and IV oxidation states. The polarizabilities have been calculated using the numerical finite field technique in a four-component relativistic framework. Methods including Fock-space coupled cluster (FSCC) and Kramers-restricted configuration interaction (KRCI) have been performed in order to account for electron correlation effects. Comparisons between polarizabilities calculated using Dirac–Hartree–Fock (DHF), FSCC, and KRCI methods have been made using both triple- and quadruple-ζ basis sets for U4+. In addition to the ground state, this study also reports the polarizability data for the first two excited states of U3+/4+, Np3+/4+, and Pu3+/4+ ions at different levels of theory. The values reported in this work are the most accurate to date calculations for the dipole polarizabilities of the hard-sphere tri- and tetravalent actinide ions and may serve as reference values, aiding in the calculation of various electronic and response properties (for example, intermolecular forces, optical properties, etc.) relevant to the nuclear fuel cycle and material science applications.
Co-reporter:J. Grant Hill and Kirk A. Peterson
Journal of Chemical Theory and Computation 2012 Volume 8(Issue 2) pp:518-526
Publication Date(Web):January 19, 2012
DOI:10.1021/ct200856f
Compact auxiliary basis sets matched to the standard aug-cc-pVnZ-PP and aug-cc-pwCVnZ-PP orbital basis sets have been developed for the coinage metals (Cu, Ag, Au) and group 12 elements (Zn, Cd, Hg) for use in the resolution of the identity (RI) approximation in explicitly correlated F12 calculations. The CCSD(T)-F12b method has been utilized with these auxiliary basis sets to carry out molecular benchmark calculations on homonuclear coinage metal diatomics (Cu2, Ag2, Au2), as well as mono- (CuF, AgF, AuF) and difluorides (CuF2, AgF2, AuF2). The resulting equilibrium geometries, harmonic vibrational frequencies, and atomization energies demonstrate that CCSD(T)-F12b calculations using double-ζ-quality basis sets produce results comparable in accuracy to conventional CCSD(T) quadruple-ζ calculations, while F12b with triple-ζ-quality sets yields results close to 5-ζ, which is near the conventional complete basis set (CBS) limit estimates. Analogous trends are observed for the group 12 monofluorides (ZnF, CdF, HgF). It is also shown that the effects of correlating the transition metal element (n – 1)s and p electrons are relatively insensitive to the size of the basis set when the F12 method is utilized.
Co-reporter:Kirk A. Peterson, David A. Dixon , Hermann Stoll
The Journal of Physical Chemistry A 2012 Volume 116(Issue 39) pp:9777-9782
Publication Date(Web):September 10, 2012
DOI:10.1021/jp3084259
Explicitly correlated CCSD(T)-F12b calculations show that the lowest energy conformer of XeF6 is the C3v structure with a stereoactive lone pair. The C3v structure is 1.08 kcal/mol below the C2v structure and 1.80 kcal/mol below the Oh structure without vibrational corrections. The C2v conformer is a transition state connecting the different C3v minima on the pseudorotation potential energy surface, and the Oh structure is a higher-order saddle point leading to the C2v transition states. The calculated vibrational frequencies for the C3v structure best fit the experimental frequencies. The calculated heats of formation for XeF6, −62.1 ± 1.4 kcal/mol at 0 K and −64.0 ± 1.4 kcal/mol at 298 K, are the best available values and show that there are serious issues with the experimental values. The results show that the explicitly correlated CCSD(T)-F12b method can be used to address important electronic structure issues with smaller basis sets.
Co-reporter:Brian P. Prascher;David E. Woon
Theoretical Chemistry Accounts 2011 Volume 128( Issue 1) pp:69-82
Publication Date(Web):2011 January
DOI:10.1007/s00214-010-0764-0
Correlation consistent basis sets of double-ζ through quintuple-ζ quality for the alkali and alkaline earth metals Li, Be, Na, and Mg have been developed, including the valence (cc-pVnZ), augmented valence (aug-cc-pVnZ), core-valence (cc-pCVnZ), and weighted core-valence (cc-pwCVnZ) basis sets. The basis sets are also re-contracted for Douglas–Kroll scalar relativistic calculations and are found to be superior to non-relativistic basis sets in recovering scalar relativistic effects. CCSD(T) computations have been performed with these basis sets, and a series of properties have been examined, including atomic ionization potentials and electron affinities, optimized molecular geometries, harmonic vibrational frequencies, atomization energies, and enthalpies of formation for the molecules Li2, LiF, BeO, BeF, BeH2, BeF2, Na2, NaF, MgO, MgF, MgH2, and MgF2.
Co-reporter:J. Grant Hill and Kirk A. Peterson
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 35) pp:10460-10468
Publication Date(Web):05 Jul 2010
DOI:10.1039/C0CP00020E
Correlation consistent basis sets for the alkali and alkaline earth metals Li, Be, Na, and Mg have been optimized for use with explicitly correlated F12 methods. These include orbital basis sets for valence-only (denoted cc-pVnZ-F12) and core–valence (cc-pCVnZ-F12) correlation, along with compact complementary auxiliary basis sets for use in the resolution of the identity approximation. Additional auxiliary basis sets that can be employed in the density fitting of two-electron integrals in both explicitly correlated methods and in more conventional correlated methods, such as density fitted second-order Møller–Plesset perturbation theory (DF-MP2), have also been developed by adding additional diffuse or core–valence functions to the cc-pVnZ/MP2FIT sets of Hättig. Explicitly correlated calculations with the approximate CCSD(T)-F12b method have been carried out with these basis sets on a series of sixteen test molecules to demonstrate their accuracy for optimized geometries, harmonic vibrational frequencies, and atomization energies. Results comparable to conventional CCSD(T) quintuple-ζ, which are near the complete basis set limits for these molecules, are obtained using CCSD(T)-F12b with just triple-ζ quality basis sets. The effects on the spectroscopic constants from correlating the outer core electrons are accurately recovered with just the cc-pCVDZ-F12 basis sets.
Co-reporter:Kazim E. Yousaf, Kirk A. Peterson
Chemical Physics Letters 2009 Volume 476(4–6) pp:303-307
Publication Date(Web):16 July 2009
DOI:10.1016/j.cplett.2009.06.003
Abstract
Compact auxiliary basis sets matched to the standard aug-cc-pVnZ and aug-cc-pV(n + d)Z orbital basis sets have been developed for use as resolution-of-the-identity (RI) sets in explicitly correlated F12 calculations. The resulting RI errors from using these sets have been benchmarked in calculations of atomization energies and electron affinities for a number of representative small molecules. These errors were always more than an order of magnitude smaller than the residual basis set error for a given choice of orbital basis set.
Co-reporter:Kirk A. Peterson, Alexander Mitrushchenkov, Joseph S. Francisco
Chemical Physics 2008 Volume 346(1–3) pp:34-44
Publication Date(Web):4 May 2008
DOI:10.1016/j.chemphys.2008.02.042
Co-reporter:J. Grant Hill and Kirk A. Peterson
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 35) pp:NaN10468-10468
Publication Date(Web):2010/07/05
DOI:10.1039/C0CP00020E
Correlation consistent basis sets for the alkali and alkaline earth metals Li, Be, Na, and Mg have been optimized for use with explicitly correlated F12 methods. These include orbital basis sets for valence-only (denoted cc-pVnZ-F12) and core–valence (cc-pCVnZ-F12) correlation, along with compact complementary auxiliary basis sets for use in the resolution of the identity approximation. Additional auxiliary basis sets that can be employed in the density fitting of two-electron integrals in both explicitly correlated methods and in more conventional correlated methods, such as density fitted second-order Møller–Plesset perturbation theory (DF-MP2), have also been developed by adding additional diffuse or core–valence functions to the cc-pVnZ/MP2FIT sets of Hättig. Explicitly correlated calculations with the approximate CCSD(T)-F12b method have been carried out with these basis sets on a series of sixteen test molecules to demonstrate their accuracy for optimized geometries, harmonic vibrational frequencies, and atomization energies. Results comparable to conventional CCSD(T) quintuple-ζ, which are near the complete basis set limits for these molecules, are obtained using CCSD(T)-F12b with just triple-ζ quality basis sets. The effects on the spectroscopic constants from correlating the outer core electrons are accurately recovered with just the cc-pCVDZ-F12 basis sets.

![Benzenesulfonamide,N-[2-[[[3-(4-chlorophenyl)-2-propen-1-yl]methylamino]methyl]phenyl]-N-(2-hydroxyethyl)-4-methoxy-](http://img.cochemist.com/ccimg/139300/139298-40-1.png)
![Benzenesulfonamide,N-[2-[[[3-(4-chlorophenyl)-2-propen-1-yl]methylamino]methyl]phenyl]-N-(2-hydroxyethyl)-4-methoxy-](http://img.cochemist.com/ccimg/139300/139298-40-1_b.png)