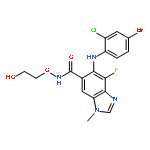
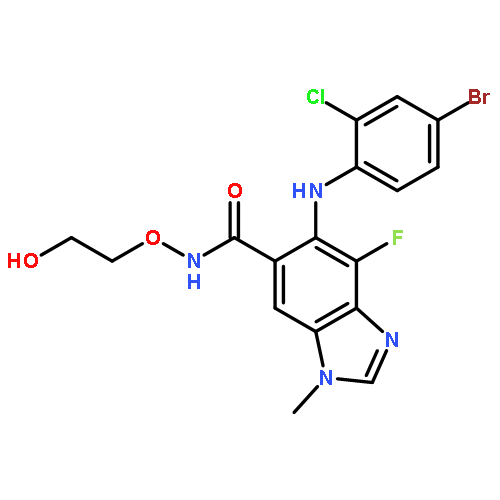
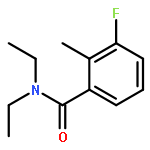
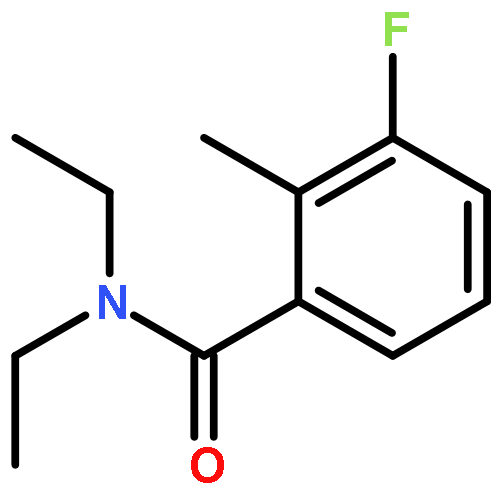
Co-reporter:A. Hooper, A. Zambon and C. J. Springer
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 3) pp:963-969
Publication Date(Web):24 Nov 2015
DOI:10.1039/C5OB01915J
The one-pot borylation/Suzuki reaction is a very efficient means of accessing cross-coupling products of two aryl-halide partners that generally requires the use of specific catalysts or ligands and/or relatively long reaction times. This new microwave-assisted method provides a quick one-pot borylation/Suzuki reaction protocol that we applied to the synthesis of various bi- or poly-aryl scaffolds, including a variety of aryl and heteroaryl ring systems and the core frameworks of kinase inhibitors vemurafenib and GDC-0879.
Co-reporter:Dan Niculescu-Duvaz, Ion Niculescu-Duvaz, Bart M.J.M. Suijkerbuijk, Delphine Ménard, Alfonso Zambon, Lawrence Davies, Jean-Francois Pons, Steven Whittaker, Richard Marais, Caroline J. Springer
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 5) pp:1284-1304
Publication Date(Web):1 March 2013
DOI:10.1016/j.bmc.2012.12.035
The RAS–RAF–MEK–ERK pathway is hyperactivated in 30% of human cancers. BRAF is a serine–threonine kinase, belonging to this pathway that is mutated with high frequency in human melanoma and other cancers thus BRAF is an important therapeutic target in melanoma. We have designed inhibitors of BRAF based on 2,4,5-trisubstituted imidazoles with naphthyl and benzothiophene-4-substituents. Two compounds were discovered to be potent BRAF inhibitors: 1-(6-{2-[4-(2-dimethylamino-ethoxy)phenyl]-5-(pyridin-4-yl)-1H-imidazol-4-yl} benzo[b]thiophen-3-yl)-2,2,2-trifluoroethanol (1i) with BRAF IC50 = 190 nM and with cellular GI50 = 2100 nM, and 6-{2-[4-(2-dimethylamino-ethoxy)-phenyl]-5-pyridin-4-yl-3H-imidazol-4-yl}-naphthalen-1-ol (1q) with IC50 = 9 nM and GI50 = 220 nM.
Co-reporter:Alfonso Zambon, Ion Niculescu-Duvaz, Dan Niculescu-Duvaz, Richard Marais, Caroline J Springer
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 2) pp:789-792
Publication Date(Web):15 January 2012
DOI:10.1016/j.bmcl.2011.11.060
Over the last few years, BRAF has emerged as a validated target in melanoma. This review summarises recent advances in the development of BRAF inhibitors, focussing on agents that have been assessed clinically.
Co-reporter:Arnaud Nourry ; Alfonso Zambon ; Lawrence Davies ; Ion Niculescu-Duvaz ; Harmen P. Dijkstra ; Delphine Ménard ; Catherine Gaulon ; Dan Niculescu-Duvaz ; Bart M. J. M. Suijkerbuijk ; Frank Friedlos ; Helen A. Manne ; Ruth Kirk ; Steven Whittaker ; Richard Marais ;Caroline J. Springer
Journal of Medicinal Chemistry 2010 Volume 53(Issue 5) pp:1964-1978
Publication Date(Web):February 11, 2010
DOI:10.1021/jm901509a
We recently reported on the development of a novel series of BRAF inhibitors based on a tripartite A−B−C system characterized by a para-substituted central aromatic core connected to an imidazo[4,5]pyridin-2-one scaffold and a substituted urea linker. Here, we present a new series of BRAF inhibitors in which the central phenyl ring connects to the hinge binder and substrate pocket of BRAF with a meta-substitution pattern. The optimization of this new scaffold led to the development of low-nanomolar inhibitors that permits the use of a wider range of linkers and terminal C rings while enhancing the selectivity for the BRAF enzyme in comparison to the para series.
Co-reporter:Bart M. J. M. Suijkerbuijk ; Ion Niculescu-Duvaz ; Catherine Gaulon ; Harmen P. Dijkstra ; Dan Niculescu-Duvaz ; Delphine Ménard ; Alfonso Zambon ; Arnaud Nourry ; Lawrence Davies ; Helen A. Manne ; Frank Friedlos ; Lesley M. Ogilvie ; Douglas Hedley ; Filipa Lopes ; Natasha P. U. Preece ; Javier Moreno-Farre ; Florence I. Raynaud ; Ruth Kirk ; Steven Whittaker ; Richard Marais ;Caroline J. Springer
Journal of Medicinal Chemistry 2010 Volume 53(Issue 7) pp:2741-2756
Publication Date(Web):March 3, 2010
DOI:10.1021/jm900607f
We describe the design, synthesis, and optimization of a series of new inhibitors of V-RAF murine sarcoma viral oncogene homologue B1 (BRAF), a kinase whose mutant form (V600E) is implicated in several types of cancer, with a particularly high frequency in melanoma. Our previously described inhibitors with a tripartite A−B−C system (where A is a hinge binding pyrido[4,5-b]imidazolone system, B is an aryl spacer group, and C is a heteroaromatic group) were potent against purified V600EBRAF in vitro but were less potent in accompanying cellular assays. Substitution of different aromatic heterocycles for the phenyl based C-ring is evaluated herein as a potential means of improving the cellular potencies of these inhibitors. Substituted pyrazoles, particularly 3-tert-butyl-1-aryl-1H-pyrazoles, increase the cellular potencies without detrimental effects on the potency on isolated V600EBRAF. Thus, compounds have been synthesized that inhibit, with low nanomolar concentrations, V600EBRAF, its downstream signaling in cells [as measured by the reduction of the phosphorylation of extracellular regulated kinase (ERK)], and the proliferation of mutant BRAF-dependent cells. Concomitant benefits are good oral bioavailability and high plasma concentrations in vivo.
Co-reporter:Dan Niculescu-Duvaz, Ion Niculescu-Duvaz, Bart M.J.M. Suijkerbuijk, Delphine Ménard, Alfonso Zambon, Arnaud Nourry, Lawrence Davies, Helen A. Manne, Frank Friedlos, Lesley Ogilvie, Douglas Hedley, Andrew K. Takle, David M. Wilson, Jean-Francois Pons, Tom Coulter, Ruth Kirk, Neus Cantarino, Steven Whittaker, Richard Marais, Caroline J. Springer
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 18) pp:6934-6952
Publication Date(Web):15 September 2010
DOI:10.1016/j.bmc.2010.06.031
V-RAF murine sarcoma viral oncogene homolog B1 (BRAF) is a serine/threonine-specific protein kinase that is mutated with high frequency in cutaneous melanoma, and many other cancers. Inhibition of mutant BRAF is an attractive therapeutic approach for the treatment of melanoma. A triarylimidazole BRAF inhibitor bearing a phenylpyrazole group (dimethyl-[2-(4-{5-[4-(1H-pyrazol-3-yl)-phenyl]-4-pyridin-4-yl-1H-imidazol-2-yl}-phenoxy)-ethyl]-amine, 1a) was identified as an active BRAF inhibitor. Based on this starting point, we synthesized a series of analogues leading to the discovery of 6-{2-[4-(4-methyl-piperazin-1-yl)-phenyl]-5-pyridin-4-yl-3H-imidazol-4-yl}-2,4-dihydro-indeno[1,2-c]pyrazole (1j), with nanomolar activity in three assays: inhibition of purified mutant BRAF activity in vitro; inhibition of oncogenic BRAF-driven extracellular regulated kinase (ERK) activation in BRAF mutant melanoma cell lines; and inhibition of proliferation in these cells.1j IC50 (BRAF) = 0.24 μM; IC50 (pERK) = 0.58 μM; GI50 (SRB) = 0.87 μM.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Dan Niculescu-Duvaz ; Catherine Gaulon ; Harmen P. Dijkstra ; Ion Niculescu-Duvaz ; Alfonso Zambon ; Delphine Ménard ; Bart M. J. M. Suijkerbuijk ; Arnaud Nourry ; Lawrence Davies ; Helen Manne ; Frank Friedlos ; Lesley Ogilvie ; Douglas Hedley ; Steven Whittaker ; Ruth Kirk ; Adrian Gill ; Richard D. Taylor ; Florence I. Raynaud ; Javier Moreno-Farre ; Richard Marais ;Caroline J. Springer
Journal of Medicinal Chemistry 2009 Volume 52(Issue 8) pp:2255-2264
Publication Date(Web):March 26, 2009
DOI:10.1021/jm801509w
BRAF is a serine/threonine kinase that is mutated in a range of cancers, including 50−70% of melanomas, and has been validated as a therapeutic target. We have designed and synthesized mutant BRAF inhibitors containing pyridoimidazolone as a new hinge-binding scaffold. Compounds have been obtained which have low nanomolar potency for mutant BRAF (12 nM for compound 5i) and low micromolar cellular potency against a mutant BRAF melanoma cell line, WM266.4. The series benefits from very low metabolism, and pharmacokinetics (PK) that can be modulated by methylation of the NH groups of the imidazolone, resulting in compounds with fewer H-donors and a better PK profile. These compounds have great potential in the treatment of mutant BRAF melanomas.
Co-reporter:Delphine Ménard ; Ion Niculescu-Duvaz ; Harmen P. Dijkstra ; Dan Niculescu-Duvaz ; Bart M. J. M. Suijkerbuijk ; Alfonso Zambon ; Arnaud Nourry ; Esteban Roman ; Lawrence Davies ; Helen A. Manne ; Frank Friedlos ; Ruth Kirk ; Steven Whittaker ; Adrian Gill ; Richard D. Taylor ; Richard Marais ;Caroline J. Springer
Journal of Medicinal Chemistry 2009 Volume 52(Issue 13) pp:3881-3891
Publication Date(Web):May 27, 2009
DOI:10.1021/jm900242c
BRAF, a serine/threonine specific protein kinase that is part of the MAPK pathway and acts as a downstream effector of RAS, is a potential therapeutic target in melanoma. We have developed a series of small-molecule BRAF inhibitors based on a 1H-imidazo[4,5-b]pyridine-2(3H)-one scaffold (ring A) as the hinge binding moiety and a number of substituted phenyl rings C that interact with the allosteric binding site. The introduction of various groups on the central phenyl ring B combined with appropriate A- and C-ring modifications afford very potent compounds that inhibit V600EBRAF kinase activity in vitro and oncogenic BRAF signaling in melanoma cells. Substitution on the central phenyl ring of a 3-fluoro, a naphthyl, or a 3-thiomethyl group improves activity to yield compounds with an IC50 of 1 nM for purified V600EBRAF and nanomolar activity in cells.
Co-reporter:Ion Niculescu-Duvaz ; Esteban Roman ; Steven R. Whittaker ; Frank Friedlos ; Ruth Kirk ; Ian J. Scanlon ; Lawrence C. Davies ; Dan Niculescu-Duvaz ; Richard Marais ;Caroline J. Springer
Journal of Medicinal Chemistry 2008 Volume 51(Issue 11) pp:3261-3274
Publication Date(Web):May 13, 2008
DOI:10.1021/jm070776b
BRAF, a serine/threonine kinase, plays a key role in the development of certain types of cancer, particularly melanoma. 2-(3,4,5-Trimethoxyphenylamino)-6-(3-acetamidophenyl)-pyrazine, 1, was identified as a low micromolar (IC50 = 3.5 µM) BRAF inhibitor from a high-throughput screen of a library of 23000 compounds. This compound was chosen as the starting point of a program aimed at developing inhibitors of mutant V600EBRAF. We have already reported on the optimization of the trimethoxyphenylamino moiety of 1. In this paper, we describe the synthesis of a series of compounds derived from 1 with the purpose of optimization of the pyrazine central core and the phenylacetamido moiety in order to increase the potency against V600EBRAF compared to CRAF. The biological activity of the new inhibitors was assessed against mutant V600EBRAF in vitro. Several compounds were identified with IC50s of 300−500 nM for V600EBRAF, and all compounds that were assessed showed selectivity for V600EBRAF compared to CRAF by 5−>86-fold.
Co-reporter:Dan Niculescu-Duvaz, Jayne Getaz and Caroline J. Springer
Bioconjugate Chemistry 2008 Volume 19(Issue 4) pp:973
Publication Date(Web):April 2, 2008
DOI:10.1021/bc060242+
A concise synthesis of long-chain poly(ethylene glycol) (PEG) of defined molecular weight up to 29 ethyleneoxy units is described. These PEG diols were converted in a two-step synthesis into Fmoc-protected PEG amino acids, suitable as long linkers and compatible with solid-phase peptide synthesis. Long PEG chains (MW > 1000) can be readily synthesized with this method, which has the advantage of defined single molecular weight products over the comparable commercial polymers. The application of these PEG linkers to the synthesis of peptide−PEG−folate conjugates on a solid support was investigated. A method for the solid support synthesis of the targeting component of the conjugate, folic acid−cysteine, was developed, resulting in improved yields with respect to literature methods. The assembly of the peptide, PEG linker, and targeting group on solid support resulted in the synthesis of a conjugate of defined molecular weight and structure.
Co-reporter:Douglas Hedley,
Lesley Ogilvie
&
Caroline Springer
Nature Reviews Cancer 2007 7(11) pp:870
Publication Date(Web):2007-11-01
DOI:10.1038/nrc2247
Gene-directed enzyme–prodrug therapy (GDEPT) aims to improve the therapeutic ratio (benefit versus toxic side-effects) of cancer chemotherapy. A gene encoding a 'suicide' enzyme is introduced into the tumour to convert a subsequently administered non-toxic prodrug into an active drug selectively in the tumour, but not in normal tissues. Significant effects can now be achieved in vitro and in targeted experimental models, and GDEPT therapies are entering the clinic. Our group has developed a GDEPT system that uses the bacterial enzyme carboxypeptidase G2 to convert nitrogen mustard prodrugs into potent DNA crosslinking agents, and a clinical trial of this system is pending.
Co-reporter:A. Hooper, A. Zambon and C. J. Springer
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 3) pp:NaN969-969
Publication Date(Web):2015/11/24
DOI:10.1039/C5OB01915J
The one-pot borylation/Suzuki reaction is a very efficient means of accessing cross-coupling products of two aryl-halide partners that generally requires the use of specific catalysts or ligands and/or relatively long reaction times. This new microwave-assisted method provides a quick one-pot borylation/Suzuki reaction protocol that we applied to the synthesis of various bi- or poly-aryl scaffolds, including a variety of aryl and heteroaryl ring systems and the core frameworks of kinase inhibitors vemurafenib and GDC-0879.

![N-[3-[3-Cyclopropyl-5-[(2-fluoro-4-iodophenyl)amino]-3,4,6,7-tetrahydro-6,8-dimethyl-2,4,7-trioxopyrido[4,3-d]pyrimidin-1(2H)-yl]phenyl]acetamide](http://img.cochemist.com/ccimg/871800/871700-17-3.png)
![N-[3-[3-Cyclopropyl-5-[(2-fluoro-4-iodophenyl)amino]-3,4,6,7-tetrahydro-6,8-dimethyl-2,4,7-trioxopyrido[4,3-d]pyrimidin-1(2H)-yl]phenyl]acetamide](http://img.cochemist.com/ccimg/871800/871700-17-3_b.png)