Co-reporter:Caleb F. Harris, Michael B. Bayless, Nicolaas P. van Leest, Quinton J. Bruch, Brooke N. Livesay, John Bacsa, Kenneth I. Hardcastle, Matthew P. Shores, Bas de Bruin, and Jake D. Soper
Inorganic Chemistry October 16, 2017 Volume 56(Issue 20) pp:12421-12421
Publication Date(Web):October 2, 2017
DOI:10.1021/acs.inorgchem.7b01906
A new family of low-coordinate Co complexes supported by three redox-noninnocent tridentate [OCO] pincer-type bis(phenolate) N-heterocyclic carbene (NHC) ligands are described. Combined experimental and computational data suggest that the charge-neutral four-coordinate complexes are best formulated as Co(II) centers bound to closed-shell [OCO]2– dianions, of the general formula [(OCO)CoIIL] (where L is a solvent-derived MeCN or THF). Cyclic voltammograms of the [(OCO)CoIIL] complexes reveal three oxidations accessible at potentials below 1.2 V vs Fc+/Fc, corresponding to generation of formally Co(V) species, but the true physical/spectroscopic oxidation states are much lower. Chemical oxidations afford the mono- and dications of the imidazoline NHC-derived complex, which were examined by computational and magnetic and spectroscopic methods, including single-crystal X-ray diffraction. The metal and ligand oxidation states of the monocationic complex are ambiguous; data are consistent with formulation as either [(SOCO)CoIII(THF)2]+ containing a closed-shell [SOCO]2– diphenolate ligand bound to a S = 1 Co(III) center, or [(SOCO•)CoII(THF)2]+ with a low-spin Co(II) ion ferromagnetically coupled to monoanionic [SOCO•]− containing a single unpaired electron distributed across the [OCO] framework. The dication is best described as [(SOCO0)CoII(THF)3]2+, with a single unpaired electron localized on the d7 Co(II) center and a doubly oxidized, charge-neutral, closed-shell SOCO0 ligand. The combined data provide for the first time unequivocal and structural evidence for [OCO] ligand redox activity. Notably, varying the degree of unsaturation in the NHC backbone shifts the ligand-based oxidation potentials by up to 400 mV. The possible chemical origins of this unexpected shift, along with the potential utility of the [OCO] pincer ligands for base-metal-mediated organometallic coupling catalysis, are discussed.
Co-reporter:Tarik J. Ozumerzifon;Indrani Bhowmick;William C. Spaller;Anthony K. Rappé
Chemical Communications 2017 vol. 53(Issue 30) pp:4211-4214
Publication Date(Web):2017/04/11
DOI:10.1039/C7CC01172E
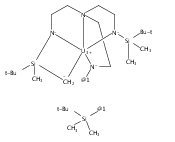
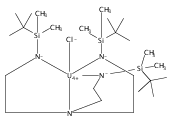
We present the syntheses and characterization of several salts of a trigonal prismatic cobalt(II) complex with a 1,3,5-triaminocyclohexane (tach)-derived ditopic ligand. The air- and moisture-stable tetraphenylborate salt (2) shows slow magnetic relaxation under both zero and applied dc fields. This complex also exhibits an unexpected ability to interact with a cationic sodium guest ion, highlighting the ambifunctional binding nature of amide groups within an iminopyridine scaffold.
Co-reporter:Robert F. Higgins; Steven M. Fatur; Samuel G. Shepard; Susan M. Stevenson; David J. Boston; Eric M. Ferreira; Niels H. Damrauer; Anthony K. Rappé
Journal of the American Chemical Society 2016 Volume 138(Issue 16) pp:5451-5464
Publication Date(Web):March 31, 2016
DOI:10.1021/jacs.6b02723
A combined experimental and theoretical investigation aims to elucidate the necessary roles of oxygen in photoredox catalysis of radical cation based Diels–Alder cycloadditions mediated by the first-row transition metal complex [Cr(Ph2phen)3]3+, where Ph2phen = bathophenanthroline. We employ a diverse array of techniques, including catalysis screening, electrochemistry, time-resolved spectroscopy, and computational analyses of reaction thermodynamics. Our key finding is that oxygen acts as a renewable energy and electron shuttle following photoexcitation of the Cr(III) catalyst. First, oxygen quenches the excited Cr3+* complex; this energy transfer process protects the catalyst from decomposition while preserving a synthetically useful 13 μs excited state and produces singlet oxygen. Second, singlet oxygen returns the reduced catalyst to the Cr(III) ground state, forming superoxide. Third, the superoxide species reduces the Diels–Alder cycloadduct radical cation to the final product and reforms oxygen. We compare the results of these studies with those from cycloadditions mediated by related Ru(II)-containing complexes and find that the distinct reaction pathways are likely part of a unified mechanistic framework where the photophysical and photochemical properties of the catalyst species lead to oxygen-mediated photocatalysis for the Cr-containing complex but radical chain initiation for the Ru congener. These results provide insight into how oxygen can participate as a sustainable reagent in photocatalysis.
Co-reporter:Janice L. Wong, Robert F. Higgins, Indrani Bhowmick, David Xi Cao, Géza Szigethy, Joseph W. Ziller, Matthew P. Shores and Alan F. Heyduk
Chemical Science 2016 vol. 7(Issue 2) pp:1594-1599
Publication Date(Web):08 Dec 2015
DOI:10.1039/C5SC03006D
A new bimetallic platform comprising a six-coordinate Fe(ONO)2 unit bound to an (ONO)M (M = Fe, Zn) has been discovered ((ONOcat)H3 = bis(3,5-di-tert-butyl-2-phenol)amine). Reaction of Fe(ONO)2 with either (ONOcat)Fe(py)3 or with (ONOq)FeCl2 under reducing conditions led to the formation of the bimetallic complex Fe2(ONO)3, which includes unique five- and six-coordinate iron centers. Similarly, the reaction of Fe(ONO)2 with the new synthon (ONOsq˙)Zn(py)2 led to the formation of the heterobimetallic complex FeZn(ONO)3, with a six-coordinate iron center and a five-coordinate zinc center. Both bimetallic complexes were characterized by single-crystal X-ray diffraction studies, solid-state magnetic measurements, and multiple spectroscopic techniques. The magnetic data for FeZn(ONO)3 are consistent with a ground state S = 3/2 spin system, generated from a high-spin iron(II) center that is antiferromagnetically coupled to a single (ONOsq˙)2− radical ligand. In the case of Fe2(ONO)3, the magnetic data revealed a ground state S = 7/2 spin system arising from the interactions of one high-spin iron(II) center, one high-spin iron(III) center, and two (ONOsq˙)2− radical ligands.
Co-reporter:Susan M. Stevenson;Dr. Matthew P. Shores;Dr. Eric M. Ferreira
Angewandte Chemie International Edition 2015 Volume 54( Issue 22) pp:6506-6510
Publication Date(Web):
DOI:10.1002/anie.201501220
Abstract
The photooxidizing capabilities of selected CrIII complexes for promoting radical cation cycloadditions are described. These complexes have sufficiently long-lived excited states to oxidize electron-rich alkenes, thereby initiating [4+2] processes. These metal species augment the spectrum of catalysts explored in photoredox systems, as they feature unique properties that can result in differential reactivity from the more commonly employed ruthenium or iridium catalysts.
Co-reporter:Kelsey A. Schulte, Stephanie R. Fiedler, Matthew P. Shores
Polyhedron 2015 Volume 85() pp:560-564
Publication Date(Web):8 January 2015
DOI:10.1016/j.poly.2014.09.007
Reported herein are the magnetic and structural properties as well as solution equilibrium investigation for a pair of iron (II) pyridyl-imidazoline complexes: [Fe(pizMe)Cl2]2 (1) and [Fe(pizMe)3](FeCl4) (2), where pizMe = 2-(2′-pyridinyl)-4,5-dihydro-1-methylimidazole. Dinuclear complex 1 contains high-spin iron (II) centers and a “flat” pizMe ligand (planar at the imidazoline N atom), while the pizMe-containing complex in 2 exhibits characteristics of a low-spin iron (II) center and a pyramidalized pizMe ligand. In solution, the two complexes are present in equilibrium as evinced through NMR and electronic absorption spectral studies. Spin-state switching between 1 and 2 in methanolic solution is a result of ligand lability.Graphical abstractTwo new Fe(II) complexes with different Fe:pyridyl-imidazoline ratios are reported, which show unique structures depending on their spin states. In methanolic solutions, the species are in equilibrium as observed from spectroscopic studies. Spin-state switching between 1 and 2 in solution is a result of ligand exchange.
Co-reporter:Susan M. Stevenson;Dr. Matthew P. Shores;Dr. Eric M. Ferreira
Angewandte Chemie 2015 Volume 127( Issue 22) pp:6606-6610
Publication Date(Web):
DOI:10.1002/ange.201501220
Abstract
The photooxidizing capabilities of selected CrIII complexes for promoting radical cation cycloadditions are described. These complexes have sufficiently long-lived excited states to oxidize electron-rich alkenes, thereby initiating [4+2] processes. These metal species augment the spectrum of catalysts explored in photoredox systems, as they feature unique properties that can result in differential reactivity from the more commonly employed ruthenium or iridium catalysts.
Co-reporter:Ashley M. McDaniel, Huan-Wei Tseng, Ethan A. Hill, Niels H. Damrauer, Anthony K. Rappé, and Matthew P. Shores
Inorganic Chemistry 2013 Volume 52(Issue 3) pp:1368-1378
Publication Date(Web):January 16, 2013
DOI:10.1021/ic302055r
We report the preparation, photophysical characterization, and computed excited state energies for a family of Cr(III) complexes based on iminopyridine (impy) Schiff base ligands: compounds 1 and 2 feature hexadentate ligands where tren (tris-(2-aminoethyl)amine) caps three impy groups; compounds 3 and 4 are tris(bidentate) analogues of 1 and 2; compounds 2 and 4 contain methyl ester substituents to alter ligand donation properties relative to 1 and 3, respectively. Cyclic voltammograms exhibit multiple reversible ligand-based reductions; the hexadentate and tris(bidentate) analogues have almost identical reduction potentials, and the addition of ester substituents shifts reduction potentials by +200 mV. The absorption spectra of the hexadentate complexes show improved absorption of visible light compared to the tris(bidentate) analogues. Over periods of several hours to days, the complexes undergo ligand-substitution-based decomposition in 1 M HCl(aq) and acetonitrile. For freshly prepared sample solutions in CH3CN, time-resolved emission and transient absorption measurements for 4 show a doublet excited state with 17–19 μs lifetime at room temperature, while no emission or transient absorption signals from the doublet states are observed for the hexadentate analogue 2 under the same conditions. The electronic structure contributions to the differences in observed photophysical properties are compared by extensive computational analyses (UB3LYP MD-DFT and TD-DFT-NTO). These studies indicate that the presence of nonligated bridgehead nitrogen atoms for 1 and 2 significantly reduce excited state doublet, quartet, and sextet energies and change the character of the low lying doublet states in comparison to species that show population of doublet excited states.
Co-reporter:Ashley M. McDaniel;Christina M. Klug
European Journal of Inorganic Chemistry 2013 Volume 2013( Issue 5-6) pp:943-950
Publication Date(Web):
DOI:10.1002/ejic.201201086
Abstract
We report the syntheses and characterizations of low-spin FeII complexes of hexadentate ligands poised for anion-triggered spin-state switching in polar solutions: [Fe(L5-OH)](BF4)2 (1) and [Fe(L5-ONHtBu)](BF4)2 (3), in which L5-OH and L5-ONHtBu are tripodal iminopyridine ligands that contain methanolic or tert-butylamide functional groups, respectively, bound meta to the pyridyl N donor atom. Solid-state evidence for strong hydrogen bonding between Cl– anions and all three amide functional groups in [Fe(L5-ONHtBu)]2+ is provided by the crystal structure of {[Fe(L5-ONHtBu)]⊂Cl}2[FeCl4] (2). In ambient-temperature acetonitrile solutions of 1 and 3, chloride ion titrations produce marked changes in the 1H NMR spectra, including large downfield shifts for the amide NH and hydroxy OH resonances, which indicates strong anion binding events. Interestingly, for amide-containing 3, we observe small changes in magnetic susceptibility as (nBu4N)Cl is added, which suggests that spin-state control by anion–cation interactions may be accessible for related compounds with weaker ligand fields.
Co-reporter:Ellen M. Matson;Mitchell D. Goshert;John J. Kiernicki;Brian S. Newell;Phillip E. Fanwick; Matthew P. Shores; Justin R. Walensky; Suzanne C. Bart
Chemistry - A European Journal 2013 Volume 19( Issue 48) pp:16176-16180
Publication Date(Web):
DOI:10.1002/chem.201303095
Co-reporter:Ashley M. McDaniel, Anthony K. Rappé, and Matthew P. Shores
Inorganic Chemistry 2012 Volume 51(Issue 22) pp:12493-12502
Publication Date(Web):November 8, 2012
DOI:10.1021/ic301909u
We report the preparation and characterization of a series of divalent 3d transition metal complexes (Cr to Zn, 1–7), featuring the multidentate, tripodal iminopyridine Schiff-base ligand trimethyl 6,6′,6″-((1E,1′E,1″E)-((nitrilotris(ethane-2,1-diyl))tris(azanylylidene))tris-(methanylylidene))trinicotinate (L5-OOMe). X-ray structural studies carried out on 1–5 and 7 reveal complex geometries ranging from local octahedral coordination to significant distortion toward trigonal prismatic geometry to heptacoordinate environments. Regardless of coordination mode, magnetic and spectroscopic studies show the ligand to provide moderately strong ligand fields: the Fe complex is low-spin, while the Co and Mn complexes are high-spin at all temperatures probed. Cyclic voltammograms exhibit multiple reversible ligand-based reductions, which are relatively consistent throughout the series; however, the electrochemical behavior of the Cr complex 1 is fundamentally different from those of the other complexes. Time-dependent (TD) density functional theory (DFT) and natural transition orbital (NTO) computational analyses are presented for the ligand, its anion, and complexes 1–7: the computed spectra reproduce the major differential features of the observed visible absorption spectra, and NTOs provide viable interpretations for the observed features. The combined studies indicate that all complexes contain neutral ligands bound to M(II) ions, except for the Cr complex 1, which is best described as a Cr(III) species bound to a radical anionic ligand.
Co-reporter:Christina M. Klug, Ashley M. McDaniel, Stephanie R. Fiedler, Kelsey A. Schulte, Brian S. Newell and Matthew P. Shores
Dalton Transactions 2012 vol. 41(Issue 40) pp:12577-12585
Publication Date(Web):07 Sep 2012
DOI:10.1039/C2DT31213A
We report the syntheses, characterisations, and spin state behaviours of salts of the tripodal-ligated Fe(II) complex [FeL6-OH]X2 (L6-OH = tris{4-[(6-methanol)-2-pyridyl]-3-aza-3-butenyl}amine, X = OTf− (1), Br− (2), I− (3), BPh4− (4)). Covalent linking of the ligand arms is imperative as a high-spin bis(tridentate) complex (5) is formed when a non-tethered ethyl iminopyridine ligand (L2 = 4-[(6-methanol)-2-pyridyl]-3-aza-3-butenyl) is used. For salts 1–4, thermally-induced spin-crossover (SCO) is observed in the solid state, with dependence on anion and solvate molecules. Salts with larger anions show more complete SCO centred at higher temperatures (1 > 3 > 2); the triflate salt 1 (T1/2 = 173 K) also shows the strongest cooperativity of the compounds examined. Hydrogen bonding appears to be critical to SCO in this family of salts: limiting interactions by use of tetraphenylborate produces a high-spin complex down to 5 K. In protic solvents such as methanol, spectra of [FeL6-OH]2+ are largely unchanged over a period of three days, but dissociate when interrogated with strong field bidentate ligands. Compounds 1–3, and 5 remain high spin in solution down to 180 K, consistent with the data obtained in the solid state.
Co-reporter:Wesley A. Hoffert, Md. Khayrul Kabir, Ethan A. Hill, Sara M. Mueller, Matthew P. Shores
Inorganica Chimica Acta 2012 380() pp: 174-180
Publication Date(Web):
DOI:10.1016/j.ica.2011.08.051
Co-reporter:Wesley A. Hoffert ; Anthony K. Rappé
Journal of the American Chemical Society 2011 Volume 133(Issue 51) pp:20823-20836
Publication Date(Web):November 8, 2011
DOI:10.1021/ja206735y
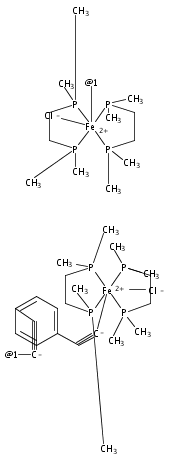
Significant variance in the magnitude of reported exchange coupling parameters (both experimental and computed) for paramagnetic transition metal–ethynylbenzene complexes suggests that nuances of the magnetostructural relationship in this class of compounds remain to be understood and controlled, toward maximizing the stability of high-spin ground states. We report the preparation, electrochemical behavior, magnetic properties, and results of computational investigations of a series of iron ethynylbenzene complexes with coordination environments suitable for metallodendrimer assembly: [(dmpe)2FeCl(C2Ph)](OTf) (1), [(dmpe)4Fe2Cl2(μ-p-DEB)](BArF4)2 (2), [(dmpe)6Fe3Cl3(TEB)] (3), [(dmpe)6Fe3Cl3(μ3-TEB)](OTf)3 (4), and [(dmpe)4Fe2Cl2(μ-m-DEB)](BArF4)2 (5) [dmpe = 1,2-bis(dimethylphosphino)ethane; p-H2DEB = 1,4-diethynylbenzene; BArF4 = tetrakis[3,5-bis(trifluoromethyl)phenyl]borate; H3TEB = 1,3,5-triethynylbenzene; m-H2DEB = 1,3-diethynylbenzene]. As expected, the ligand topology drives the antiferromagnetic coupling in 2 (J = −134 cm–1 using the Ĥ = −2JŜ1·Ŝ2 convention) and the ferromagnetic coupling in 4 and 5 (J = +37 cm–1, J′ = +5 cm–1 for 4; J = +11 cm–1 for 5); the coupling is comparable to but deviates significantly from values reported for related Cp*-containing species (Cp* = η5-C5Me5). The origins of these differences are explored computationally: a density functional theory (DFT) approach for treating the coupling of three spin centers as a linear combination of single-determinantal descriptions is developed and described, and the results of these computations can be generalized to other paramagnetic systems. Unrestricted B3LYP hybrid DFT calculations performed on rotamers of 4 and 5 and related complexes, as well as Cp* analogues, provide J values that correlate with the experimental values. We find that geometric considerations dominate the magnetism of the Cp* complexes, while topology and alkynyl ligand electronics combine more subtly to drive the magnetism of the new complexes reported here. These calculations imply that substantial magnetic exchange parameters, with accompanying well-isolated high-spin ground states, are achievable for ethynylbenzene-bridged paramagnetic metallodendrimers.
Co-reporter:Ashley M. McDaniel, Huan-Wei Tseng, Niels H. Damrauer and Matthew P. Shores
Inorganic Chemistry 2010 Volume 49(Issue 17) pp:7981-7991
Publication Date(Web):August 9, 2010
DOI:10.1021/ic1009972
We report the preparation and characterization of Cr(III) coordination complexes featuring the dimethyl 2,2′-bipyridine-4,4′-dicarboxylate (4-dmcbpy) ligand: [(phen)2Cr(4-dmcbpy)](OTf)3 (1), [(Ph2phen)2Cr(4-dmcbpy)](OTf)3 (4), [(Me2bpy)2Cr(4-dmcbpy)](OTf)3 (7), and [Cr(4-dmcbpy)3](BF4)3 (8), where phen is 1,10-phenanthroline, Ph2phen is 4,7-diphenyl-1,10-phenanthroline, and Me2bpy is 4,4′-dimethyl-2,2′-bipyridine. Static and nanosecond time-resolved absorption and emission properties of these complexes dissolved in acidic aqueous (1 M HCl) solutions are reported. Emission spectra collected at 297 K show a narrow spectrum with an emission maximum ranging from 732 nm (1) to 742 nm (4). The emissive state is thermally activated and decays via first order kinetics at all temperatures explored (283 to 353 K). At 297 K the observed lifetime ranges from 7.7 μs (8) to 108 μs (4). The photophysical data suggest that in these acidic aqueous environments these complexes store ∼1.7 eV for multiple microseconds at room temperature. Of the heteroleptic species, complex 4 shows the greatest absorption of visible wavelengths (ε = 1270 M−1 cm−1 at 491 nm), and homoleptic complex 8 has improved absorption at visible wavelengths over [Cr(bpy)3]3+. The electrochemical properties of 1, 4, 7, and 8 were investigated by cyclic voltammetry. It is found that inclusion of 4-dmcbpy shifts the “CrIII/II” E1/2 by +0.22 V compared to those of homoleptic parent complexes, with the first reduction event occurring at −0.26 V versus Fc+/Fc for 8. The electrochemical and photophysical data allow for excited state potentials to be determined: for 8, CrIII*/II lies at +1.44 V versus ferrocenium/ferrocene (∼+2 V vs NHE), placing it among the most powerful photooxidants reported.
Co-reporter:Christina M. Klug, Ashley M. McDaniel, Stephanie R. Fiedler, Kelsey A. Schulte, Brian S. Newell and Matthew P. Shores
Dalton Transactions 2012 - vol. 41(Issue 40) pp:NaN12585-12585
Publication Date(Web):2012/09/07
DOI:10.1039/C2DT31213A
We report the syntheses, characterisations, and spin state behaviours of salts of the tripodal-ligated Fe(II) complex [FeL6-OH]X2 (L6-OH = tris{4-[(6-methanol)-2-pyridyl]-3-aza-3-butenyl}amine, X = OTf− (1), Br− (2), I− (3), BPh4− (4)). Covalent linking of the ligand arms is imperative as a high-spin bis(tridentate) complex (5) is formed when a non-tethered ethyl iminopyridine ligand (L2 = 4-[(6-methanol)-2-pyridyl]-3-aza-3-butenyl) is used. For salts 1–4, thermally-induced spin-crossover (SCO) is observed in the solid state, with dependence on anion and solvate molecules. Salts with larger anions show more complete SCO centred at higher temperatures (1 > 3 > 2); the triflate salt 1 (T1/2 = 173 K) also shows the strongest cooperativity of the compounds examined. Hydrogen bonding appears to be critical to SCO in this family of salts: limiting interactions by use of tetraphenylborate produces a high-spin complex down to 5 K. In protic solvents such as methanol, spectra of [FeL6-OH]2+ are largely unchanged over a period of three days, but dissociate when interrogated with strong field bidentate ligands. Compounds 1–3, and 5 remain high spin in solution down to 180 K, consistent with the data obtained in the solid state.
Co-reporter:Janice L. Wong, Robert F. Higgins, Indrani Bhowmick, David Xi Cao, Géza Szigethy, Joseph W. Ziller, Matthew P. Shores and Alan F. Heyduk
Chemical Science (2010-Present) 2016 - vol. 7(Issue 2) pp:NaN1599-1599
Publication Date(Web):2015/12/08
DOI:10.1039/C5SC03006D
A new bimetallic platform comprising a six-coordinate Fe(ONO)2 unit bound to an (ONO)M (M = Fe, Zn) has been discovered ((ONOcat)H3 = bis(3,5-di-tert-butyl-2-phenol)amine). Reaction of Fe(ONO)2 with either (ONOcat)Fe(py)3 or with (ONOq)FeCl2 under reducing conditions led to the formation of the bimetallic complex Fe2(ONO)3, which includes unique five- and six-coordinate iron centers. Similarly, the reaction of Fe(ONO)2 with the new synthon (ONOsq˙)Zn(py)2 led to the formation of the heterobimetallic complex FeZn(ONO)3, with a six-coordinate iron center and a five-coordinate zinc center. Both bimetallic complexes were characterized by single-crystal X-ray diffraction studies, solid-state magnetic measurements, and multiple spectroscopic techniques. The magnetic data for FeZn(ONO)3 are consistent with a ground state S = 3/2 spin system, generated from a high-spin iron(II) center that is antiferromagnetically coupled to a single (ONOsq˙)2− radical ligand. In the case of Fe2(ONO)3, the magnetic data revealed a ground state S = 7/2 spin system arising from the interactions of one high-spin iron(II) center, one high-spin iron(III) center, and two (ONOsq˙)2− radical ligands.
Co-reporter:Jesse V. Gavette, Christina M. Klug, Lev N. Zakharov, Matthew P. Shores, Michael M. Haley and Darren W. Johnson
Chemical Communications 2014 - vol. 50(Issue 54) pp:NaN7175-7175
Publication Date(Web):2014/05/15
DOI:10.1039/C4CC02297A
A bipyridyl-based anion receptor is utilized as a ligand in a tetrahedral FeCl2 complex and demonstrates secondary coordination sphere influence through intramolecular hydrogen bonding to the chloride ligands as evidenced by X-ray crystallography.
Co-reporter:Tarik J. Ozumerzifon, Indrani Bhowmick, William C. Spaller, Anthony K. Rappé and Matthew P. Shores
Chemical Communications 2017 - vol. 53(Issue 30) pp:NaN4214-4214
Publication Date(Web):2017/03/13
DOI:10.1039/C7CC01172E
We present the syntheses and characterization of several salts of a trigonal prismatic cobalt(II) complex with a 1,3,5-triaminocyclohexane (tach)-derived ditopic ligand. The air- and moisture-stable tetraphenylborate salt (2) shows slow magnetic relaxation under both zero and applied dc fields. This complex also exhibits an unexpected ability to interact with a cationic sodium guest ion, highlighting the ambifunctional binding nature of amide groups within an iminopyridine scaffold.
.png)
![1-methoxy-2-[(1E)-prop-1-en-1-yl]benzene](http://img.cochemist.com/ccimg/2100/2077-36-3.png)
![1-methoxy-2-[(1E)-prop-1-en-1-yl]benzene](http://img.cochemist.com/ccimg/2100/2077-36-3_b.png)

























![[6-(HYDROXYMETHYL)PYRIDIN-3-YL]METHANOL](/data/chemimg/417700/21514-99-8.png)
![[6-(HYDROXYMETHYL)PYRIDIN-3-YL]METHANOL](/data/chemimg/417700/21514-99-8_b.png)