Co-reporter:Liang Liu, Miaomiao Zhu, Hai-Tao Yu, Wen-Xiong Zhang, and Zhenfeng Xi
Journal of the American Chemical Society October 4, 2017 Volume 139(Issue 39) pp:13688-13688
Publication Date(Web):September 21, 2017
DOI:10.1021/jacs.7b08803
Reductive elimination of Cu(III) intermediates is often proposed as a key step in many copper-catalyzed or -mediated formation of C–C or C–heteroatom bonds. However, there still lacks concrete evidence on this key step, mainly because Cu(III) complexes are usually too unstable to be isolated and structurally characterized. In this work, novel organocopper(III) spiro complexes and their analogous organocopper(I) spiro complexes were synthesized and structurally characterized. Single-crystal X-ray structural analysis revealed that the spiro Cu(III) atom adopted a distorted square-planar geometry while its corresponding spiro Cu(I) atom was tetrahedrally coordinated. A redox transformation between these spiro Cu(I) and Cu(III) complexes was experimentally observed by reacting with reductants or oxidants, respectively. As concrete evidence, the organocopper(III) spiro compounds were found to form C–C bonds intramolecularly via reductive elimination.
Co-reporter:Yongliang Zhang, Yue Chi, Junnian Wei, Qi Yang, Zhenqiang Yang, Hui Chen, Ruina Yang, Wen-Xiong Zhang, and Zhenfeng Xi
Organometallics August 14, 2017 Volume 36(Issue 15) pp:2982-2982
Publication Date(Web):July 30, 2017
DOI:10.1021/acs.organomet.7b00447
Aromatic 1,1,2,2-tetralithiodigalloles 5 with a Ga–Ga bond were realized. Reaction of 1,4-dilithio-1,3-butadienes 6 with 0.5 equiv of GaCl3 afforded lithium spirogallane ate complexes 7 in excellent isolated yields. Reduction of the above ate complexes 7 with an excess amount of lithium generated the 1,1,2,2-tetralithiodigalloles 5 in moderate isolated yields. The structures of compounds 5 and 7 were both determined by single-crystal X-ray structural analysis. The 1,1,2,2-tetralithiodigalloles 5 contained two aromatic dilithiogalloles connected by a Ga–Ga bond. The aromaticity of 5 was confirmed by both experimental measurements (X-ray structural analysis, NMR spectroscopy) and theoretical analysis (ISE, NICS).
Co-reporter:Yongliang Zhang, Junnian Wei, Yue Chi, Xuan Zhang, Wen-Xiong Zhang, and Zhenfeng Xi
Journal of the American Chemical Society April 12, 2017 Volume 139(Issue 14) pp:5039-5039
Publication Date(Web):March 31, 2017
DOI:10.1021/jacs.7b02039
Since the concept of aromaticity represents one of the most fundamental principles in chemistry, the search for unprecedented and exciting aromatic systems, therefore, continues to drive research in this area. Herein we report the synthesis and characterization of spiro metalla-aromatics, in which the transition metal (Pd, Pt, or Rh) is the spiro atom, that cross-conjugates two aromatic five-membered metallacycles. These spiro metalla-aromatics tend to take square planar geometries, with the dihedral angle being influenced by the steric repulsion between the α-positioned substituents. Rationalized and classified via both experimental measurements (X-ray structural analysis, NMR spectroscopy, XPS, etc.) and theoretical analysis (DFT calculation, ISE, AICD, NICS, and CMOs), all these fundamental observations extend the concept of aromaticity and organometallic chemistry.
Co-reporter:Jianhao Yin, Qingyu Ye, Wei Hao, Shuaijing Du, Yucheng Gu, Wen-Xiong Zhang, and Zhenfeng Xi
Organic Letters 2017 Volume 19(Issue 1) pp:138-141
Publication Date(Web):December 12, 2016
DOI:10.1021/acs.orglett.6b03431
Reactions between 1,4-dibromo-1,3-butadienes and 2,5-disubstituted pyrroles afforded cyclopenta[c]pyridine derivatives in high yield, catalyzed by palladium and a cyclopentadiene-phosphine ligand (L1). Insertion of one terminal carbon of the butadienyl skeleton into one C═C double bond in the pyrrole ring resulted in ring expansion, along with a 1,2-shift of an alkyl or an aryl substituent on the butadienes.
Co-reporter:Zhe Huang;Ming Zhan;Shaoguang Zhang;Qian Luo;Wen-Xiong Zhang
Organic Chemistry Frontiers 2017 vol. 4(Issue 9) pp:1785-1788
Publication Date(Web):2017/08/22
DOI:10.1039/C7QO00287D
Treatment of Δ1-dipyrrolines with NBS afforded α,α′-dibromo-Δ1-bipyrrolines and α,α,α′,α′-tetrabromo-Δ1-bipyrrolines respectively with excellent selectivity depending on the amount of NBS. All these multibromo-substituted Δ1-bipyrrolines could be efficiently transformed into their corresponding 2,6-diazasemibullvalene derivatives via reduction with lithium. An unprecedented rearrangement of 4,8-dibromo-2,6-diazasemibullvalene afforded a new type of bipyrroline derivative.
Co-reporter:Dr. Yue Chi;Haihan Yan; Dr. Wen-Xiong Zhang; Dr. Zhenfeng Xi
Chemistry - A European Journal 2017 Volume 23(Issue 4) pp:757-761
Publication Date(Web):2017/01/18
DOI:10.1002/chem.201604739
AbstractThe selective C(sp3)−H bond functionalization is an ideal and atom-economical method in organic synthesis. In this work, 2-aminopyrimidines are generated from a Cu-catalyzed reaction between carbodiimides and diaryliodonium salts, by cleavage of four C(sp3)−H, one C−N, and one C=N bonds in the carbodiimides. It is the first triple C(sp3)−H bond functionalization neighboring a C=N bond. The selective synthesis of 2-aminopyrimidines is controlled by the amount of the diaryliodonium salts. The novel mechanism involving a C−N formation/1,5-H shift/1,7-H shift/6 π-electrocyclic ring-closing/aromatization is well elucidated by the detection of important intermediates and DFT calculations.
Co-reporter:Dr. Yue Chi;Haihan Yan; Dr. Wen-Xiong Zhang; Dr. Zhenfeng Xi
Chemistry - A European Journal 2017 Volume 23(Issue 4) pp:977-977
Publication Date(Web):2017/01/18
DOI:10.1002/chem.201605446
This cover picture mainly focuses on the Copper-catalyzed reaction between carbodiimides and diaryliodonium salts by cleavage of four C(sp3)−H, one C−N, and one C=N bonds. The core creativity is to demonstrate the various interesting bond functionalizations. All these functionalities are located around the Cu catalyst, and the edge of the flask marks all the broken bonds; the backbone fragments in the flask can be selectively combined and form the final products. The Cu-catalyst “flask” selectively cuts chemical bonds and pours only the 2-aminopryimidine products. More information can be found in the Communication by W.-X. Zhang, Z. Xi et al. on page 757 ff.
Co-reporter:Baosheng Wei, Heng Li, Wen-Xiong Zhang, and Zhenfeng Xi
Organometallics 2016 Volume 35(Issue 10) pp:1458-1463
Publication Date(Web):March 14, 2016
DOI:10.1021/acs.organomet.6b00073
The development of Ca-mediated reactions is of great importance and is in great demand in both organometallic and organic chemistry. In this paper, we report that calcium iodide efficiently promotes the transformation of various 1,4-dilithio-1,3-butadienes into indenes or perfluorodibenzopentalenes, via an intramolecular C–H or C–F bond cleavage, respectively. In the reaction process, the Ca-induced s-cis/trans configurational isomerization of 1,4-dilithio-1,3-butadienes is followed by the subsequent nucleophilic C–H or C–F bond activation.
Co-reporter:Shaoguang Zhang, Wen-Xiong Zhang, and Zhenfeng Xi
Accounts of Chemical Research 2015 Volume 48(Issue 7) pp:1823
Publication Date(Web):June 10, 2015
DOI:10.1021/acs.accounts.5b00190
Semibullvalene (SBV) and its aza analogue 2,6-diazasemibullvalene (NSBV) are theoretically interesting and experimentally challenging organic molecules because of four unique features: highly strained ring systems, intramolecular skeletal rearrangement, extremely rapid degenerate (aza-)Cope rearrangement, and the predicted existence of neutral homoaromatic delocalized structures. SBV has received much attention in the past 50 years. In contrast, after NSBV was predicted in 1971 and the first in situ synthesis was realized in 1982, no progress on NSBV chemistry was made until our results in 2012. We have been interested in the reaction chemistry of 1,4-dilithio-1,3-butadienes (dilithio reagents for short), especially for their applications in the synthesis of SBV and NSBV, because (i) the cyclodimerization of dilithio reagents could provide the potential eight-carbon skeleton of SBV from four-carbon butadiene units and (ii) the insertion reaction of dilithio reagents with C≡N bonds of two nitriles could provide a 6C + 2N skeleton that might be a good precursor for the synthesis of NSBV. Therefore, we initiated a journey into the synthesis and reaction chemistry of SBV and NSBV starting from dilithio reagents that has been ongoing since 2006. In this Account, we outline mainly our recent achievements in the synthesis, structural characterization, reaction chemistry, synthetic application, and theoretical/computational analysis of NSBV.Two efficient strategies for the synthesis of NSBV from dilithio reagents and nitriles via oxidant-induced C–N bond formation are described. Structural investigations of NSBV, including X-ray crystal structure analysis, determination of the activation barrier for the aza-Cope rearrangement, and theoretical analysis, show that the localized structure of NSBV is the predominant form and that the homoaromatic delocalized structure exists as a minor component in the equilibrium. We also discuss the reaction chemistry and synthetic applications of NSBV. Several novel reaction patterns have been explored, including thermolysis, C–N bond insertion, rearrangement–cycloaddition, oxidation, and nucleophilic ring-opening reactions. Diverse and interesting N-containing polycyclic skeletons can be constructed, such as nickelaazetidine, 1,5-diazatriquinacenes, and triazabrexadienes, which are not available by other means.Our results show that NSBV not only features a rapid aza-Cope rearrangement with a low activation barrier but also acts as unique synthetic reagent that is significantly different from aziridine. The strained rigid ring systems as a whole can be involved in the reactions. Our achievements highlight two significant advances: (i) the well-established efficient synthesis and isolation of NSBV has greatly accelerated the development of NSBV chemistry, and (ii) the previously unattainable molecules have become “normal” and routine starting materials for the synthesis of otherwise unavailable but interesting structures. We expect that our pursuits will inspire and help direct future chemical and physical research on NSBV.
Co-reporter:Junnian Wei; Yongliang Zhang; Yue Chi; Liang Liu; Wen-Xiong Zhang
Journal of the American Chemical Society 2015 Volume 138(Issue 1) pp:60-63
Publication Date(Web):December 28, 2015
DOI:10.1021/jacs.5b11317
Metal-containing aromatic systems (metalla-aromatics) are unique and important both experimentally and theoretically. Among metalla-aromatics, six-membered metallabenzenes and metallabenzynes have attracted much attention in recent years. However, reports on their superior homologues are rare. In this work, the first series of aromatic dicupra[10]annulenes were isolated from the reaction of dilithio reagents and copper salts. Single-crystal X-ray structural analysis revealed dicupra[10]annulenes with averaged bond lengths. 7Li NMR spectra and theoretical calculations revealed considerable aromatic character. XPS data suggested that the oxidation state of Cu atom in dicupra[10]annulenes was more likely to be Cu(I), indicating that the dilithio moieties in dicupra[10]annulenes participated as noninnocent ligands. This work demonstrates a novel approach to construct macrocyclic metalla-aromatics.
Co-reporter:Wei Hao, Han Wang, Qingyu Ye, Wen-Xiong Zhang, and Zhenfeng Xi
Organic Letters 2015 Volume 17(Issue 22) pp:5674-5677
Publication Date(Web):November 2, 2015
DOI:10.1021/acs.orglett.5b02959
The cyclopentadiene–phosphine ligand (L1) and palladium were found to be an efficient catalyst system to activate the α-C(sp2)–H bond of pyrrole and indole derivatives. Various alkenyl or aryl dibromides could be used to react with pyrrole and indole derivatives to afford multisubstituted indolizines in high yields.
Co-reporter:Ming Zhan, Shaoguang Zhang, Zhe Huang, and Zhenfeng Xi
Organic Letters 2015 Volume 17(Issue 4) pp:1026-1029
Publication Date(Web):February 9, 2015
DOI:10.1021/acs.orglett.5b00136
A series of 4,8-dichloro-2,6-diazasemibullvalenes were synthesized and isolated from the reaction of α,α,α′,α′-tetrachloro-Δ1-bipyrrolines with lithium via C–N bond formation. All those dichlorodiazasemibullvalene derivatives demonstrated extremely rapid aza-Cope rearrangement in solution. An unprecedented skeletal rearrangement of 4,8-dichloro-2,6-diazasemibullvalene derivatives took place, resulting in the formation of a new bipyrroline skeleton.
Co-reporter:Yongliang Zhang; Junnian Wei; Wen-Xiong Zhang
Inorganic Chemistry 2015 Volume 54(Issue 22) pp:10695-10700
Publication Date(Web):October 26, 2015
DOI:10.1021/acs.inorgchem.5b01551
A series of lithium aluminate complexes and alumoles were synthesized from 1,4-dilithio-1,3-butadienes 1 and AlEt2Cl. Their structures were characterized using single-crystal X-ray structural analysis and NMR spectroscopy. The structure of the lithium aluminate complex 2-TMEDA showed that the Al atom adopted a tetra-coordinated mode bonded with two butadienyl Csp2 atoms and two ethyl Csp3 atoms. The lithium cation was located above the alumole ring. The structure of 3a revealed a dimeric 1-ethylalumole in the solid state. Diffusion ordered spectroscopy NMR spectra showed that 3a was also a dimer in C6D6 solvent. However, in tetrahydrofuran (THF) solution, the dimeric 3a dissociated into the 1-ethylalumole–THF adduct. The lithium aluminate complex 2 transformed into 3a-THF when treated with 1.0 equiv of AlEt2Cl. Preliminary reaction chemistry and synthetic applications of the lithium aluminate complex were also investigated.
Co-reporter:Ming Zhan;Dr. Shaoguang Zhang;Zhe Huang;Dr. Zhenfeng Xi
Chemistry – An Asian Journal 2015 Volume 10( Issue 4) pp:862-864
Publication Date(Web):
DOI:10.1002/asia.201403030
Abstract
The reaction between 2,6-diazasemibullvalenes and nitroso compounds was investigated. Aza-triquinacene derivatives of interesting structural and synthetic chemistry were generated highly selectively in good to excellent isolated yields. This reaction, which was rarely found between common aziridine derivatives and nitroso compounds, could be attributed to the rigid polycyclic ring system and the substitution patterns of 2,6-diazasemibullvalenes. Δ1-Bipyrroline derivatives were formed in excellent yields when these aza-triquinacene derivatives were treated with SmI2.
Co-reporter:Junnian Wei;Yongliang Zhang;Dr. Wen-Xiong Zhang ;Dr. Zhenfeng Xi
Angewandte Chemie 2015 Volume 127( Issue 34) pp:10124-10128
Publication Date(Web):
DOI:10.1002/ange.201504521
Abstract
Herein we report that 1,4-dilithio-1,3-butadienes, a type of 1,3-butadienyl dianion, can act as non-innocent ligands, taking electrons from low-valent transition metals. Dilithio reagents reacted with [{RhCl(cod)}2] to give dilithio rhodacycle 3 a. Single-crystal X-ray structural analysis revealed the structure of 3 a with averaged bond lengths. XPS data suggested that the oxidation state of Rh in 3 a was more likely to be Rh3+. CDA/ECDA confirmed the electron-transfer process. 7Li NMR spectra of 3 a and theoretical calculations revealed a considerable aromatic character. In this process, the dilithio compounds behaved as non-innocent ligands and formal oxidants. These results demonstrated that organolithium compounds with suitable π-conjugation could be used as electron acceptor.
Co-reporter:Junnian Wei;Yongliang Zhang;Dr. Wen-Xiong Zhang ;Dr. Zhenfeng Xi
Angewandte Chemie International Edition 2015 Volume 54( Issue 34) pp:9986-9990
Publication Date(Web):
DOI:10.1002/anie.201504521
Abstract
Herein we report that 1,4-dilithio-1,3-butadienes, a type of 1,3-butadienyl dianion, can act as non-innocent ligands, taking electrons from low-valent transition metals. Dilithio reagents reacted with [{RhCl(cod)}2] to give dilithio rhodacycle 3 a. Single-crystal X-ray structural analysis revealed the structure of 3 a with averaged bond lengths. XPS data suggested that the oxidation state of Rh in 3 a was more likely to be Rh3+. CDA/ECDA confirmed the electron-transfer process. 7Li NMR spectra of 3 a and theoretical calculations revealed a considerable aromatic character. In this process, the dilithio compounds behaved as non-innocent ligands and formal oxidants. These results demonstrated that organolithium compounds with suitable π-conjugation could be used as electron acceptor.
Co-reporter:Baosheng Wei, Heng Li, Wen-Xiong Zhang, and Zhenfeng Xi
Organometallics 2015 Volume 34(Issue 7) pp:1339-1344
Publication Date(Web):March 31, 2015
DOI:10.1021/acs.organomet.5b00059
Direct deprotonation of 9-fluorenol with Ca[N(SiMe3)2]2 efficiently generated a fluorene-based calcium oxycyclopentadienide complex, which is the first alkaline earth metal complex containing an oxycyclopentadienyl dianion ligand. The η5, η1, and η2 interactions between the oxycyclopentadienyl dianion ligand and the calcium center were observed in the solid state. The calcium oxycyclopentadienide complex displayed reductive character and multiple reactive sites. Its reactivity toward a range of electrophiles, especially acid chlorides, was investigated and discussed in detail. A wide variety of fluorene derivatives were synthesized.
Co-reporter:Liang Liu, Weizhi Geng, Qi Yang, Wen-Xiong Zhang, and Zhenfeng Xi
Organometallics 2015 Volume 34(Issue 17) pp:4198-4201
Publication Date(Web):August 31, 2015
DOI:10.1021/acs.organomet.5b00598
Two examples of mixed alkenyl/aryl organocopper(I) aggregates possessing conjugated alkenyl/aryl C–Cu bonds were synthesized by transmetalation of zirconaindenes with CuCl. Single-crystal X-ray structural analysis revealed tetrameric and hexameric structures. Butadienyl copper(I) aggregates were also synthesized and structurally characterized for further comparison. The preliminary reaction chemistry of the copper(I) aggregates was studied.
Co-reporter:Junnian Wei;Dr. Wen-Xiong Zhang ;Dr. Zhenfeng Xi
Angewandte Chemie 2015 Volume 127( Issue 20) pp:6097-6100
Publication Date(Web):
DOI:10.1002/ange.201411009
Abstract
Organolithium compounds can behave as reductants but never as oxidants in redox reactions. Reported herein is that 1,4-dilithio-1,3-butadienes reacted with [Ni(cod)2] (cod=1,5-cyclooctadiene) to deliver dilithionickeloles. Single-crystal X-ray structural analysis revealed a coplanar structure of dilithionickeloles with an averaging of bond lengths. XPS data confirmed the oxidation state of Ni in dilithionickeloles was Ni2+. 7Li NMR spectra of dilithionickeloles and theoretical calculations revealed a considerable aromatic character. In this redox reaction, the dilithio dianionic compounds behaved as formal oxidants, thus oxidizing Ni0 into Ni2+. These results demonstrated that organolithium compounds with π-conjugation could be used as oxidants and could continue to accept extra electrons.
Co-reporter:Baosheng Wei, Heng Li, Jianhao Yin, Wen-Xiong Zhang, and Zhenfeng Xi
The Journal of Organic Chemistry 2015 Volume 80(Issue 17) pp:8758-8762
Publication Date(Web):August 13, 2015
DOI:10.1021/acs.joc.5b01595
The reaction chemistry between 1,4-dilithio-1,3-butadienes (dilithio reagents for short) and PhSiH3 has been investigated. Direct substitution of two hydride ions from PhSiH3 with the dilithio reagents led to multisubstituted siloles (silacyclopentadienes) in diethyl ether solution, with the concomitant generation of LiH. When THF was used as the solvent, the reaction between PhSiH3 and 1,4-bis(silyl) dilithio reagents afforded cis-3-silacyclopentenes stereoselectively. Experimental results demonstrated that reactive LiH was generated in situ in the reaction system. Formal syn addition of LiH to silacyclopentadiene intermediates would afford silacyclopentenes, most likely via pentavalent organosilicates.
Co-reporter:Junnian Wei;Dr. Wen-Xiong Zhang ;Dr. Zhenfeng Xi
Angewandte Chemie International Edition 2015 Volume 54( Issue 20) pp:5999-6002
Publication Date(Web):
DOI:10.1002/anie.201411009
Abstract
Organolithium compounds can behave as reductants but never as oxidants in redox reactions. Reported herein is that 1,4-dilithio-1,3-butadienes reacted with [Ni(cod)2] (cod=1,5-cyclooctadiene) to deliver dilithionickeloles. Single-crystal X-ray structural analysis revealed a coplanar structure of dilithionickeloles with an averaging of bond lengths. XPS data confirmed the oxidation state of Ni in dilithionickeloles was Ni2+. 7Li NMR spectra of dilithionickeloles and theoretical calculations revealed a considerable aromatic character. In this redox reaction, the dilithio dianionic compounds behaved as formal oxidants, thus oxidizing Ni0 into Ni2+. These results demonstrated that organolithium compounds with π-conjugation could be used as oxidants and could continue to accept extra electrons.
Co-reporter:Junnian Wei;Liang Liu;Ming Zhan;Ling Xu;Dr. Wen-Xiong Zhang;Dr. Zhenfeng Xi
Angewandte Chemie International Edition 2014 Volume 53( Issue 22) pp:5634-5638
Publication Date(Web):
DOI:10.1002/anie.201310116
Abstract
Metallacyclopentadienes have attracted much attention as building blocks for synthetic chemistry as well as key intermediates in many metal-mediated or metal-catalyzed reactions. However, metallacyclopentadienes of the alkaline-earth metals have not been reported, to say nothing of their structures, reaction chemistry, and synthetic applications. In this work, the first series of magnesiacyclopentadienes, spiro-dilithio magnesiacyclopentadienes, and dimagnesiabutadiene were synthesized from 1,4-dilithio 1,3-butadienes. Single-crystal X-ray structural analysis of these magnesiacycles revealed unique structural characteristics and bonding modes. Their reaction chemistry and synthetic application were preliminarily studied and efficient access to amino cyclopentadienes was established through their reaction with thioformamides. Experimental and DFT calculations demonstrated that these magnesiacyclopentadienes could be regarded as bis(Grignard) reagents wherein the two MgC(sp2) bonds have a synergetic effect when reacting with substrates.
Co-reporter:Wei Hao;Junnian Wei;Weizhi Geng;Dr. Wen-Xiong Zhang;Dr. Zhenfeng Xi
Angewandte Chemie International Edition 2014 Volume 53( Issue 52) pp:14533-14537
Publication Date(Web):
DOI:10.1002/anie.201408341
Abstract
β-Hydride abstraction is a well-accepted elementary step for catalytic cycles in organometallic chemistry. It is usually anticipated that alkylpalladium halides containing syn-β-hydrogen atoms will undergo β-hydride abstraction to afford the Heck-type products. However, this study discloses that the above general knowledge is only conditionally correct. Our experimental results demonstrate that the reductive elimination of alkylhalides from alkylpalladium halides containing syn-β-hydrogen atoms may surpass the β-hydride abstraction or even become exclusive in certain cases.
Co-reporter:Wei Hao;Weizhi Geng;Dr. Wen-Xiong Zhang;Dr. Zhenfeng Xi
Chemistry - A European Journal 2014 Volume 20( Issue 9) pp:2605-2612
Publication Date(Web):
DOI:10.1002/chem.201304215
Abstract
An efficient synthesis of N-substituted indole derivatives was realized by combining the Pd-catalyzed one-pot multicomponent coupling approach with cleavage of the C(sp3)N bonds. Three or four components of aryl iodides, alkynes, and amines were involved in this coupling process. The cyclopentadiene–phosphine ligand showed high efficiency. A variety of aryl iodides, including cyclic and acyclic tertiary amino aryl iodides, and substituted 1-bromo-2-iodobenzene derivatives could be used. Both symmetric and unsymmetric alkynes substituted with alkyl, aryl, or trimethylsilyl groups could be applied. Cyclic secondary amines such as piperidine, morpholine, 4-methylpiperidine, 1-methylpiperazine, 2-methylpiperidine, and acyclic amines including secondary and primary amines all showed good reactivity. Further application of the resulting indole derivatives was demonstrated by the synthesis of benzosilolo[2,3-b]indole.
Co-reporter:Dr. Shaoguang Zhang;Ming Zhan;Dr. Wen-Xiong Zhang;Dr. Zhenfeng Xi
Chemistry - A European Journal 2014 Volume 20( Issue 31) pp:9744-9752
Publication Date(Web):
DOI:10.1002/chem.201402911
Abstract
Nucleophilic ring-opening reactions of 2,6-diazasemibullvalenes (NSBVs) were investigated. Different types of nucleophile (alcohols, phenols, thiols, carboxylic acids, water, enols, amines, indoles, metal-halide salts, sodium azide, organozinc compounds, lithium alkynethiolate, and sulfoxonium ylides) were used to afford diverse functionalized Δ1-bipyrroline derivatives in good yields with high regio- and diastereoselectivity. Most of the reactions featured milder conditions and higher reactivity relative to those for common aziridine derivatives, probably because of the rigid ring system and substitution patterns of NSBVs.
Co-reporter:Jing Zhao, Shaoguang Zhang, Wen-Xiong Zhang, Zhenfeng Xi
Coordination Chemistry Reviews 2014 s 270–271() pp: 2-13
Publication Date(Web):
DOI:10.1016/j.ccr.2013.08.032
Co-reporter:Junnian Wei;Liang Liu;Ming Zhan;Ling Xu;Dr. Wen-Xiong Zhang;Dr. Zhenfeng Xi
Angewandte Chemie 2014 Volume 126( Issue 22) pp:5740-5744
Publication Date(Web):
DOI:10.1002/ange.201310116
Abstract
Metallacyclopentadienes have attracted much attention as building blocks for synthetic chemistry as well as key intermediates in many metal-mediated or metal-catalyzed reactions. However, metallacyclopentadienes of the alkaline-earth metals have not been reported, to say nothing of their structures, reaction chemistry, and synthetic applications. In this work, the first series of magnesiacyclopentadienes, spiro-dilithio magnesiacyclopentadienes, and dimagnesiabutadiene were synthesized from 1,4-dilithio 1,3-butadienes. Single-crystal X-ray structural analysis of these magnesiacycles revealed unique structural characteristics and bonding modes. Their reaction chemistry and synthetic application were preliminarily studied and efficient access to amino cyclopentadienes was established through their reaction with thioformamides. Experimental and DFT calculations demonstrated that these magnesiacyclopentadienes could be regarded as bis(Grignard) reagents wherein the two MgC(sp2) bonds have a synergetic effect when reacting with substrates.
Co-reporter:Wei Hao;Junnian Wei;Weizhi Geng;Dr. Wen-Xiong Zhang;Dr. Zhenfeng Xi
Angewandte Chemie 2014 Volume 126( Issue 52) pp:14761-14765
Publication Date(Web):
DOI:10.1002/ange.201408341
Abstract
β-Hydride abstraction is a well-accepted elementary step for catalytic cycles in organometallic chemistry. It is usually anticipated that alkylpalladium halides containing syn-β-hydrogen atoms will undergo β-hydride abstraction to afford the Heck-type products. However, this study discloses that the above general knowledge is only conditionally correct. Our experimental results demonstrate that the reductive elimination of alkylhalides from alkylpalladium halides containing syn-β-hydrogen atoms may surpass the β-hydride abstraction or even become exclusive in certain cases.
Co-reporter:Jing Zhao, Shaoguang Zhang, Wen-Xiong Zhang, and Zhenfeng Xi
Organometallics 2014 Volume 33(Issue 1) pp:8-11
Publication Date(Web):December 17, 2013
DOI:10.1021/om401065c
The reaction of titanacyclobutene–silacyclobutene fused-ring complexes with nitriles was investigated. Formal insertion of the C–N triple bond of nitriles into the silacyclobutene ring was observed, affording their corresponding titanacyclobutene and six-membered azasilacycle fused-ring complexes. This reaction pattern with nitriles is totally different from that of their analogous zirconacyclobutene–silacyclobutene fused-ring complexes. The size effect of the central metals (Ti vs Zr) is considered as the major reason for the different reaction patterns.
Co-reporter:Peng-Hui Wei, Ling Xu, Li-Cheng Song, Wen-Xiong Zhang, and Zhenfeng Xi
Organometallics 2014 Volume 33(Issue 11) pp:2784-2789
Publication Date(Web):May 27, 2014
DOI:10.1021/om5002793
A series of mixed Cp′/bis(amidinato) (Cp′ = η5-C5Me4(SiMe3)) lanthanide complexes were synthesized by the 1:2 acid–base reaction between Cp′Ln(CH2SiMe3)2(THF) (Ln = Y, Dy, Er, Lu) and amidines. These Cp′/bis(amidinato) complexes showed excellent catalytic activity for the addition of amines to carbodiimides, yielding the corresponding guanidines. Isolation, structural characterization, and catalytic application of the binuclear lutetium amido complex showed clearly that the catalytic cycle was initiated by the dissociation of Cp′. These results demonstrated that Cp′, for the first time, acted as a reactive site to yield the active Ln–N species.
Co-reporter:Ling Xu, Yu-Chen Wang, Wangyang Ma, Wen-Xiong Zhang, and Zhenfeng Xi
The Journal of Organic Chemistry 2014 Volume 79(Issue 24) pp:12004-12009
Publication Date(Web):October 14, 2014
DOI:10.1021/jo501865b
Cleavage of the N—N bond in 1,2-diarylhydrazine was achieved through an alkyllithium-catalyzed guanylation reaction of 1,2-diarylhydrazine with carbodiimide, affording guanidine and azo compounds. This N—N bond cleavage via thermal rearrangement was driven by an intramolecular proton shift. No reductants, oxidants, bases, or external protons were needed. The proposed mechanism has been well elucidated by the isolation, characterization, and reaction studies of two important amido lithium intermediates and an ArHN-substituted guanidine.
Co-reporter:Weizhi Geng ; Junnian Wei ; Wen-Xiong Zhang
Journal of the American Chemical Society 2013 Volume 136(Issue 2) pp:610-613
Publication Date(Web):December 24, 2013
DOI:10.1021/ja4114243
Four types of alkenyl organocopper(I) aggregates linked by 1,3-butadienyl and/or 1,3,5,7-octatetraenyl moieties were selectively realized in good isolated yields. All these organocopper(I) aggregates were structurally characterized by single-crystal X-ray structural analysis. These unprecedented aggregates, stabilized by multiple Cu–Cu interactions and the conjugated 1,3-butadienyl or 1,3,5,7-octatetraenyl bridges, could undergo controlled structural transformations. The 1,4-dicopper 1,3-butadienyl aggregate 3 could be efficiently transformed to aggregate 2, while LiI could disaggregate the 1,3-butadienyl-1,3,5,7-octatetraenyl aggregate 4 to 1,3,5,7-octatetraenyl aggregate 5 and 1,3-butadienyl aggregate 2. Preliminary reaction chemistry and synthetic applications of these organocopper(I) aggregates were also investigated.
Co-reporter:Fei Zhao, Yun-Fei Zhang, Jing Wen, Da-Gang Yu, Jiang-Bo Wei, Zhenfeng Xi, and Zhang-Jie Shi
Organic Letters 2013 Volume 15(Issue 13) pp:3230-3233
Publication Date(Web):June 21, 2013
DOI:10.1021/ol4011757
A variety of important multiarylated benzenes were efficiently synthesized from phloroglucinol derivatives 1 through sequential cross-couplings via Pd-catalyzed C–OTs, Ni-catalyzed C–OC(O)NEt2, and C–OMe bond activation. High selectivity was achieved based on the rational design and inherent diversity in the reactivity of different C–O bonds.
Co-reporter:Junnian Wei, Zitao Wang, Wen-Xiong Zhang, and Zhenfeng Xi
Organic Letters 2013 Volume 15(Issue 6) pp:1222-1225
Publication Date(Web):March 1, 2013
DOI:10.1021/ol400140n
Lithium iodide-assisted linear dimerization of 1,4-dicopper-1,3-butadienes and subsequent Pd-catalyzed cross-coupling reaction with halides provide an efficient way to construct octaalkyl-substituted and decasubstituted all-cis octatetraenes.
Co-reporter:Ming Zhan, Shaoguang Zhang, Wen-Xiong Zhang, and Zhenfeng Xi
Organic Letters 2013 Volume 15(Issue 16) pp:4182-4185
Publication Date(Web):August 7, 2013
DOI:10.1021/ol401878q
1-Imino-pyrrole and indole derivatives were synthesized in high yields from the reaction of diaryl diazomethanes with 1,4-dilithio-1,3-dienes. Diaryl diazomethanes reacted as electrophiles in this reaction. An unprecedented Zn-complex was formed via transmetalation of the above reaction intermediate with ZnCl2 and was structurally characterized. The trans-μ2-η1:η1 coordination mode in the solid state for this azaallylzinc compound was observed.
Co-reporter:Shaoguang Zhang, Ming Zhan, Qian Luo, Wen-Xiong Zhang and Zhenfeng Xi
Chemical Communications 2013 vol. 49(Issue 55) pp:6146-6148
Publication Date(Web):20 May 2013
DOI:10.1039/C3CC43061H
2,6-Diazasemibullvalenes (NSBVs) readily reacted with oxygen under mild conditions, affording Δ1-bipyrrolinones via C–N bond cleavage and C–H bond oxidation. Pyrrolino[3,2-b]pyrrolinone derivatives were efficiently generated when NSBVs were treated with N-oxides in the presence of Lewis acids. Δ1-Bipyrrolinones thus obtained could be readily transformed into other heterocycles.
Co-reporter:Tianhao Meng, Kunbing Ouyang and Zhenfeng Xi
RSC Advances 2013 vol. 3(Issue 34) pp:14273-14276
Publication Date(Web):26 Jun 2013
DOI:10.1039/C3RA42910E
An efficient Pd-catalyzed cleavage of the Me–Si bond in ortho-trimethylsilyl aryltriflates was realized and synthetically applied. Most of the commercially available ortho-trimethylsilyl aryltriflates could undergo the Pd-catalyzed intermolecular coupling with alkynes via cleavage of the Me–Si bond, which represents a new reaction pattern of ortho-trimethylsilyl aryltriflates. Potassium bromide (KBr) was found effective for this process. A variety of benzosilole derivatives were thus generated in high yields.
Co-reporter:Weizhi Geng;Dr. Chao Wang;Jie Guang;Wei Hao;Dr. Wen-Xiong Zhang;Dr. Zhenfeng Xi
Chemistry - A European Journal 2013 Volume 19( Issue 26) pp:8657-8664
Publication Date(Web):
DOI:10.1002/chem.201300416
Abstract
1,2,3,4-Tetrasubstituted cyclopentadienes and indene derivatives with identical or different substituents were obtained in good to excellent isolated yields through a zirconocene- and CuCl-mediated intermolecular coupling process. This synthetic procedure involved three organic partners, including one CH2I2, and two different or identical alkynes. Two alkynes or one diyne undergo Cp2ZrII-mediated (Cp=η5-C5H5) pair-selective reductive coupling to afford the corresponding zirconacyclopentadiene derivatives, which react, in the presence of CuCl and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1 H)-pyrimidinone (DMPU), with CH2I2 through intermolecular followed by intramolecular coupling to afford the cyclopentadiene derivatives. An application of the prepared tetrasubstituted cyclopentadiene derivatives was demonstrated by the facile synthesis of the corresponding zirconocene complexes [(4RCp)2ZrCl2] and [(4RCp)2ZrR′2] (R′=Me, Et, or nBu). The unique 1,2,3,4-tetrasubstituted cyclopentadiene ligands and the corresponding metallocenes are expected to have further applications in organometallic chemistry and organic synthesis.
Co-reporter:Heng Li;Dr. Wen-Xiong Zhang;Dr. Zhenfeng Xi
Chemistry - A European Journal 2013 Volume 19( Issue 38) pp:12859-12866
Publication Date(Web):
DOI:10.1002/chem.201301618
Abstract
A variety of ester-substituted cyclopentadiene derivatives have been synthesized by one-pot reactions of 1,4-dilithio-1,3-butadienes, CO, and acid chlorides. Direct deprotonation of the ester-substituted cyclopentadienes with Ae[N(SiMe3)2]2 (Ae=Ca, Sr, Ba) efficiently generated members of a new class of heavier alkaline earth (Ca, Sr, Ba) metallocenes in good to excellent yields. Single-crystal X-ray structural analysis demonstrated that these heavier alkaline earth metallocenes incorporated two intramolecularly coordinated ester pendants and multiply-substituted cyclopentadienyl ligands. The corresponding transition metal metallocenes, such as ferrocene derivatives and half-sandwich cyclopentadienyl tricarbonylrhenium complexes, could be generated highly efficiently by metathesis reactions. The multiply-substituted cyclopentadiene ligands bearing an ester pendant, and the corresponding heavier alkaline earth and transition-metal metallocenes, may have further applications in coordination chemistry, organometallic chemistry, and organic synthesis.
Co-reporter:Shaoguang Zhang;Dr. Wen-Xiong Zhang;Dr. Zhenfeng Xi
Angewandte Chemie International Edition 2013 Volume 52( Issue 12) pp:3485-3489
Publication Date(Web):
DOI:10.1002/anie.201210126
Co-reporter:Heng Li;Baosheng Wei;Ling Xu;Dr. Wen-Xiong Zhang;Dr. Zhenfeng Xi
Angewandte Chemie International Edition 2013 Volume 52( Issue 41) pp:10822-10825
Publication Date(Web):
DOI:10.1002/anie.201305254
Co-reporter:Heng Li;Baosheng Wei;Ling Xu;Dr. Wen-Xiong Zhang;Dr. Zhenfeng Xi
Angewandte Chemie International Edition 2013 Volume 52( Issue 41) pp:
Publication Date(Web):
DOI:10.1002/anie.201307184
Co-reporter:Heng Li;Baosheng Wei;Ling Xu;Dr. Wen-Xiong Zhang;Dr. Zhenfeng Xi
Angewandte Chemie 2013 Volume 125( Issue 41) pp:11022-11025
Publication Date(Web):
DOI:10.1002/ange.201305254
Co-reporter:Heng Li;Baosheng Wei;Ling Xu;Dr. Wen-Xiong Zhang;Dr. Zhenfeng Xi
Angewandte Chemie 2013 Volume 125( Issue 41) pp:
Publication Date(Web):
DOI:10.1002/ange.201307184
Co-reporter:Fei Zhao, Ming Zhan, Wen-Xiong Zhang, and Zhenfeng Xi
Organometallics 2013 Volume 32(Issue 7) pp:2059-2068
Publication Date(Web):March 19, 2013
DOI:10.1021/om300869t
Mechanisms for the reactions of 1,4-dilithio-1,3-butadienes and nitriles are explored through both experiments and DFT calculations. The computational results suggest that the selectivity of these reaction systems is strongly affected by the structures of the substrates. As the first step of all reaction pathways, the addition intermediate of one C–Li bond to the nitrile is formed. When tetraalkyl-substituted 1,4-dilithio 1,3-dienes and 2-cyanopyridine are used, the intermediate gives the cyclopentadienyl amine product as the kinetic product because of the coordination of the pyridyl N atom to the lithium atoms (system B). This addition intermediate also undergoes a second nitrile insertion into the C–Li bond, giving the dilithio ketimine intermediate. When tetraalkyl-substituted 1,4-dilithio 1,3-dienes and aryl nitriles are used, the dilithio ketimine intermediate undergoes a 1,6-cyclization, generating pyridine and triazine products through thermodynamically favored pathways (systems A and B). When cyclic 1,4-dilithiobutadiene and tertiary aliphatic nitriles are used, the dilithio ketimine intermediate undergoes two sequential 1,5-cyclization steps with a lower energy barrier, generating tricyclic Δ1-bipyrrolines (system C). Experimentally, the lithium-containing triazine intermediate 10 and Δ1-bipyrroline intermediate 19 have been isolated and their structures investigated through NMR and quenching experiments. The calculation results clearly show the mechanism details of these reactions and are in good agreement with the experimental observations.
Co-reporter:Shaoguang Zhang, Ming Zhan, Wen-Xiong Zhang, and Zhenfeng Xi
Organometallics 2013 Volume 32(Issue 14) pp:4020-4023
Publication Date(Web):July 2, 2013
DOI:10.1021/om400531h
The synthesis and X-ray crystallographic structure of a 3-D brick-wall coordination polymer of TMEDA-supported dimeric 2,3-dialkyl-1,4-dilithio-1,3-butadiene are reported. TMEDAs behave as bridging coordinating ligands rather than intramolecular chelating ligands. The Li4-tetrahedron core was stable toward the deaggregation of TMEDA because of the small steric hindrance on the butadiene skeleton.
Co-reporter:Shaoguang Zhang;Dr. Wen-Xiong Zhang;Dr. Zhenfeng Xi
Angewandte Chemie 2013 Volume 125( Issue 12) pp:3569-3573
Publication Date(Web):
DOI:10.1002/ange.201210126
Co-reporter:Shaoguang Zhang ; Junnian Wei ; Ming Zhan ; Qian Luo ; Chao Wang ; Wen-Xiong Zhang
Journal of the American Chemical Society 2012 Volume 134(Issue 29) pp:11964-11967
Publication Date(Web):July 11, 2012
DOI:10.1021/ja305581f
A series of 2,6-diazasemibullvalenes (NSBVs) were synthesized and isolated from the reaction of 1,4-dilithio-1,3-dienes with nitriles via oxidant-induced C–N bond formation. For the first time, the activation barrier and an X-ray crystal structure of a substituted 2,6-diazasemibullvalene were determined. All NSBVs show extremely rapid aza-Cope rearrangement in solution, but the rapid aza-Cope rearrangement is “frozen” in the solid state, as shown by solid-state NMR measurements and X-ray single-crystal structural analysis. Insertion of unsaturated compounds or a low-valent metal center into the NSBV C–N bond gave diverse and interesting ring-expansion products. Theoretical analysis showed that the localized structure is predominant and that the homoaromatic delocalized structure exists as a minor component in the equilibrium.
Co-reporter:Weizhi Geng ; Wen-Xiong Zhang ; Wei Hao
Journal of the American Chemical Society 2012 Volume 134(Issue 50) pp:20230-20233
Publication Date(Web):December 3, 2012
DOI:10.1021/ja308950d
An efficient Pd-catalyzed cleavage of C(sp3)–N bonds in secondary amines and a consequent C(sp2)–N and C(sp3)–N coupling process was developed. Various secondary amines could be used to react with alkenyl or aryl dibromides, affording pyrroles and indoles in high yields. Cyclopentadiene–phosphine ligands, a new type of P–olefin ligand, were found to be able to promote the efficiency of this Pd-catalyzed process remarkably. A reactive Pd complex coordinated with a cyclopentadiene–phosphine ligand was successfully isolated and structurally characterized.
Co-reporter:Yang Wang ; Yue Chi ; Wen-Xiong Zhang
Journal of the American Chemical Society 2012 Volume 134(Issue 6) pp:2926-2929
Publication Date(Web):February 3, 2012
DOI:10.1021/ja211486f
A highly regioselective base-mediated ring expansion of 2,4-diiminoazetidines via cleavage of C–N and C(sp3)–H bonds is achieved for the first time to afford efficiently 2,3-dihydropyrimidinesulfonamides. The mechanism of the ring expansion via tandem 4π electrocyclic ring-opening/1,5-H shift/6π electrocyclic ring-closing is well confirmed by the trapping experiments of two key intermediates and deuterium labeling studies.
Co-reporter:Kunbing Ouyang, Yun Liang, and Zhenfeng Xi
Organic Letters 2012 Volume 14(Issue 17) pp:4572-4575
Publication Date(Web):August 10, 2012
DOI:10.1021/ol302040j
A variety of silacycles including benzosiloles, six- and eight-membered silacyclic skeletons, were efficiently synthesized via a Pd-catalyzed intramolecular Mizoroki–Heck reaction of vinylsilanes.
Co-reporter:Ling Xu, Zitao Wang, Wen-Xiong Zhang, and Zhenfeng Xi
Inorganic Chemistry 2012 Volume 51(Issue 21) pp:11941-11948
Publication Date(Web):October 24, 2012
DOI:10.1021/ic3018369
Synthesis and structural characterization of half-sandwich rare-earth metal tris(trimethylsilylmethyl) anionic complexes bearing one 1-phenyl-2,3,4,5-tetrapropylcyclopentadienyl ligand are achieved. These soluble anionic compounds show good reactivity in the stoichiometric reaction with dibenzoylmethane (DBM) to give the salt-free half-sandwich complex bearing two chelate DBM ligands. More importantly, they can serve as excellent and general catalyst precursors for the addition of different types of amines including primary aromatic amines (ArNH2), acyclic (RR′NH, ArRNH, and ArAr′NH; R, R′ = Alkyl group, Ar, Ar′ = Aromatic group), or cyclic secondary aliphatic amines to carbodiimides yielding efficiently guanidines. Acyclic secondary amines of the general formula ArAr′NH and ArRNH cannot be achieved efficiently by the previous rare-earth catalysts because of their weak nucleophilicity and steric hindrance. Our results show clearly the reactivities of anionic trialkyl precursors are comparable with the corresponding neutral alkyl complex in the stoichimetric reaction but exhibit better catalytic activity than the known catalysts.
Co-reporter:Yun Liang, Weizhi Geng, Junnian Wei, Kunbing Ouyang and Zhenfeng Xi
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 8) pp:1537-1542
Publication Date(Web):05 Jan 2012
DOI:10.1039/C2OB06941E
The first transition-metal-catalyzed activation of silyl C(sp3)–H bond was realized and synthetically applied. A variety of organic skeletons substituted with SiMe3 groups could undergo the Pd-catalyzed intramolecular coupling reaction, resulting in an unprecedented synthetic method for yielding six-membered silacycles. It was found that the adjacent Si atom played an essential role for the activation of the C(sp3)–H bond of the SiMe3 group; no activation reaction of the C(sp3)–H bond of the CMe3 group took place under the same reaction conditions.
Co-reporter:Tianhao Meng, Hui-Jun Zhang, Zhenfeng Xi
Tetrahedron Letters 2012 Volume 53(Issue 34) pp:4555-4557
Publication Date(Web):22 August 2012
DOI:10.1016/j.tetlet.2012.06.060
Co-reporter:Heng Li, Lantao Liu, Fei Zhao, Congyang Wang, Chao Wang, Qiuling Song, Wen-Xiong Zhang, and Zhenfeng Xi
The Journal of Organic Chemistry 2012 Volume 77(Issue 10) pp:4793-4800
Publication Date(Web):April 23, 2012
DOI:10.1021/jo300631d
The carbonylation of a 1-lithio-1,3-butadiene derivative with CO gave rise to a butadienyl acyllithio intermediate, which underwent an immediate intramolecular acyllithiation of the C═C double bond, affording a lithio cyclopentadienyl enolate. The X-ray structural analysis of the enolate revealed a dimer connected with a “Li2O2” four-membered ring. Subsequent intermolecular acylation of this enolate with acid chlorides afforded β-keto-3-cyclopentenones, γ-keto-2-cyclopentenones, or cyclopentadienyl ester derivatives. The stereo- and regioselectivity of the in situ generated lithio cyclopentadienyl enolate with various acid chlorides was investigated and analyzed, showing that the formation of the above products was significantly dependent on both the substituents on the butadienyl skeleton and the bulkiness of acid chlorides.
Co-reporter:Weizhi Geng, Hanliu Wang, Zitao Wang, Shaoguang Zhang, Wen-Xiong Zhang, Zhenfeng Xi
Tetrahedron 2012 68(26) pp: 5283-5289
Publication Date(Web):
DOI:10.1016/j.tet.2012.01.088
Co-reporter:Yi Zhou ; Wen-Xiong Zhang
Organometallics 2012 Volume 31(Issue 15) pp:5546-5550
Publication Date(Web):July 16, 2012
DOI:10.1021/om3005004
The first well-defined 1,3-butadienylzinc trimers have been synthesized by transmetalation of 1,4-dilithio-1,3-butadienes with 1 equiv of ZnBr2. Their structures have been determined by single-crystal X-ray structural analysis. Their reaction chemistry has been demonstrated by Pd-catalyzed Negishi cross-coupling with iodobenzenes.
Co-reporter:Jing Zhao, Shaoguang Zhang, Wen-Xiong Zhang, and Zhenfeng Xi
Organometallics 2012 Volume 31(Issue 23) pp:8370-8374
Publication Date(Web):November 29, 2012
DOI:10.1021/om300949a
The reaction of the seven-membered azazirconacycloallene 2 with Ph2CHCN was carried out to yield a zirconocene complex with three fused rings and a keteniminate ligand. Further reactivity toward propargyl bromide or propionyl chloride shows that the keteniminate ligand can be replaced by halogen atoms (Br or Cl). In addition, the reaction of the strained four-membered zirconacycle 1 with Ph2CHCN gives zirconocenes possessing vinyl-imine and keteniminate species.
Co-reporter:Dr. Yun Liang;Weizhi Geng;Junnian Wei;Dr. Zhenfeng Xi
Angewandte Chemie International Edition 2012 Volume 51( Issue 8) pp:1934-1937
Publication Date(Web):
DOI:10.1002/anie.201108154
Co-reporter:Dr. Yun Liang;Weizhi Geng;Junnian Wei;Dr. Zhenfeng Xi
Angewandte Chemie 2012 Volume 124( Issue 8) pp:1970-1973
Publication Date(Web):
DOI:10.1002/ange.201108154
Co-reporter:Wen-Xiong Zhang, Shaoguang Zhang, and Zhenfeng Xi
Accounts of Chemical Research 2011 Volume 44(Issue 7) pp:541
Publication Date(Web):May 25, 2011
DOI:10.1021/ar200078e
Characterizing reactive organometallic intermediates is critical for understanding the mechanistic aspects of metal-mediated organic reactions. Moreover, the isolation of reactive organometallic intermediates can often result in the ability to design new synthetic methods. In this Account, we outline synthetic methods that we developed for a variety of diverse Zr/Si organo-bimetallic compounds and Si/N heteroatom–organic compounds through the detailed study of zirconacyclobutene–silacyclobutene fused compounds.Two basic components are involved in this chemistry. The first is the Si-tethered diyne, which owes its rich reactive palette to the combination of the Si–C bond and the C≡C triple bond. The second is the low-valent zirconocene species Cp2Zr(II), which has proven very useful in organic synthesis. The reaction of these two components affords the zirconacyclobutene–silacyclobutene fused compound, which is the key reactive Zr/Si organo-bimetallic intermediate discussed here.We discuss the three types of reactions that have been developed for the zirconacyclobutene–silacyclobutene fused intermediate. The reaction with nitriles (the C≡N triple bond) is introduced in the first section. In this one-pot reaction, up to four different components can be combined: the Si-tethered diyne can be reacted with three identical nitriles, with differing nitriles, or with a nitrile and other unsaturated organic substrates such as formamides, isocyanides, acid chlorides, aldehydes, carbodiimides, and azides. Several unexpected multiring, fused Zr/Si organo-bimetallic intermediates were isolated and characterized. A wide variety of N-heterocycles, such as 5-azaindole, pyrrole, and pyrroloazepine derivatives, were obtained. We then discuss the reaction with alkynes (the C≡C triple bond). A consecutive skeletal rearrangement, differing from that observed in the reactions with nitriles, takes place in this reaction. Finally, we discuss the reaction with the C═X substrates (where X is O or N), including ketones, aldehydes, and isocyanides. Oxa- and azazirconacycles are formed via a new skeletal rearrangement.Our results show that the zirconocene and the Si-tethered diyne cooperate as a “chemical transformer” after treatment with various substrates, leading to a diverse range of cyclic Zr/Si organo-bimetallic compounds. This mechanism-derived synthesis of organometallic and organic compounds demonstrates that the investigation of metal-mediated reactions and the isolation of reactive organometallic intermediates not only contribute to the understanding of complex reactions but can also lead to the discovery of synthetically useful methods.
Co-reporter:Wen-Xiong Zhang ; Zitao Wang ; Masayoshi Nishiura ; Zhenfeng Xi ;Zhaomin Hou
Journal of the American Chemical Society 2011 Volume 133(Issue 15) pp:5712-5715
Publication Date(Web):March 29, 2011
DOI:10.1021/ja200540b
Tetranuclear cubane-type rare-earth methylidene complexes consisting of four “Cp′LnCH2” units, [Cp′Ln(μ3-CH2)]4 (4-Ln; Ln = Tm, Lu; Cp′ = C5Me4SiMe3), have been obtained for the first time through CH4 elimination from the well-defined polymethyl complexes [Cp′Ln(μ2-CH3)2]3 (2-Ln) or mixed methyl/methylidene precursors such as [Cp′3Ln3(μ2-Me)3(μ3-Me)(μ3-CH2)] (3-Ln). The reaction of the methylidene complex 4-Lu with benzophenone leads to C═O bond cleavage and C═C bond formation to give the cubane-type oxo complex [Cp′Lu(μ3-O)]4 and CH2═CPh2, while the methyl/methylidene complex 3-Tm undergoes sequential methylidene addition to the C═O group and ortho C−H activation of the two phenyl groups of benzophenone to afford the bis(benzo-1,2-diyl)ethoxy-chelated trinuclear complex [Cp′3Tm3(μ2-Me)3{(C6H4)2C(O)Me}] (6-Tm).
Co-reporter:Yun Liang ; Shaoguang Zhang
Journal of the American Chemical Society 2011 Volume 133(Issue 24) pp:9204-9207
Publication Date(Web):April 25, 2011
DOI:10.1021/ja2024959
An efficient process involving Pd-catalyzed selective cleavage of a C(sp3)–Si bond and consequent intramolecular C(sp2)–Si coupling has been developed, affording benzosilolo[2,3-b]indoles as a new type of silicon-bridged polyheteroarene in excellent yields. Aldehyde was found for the first time to be able to promote the efficiency of the catalytic process remarkably.
Co-reporter:Qian Luo, Chao Wang, Yuexing Li, Kunbing Ouyang, Li Gu, Masanobu Uchiyama and Zhenfeng Xi
Chemical Science 2011 vol. 2(Issue 11) pp:2271-2274
Publication Date(Web):26 Aug 2011
DOI:10.1039/C1SC00356A
An efficient and specific cleavage of the endo-C(sp2)-Si bond of silole rings was developed. The stable silole ring was effectively opened in an unexpected regioselectivity by using an AcOH/ROH system, in which the AcOH behaved as a catalyst and the ROH as a nucleophile. Depending on the nature of the substituents at the 2- and 5-positions of the silole ring, the cooperative effect of the AcOH/ROH system exclusively cleaved one of the two endo-C-Si bonds to afford silylbutadiene derivatives.
Co-reporter:Fei Zhao, Shaoguang Zhang and Zhenfeng Xi
Chemical Communications 2011 vol. 47(Issue 15) pp:4348-4357
Publication Date(Web):21 Feb 2011
DOI:10.1039/C0CC05665K
Silyl-substituted 1,3-butadienes are useful building blocks and are readily applied in several types of reactions such as Diels–Alder reaction, ene reaction and allylation. They can also participate in different tandem reactions such as Diels–Alder/allylation, ene/allylation, ene/allylation/Diels–Alder reaction, ene/allylation/ene reaction and ene/allylation/Diels–Alder/allylation reaction. This feature article reviews the synthesis of silyl-substituted 1,3-butadienes, and their applications in the reaction types mentioned above, involving a tandem Diels–Alder/ene/allylation process. This article also introduces some reactions of alkenylsilanes and allylsilanes for comparison and discussion about the tandem reaction. The tandem reactions described in this article are a powerful tool to construct complicated multicyclic compounds with high selectivity and high efficiency.
Co-reporter:Zitao Wang, Weizhi Geng, Hanliu Wang, Shaoguang Zhang, Wen-Xiong Zhang, Zhenfeng Xi
Tetrahedron Letters 2011 Volume 52(Issue 51) pp:6997-6999
Publication Date(Web):21 December 2011
DOI:10.1016/j.tetlet.2011.10.098
Benzothiophene derivatives were synthesized in high yields from readily available o-(alkynyllithio)aryllithio compounds, sulfur, and 2 equiv of acid chlorides or other electrophiles. An acid chloride-induced electrophilic cyclization resulted in the formation of the thiophene ring.
Co-reporter:Yang Wang;Dr. Wen-Xiong Zhang;Zitao Wang;Dr. Zhenfeng Xi
Angewandte Chemie International Edition 2011 Volume 50( Issue 35) pp:8122-8126
Publication Date(Web):
DOI:10.1002/anie.201101948
Co-reporter:Dongzhen Li;Yang Cao;An Shi;Dr. Zhenfeng Xi
Chemistry – An Asian Journal 2011 Volume 6( Issue 2) pp:392-395
Publication Date(Web):
DOI:10.1002/asia.201000257
Co-reporter:Jing Zhao, Shaoguang Zhang, Wen-Xiong Zhang, and Zhenfeng Xi
Organometallics 2011 Volume 30(Issue 13) pp:3464-3467
Publication Date(Web):June 15, 2011
DOI:10.1021/om200404p
The first well-defined zirconocene complexes processing vinyl-imine and keteniminate species have been achieved from zirconacyclopentenes or zirconacyclopentadienes and 2 equiv of Ph2CHCN. Multifunctional effect of Ph2CHCN is observed for the first time in one system. This process proceeds via the azazirconacyclopentadiene intermediate and its intramolecular proton transfer. Moreover, β,β′-C–C bond cleavage of tetra-alkyl-substituted zirconacyclopentadienes is observed for the first time under the appropriate conditions.
Co-reporter:Dongzhen Li, Yang Wang, Wen-Xiong Zhang, Shaoguang Zhang, Jie Guang, and Zhenfeng Xi
Organometallics 2011 Volume 30(Issue 19) pp:5278-5283
Publication Date(Web):September 9, 2011
DOI:10.1021/om2006613
The first synthesis of trisubstituted guanidinium triflates has been achieved by a simple one-step procedure from readily available amines, carbodiimides, and Zn(OTf)2. X-ray analysis revealed they adopt 1-D chain-like or novel 3-D hydrogen-bonded networks, which have not been reported previously in the solid structure of guanidinium salts. Isolation of a binuclear zinc complex and its subsequent carbodiimide insertion provide the insight into the mechanism for the formation of guanidinium triflates.
Co-reporter:Shaoguang Zhang;Dr. Wen-Xiong Zhang;Jing Zhao;Dr. Zhenfeng Xi
Chemistry - A European Journal 2011 Volume 17( Issue 8) pp:2442-2449
Publication Date(Web):
DOI:10.1002/chem.201003119
Abstract
5-Azaindoles either with three different substituents at their 2-, 4-, and 6-positions or with two identical substituents at their 2- and 6-positions and a different one at the 4-position, were obtained in good to excellent isolated yields by a zirconocene-mediated multicomponent process. Each reaction involved four organic partners, comprising a Si-tethered diyne, one tBuCN component, and two (either different or identical) nitriles. All these four components were combined through the action of a Cp2ZrII species into a three-ring fused Zr/Si-containing organometallic complex in a perfectly chemo- and regioselective manner. This multicomponent reaction process consisted of three reaction steps, all of which were made clear through the isolation and characterization of their corresponding organometallic intermediates: the zirconacyclopropene-azasilacyclopentadienes 2, the allenyl-aza-zirconacycles 3, and the three-ring fused complexes 6. X-ray single-crystal structural analyses of two three-ring fused Zr/Si-containing intermediates and two 5-azaindoles unambiguously showed the positions of the different substituents and the regioselectivity. Iminopyrrole derivatives could be also highly selectively prepared from a Si-tethered diyne and two different nitriles.
Co-reporter:Heng Li;Dr. Lantao Liu;Zitao Wang;Fei Zhao;Shaoguang Zhang;Dr. Wen-Xiong Zhang;Dr. Zhenfeng Xi
Chemistry - A European Journal 2011 Volume 17( Issue 27) pp:7399-7403
Publication Date(Web):
DOI:10.1002/chem.201100830
Co-reporter:Zhenfeng Xi
Accounts of Chemical Research 2010 Volume 43(Issue 10) pp:1342
Publication Date(Web):July 15, 2010
DOI:10.1021/ar1000583
The development of organometallic reagents remains one of the most important frontiers in synthetic chemistry. Commonly used organometallic reagents (such as RLi and RMgBr) are typically monometallic compounds, although they aggregate in many cases. When two carbon−metal bonds are in the same molecule in close proximity, however, these two carbon−metal moieties may exhibit novel reactivity. In this Account, we outline our work on new reactions and synthetic applications of the organo-dilithio reagents 1,4-dilithio-1,3-butadienes. The 1,4-dilithio-1,3-butadienes can be accessed readily in high efficiency with a wide variety of substitution patterns on the butadienyl skeleton. The configuration has been predicted and demonstrated to favor a double dilithium bridging structure in both solution and solid states. The two Li atoms are bridged by a butadiene moiety and are in close proximity. By taking advantage of this unique configuration, we have developed useful and interesting synthetic methodologies. Three types of reactions of 1,4-dilithio-1,3-butadienes, termed dilithio reagents here, have been developed and are discussed. An intramolecular reaction is introduced in the first section. The reaction is a result of the intracooperative effect among the two C−Li moieties, the butadienyl bridge, and the substituents. A useful transformation from silylated 1,4-dilithio-1,3-butadienes to α-lithio siloles is described. Second, we discuss an intermolecular reaction that results from the intercooperative effect of the two C−Li moieties toward substrates. As an example of the formation of functionalized cyclic dianions from the linear dianions of the dilithio reagents and organic substrates, we describe the isolation and structural characterization of a novel type of cyclic dianion; that is, fully substituted oxy-cyclopentadienyl dilithium formed via the reaction of dilithio reagents with CO. We also describe diverse reactions of dilithio reagents with nitriles to form substituted pyridines, tricyclic 1-bipyrrolines, and siloles, demonstrating the remarkable effect of substituents on the butadienyl skeleton. Third, we discuss transmetalation of dilithio reagents to generate other organo-dimetallic compounds. This section focuses on organo-dicopper compounds and their reactivity toward the synthesis of strained ring systems, such as semibullvalenes and twisted four-membered rings, with the metal-mediated C−C bond-forming approach. In addition to these three representative reactions, other useful applications are also briefly introduced. The dimetallic 1,4-dilithio-1,3-butadienes and their transmetalated derivatives provide unique synthetic organometallic reagents that are very different from monometallic reagents, both in terms of reactivity and synthetic application. These organo-dimetallic reagents provide access to interesting and useful compounds that are not available by other means. Moreover, given the possibilities afforded, the study of organo-dimetallic and organo-polymetallic compounds should yield further synthetic applications in the near future.
Co-reporter:Hui-jun Zhang, Junnian Wei, Fei Zhao, Yun Liang, Zitao Wang and Zhenfeng Xi
Chemical Communications 2010 vol. 46(Issue 39) pp:7439-7441
Publication Date(Web):06 Sep 2010
DOI:10.1039/C0CC02380A
Dibenzo[a,c]cyclooctatetraenes were selectively generated in good yields from the palladium-catalyzed [4+4] cyclic homocoupling of borylvinyl iodobenzene derivatives, which were prepared via the selective Suzuki–Miyaura cross-coupling of the gem-diboryl reagents with iodo compounds.
Co-reporter:Qian Luo;Chao Wang Dr.;Li Gu;Wen-Xiong Zhang Dr. Dr.
Chemistry – An Asian Journal 2010 Volume 5( Issue 5) pp:1120-1128
Publication Date(Web):
DOI:10.1002/asia.200900287
Abstract
A full account of a useful transformation from silylated 1,4-dilithio-1,3-butadienes to α-lithio siloles is described. These lithio siloles formed by this procedure are general, in terms of substitution patterns and synthetic methods, affording diversified silole derivatives. Notably, some structurally complex molecules, such as bridged bis-silole compounds, have been synthesized easily and successfully by applying our protocol. The structure of the α-lithio silole, which adopts a dimeric fashion through two lithium bridges, was confirmed by X-ray analysis. Furthermore, a possible mechanism of the skeleton rearrangements via E/Z isomerization of 1-silyl-1-lithio alkene and nucleophilic attack on silicon is proposed, and is also proved by experimental investigations.
Co-reporter:QiFeng Wang;WenXiong Zhang;Chen Lin
Science Bulletin 2010 Volume 55( Issue 26) pp:2915-2923
Publication Date(Web):2010 September
DOI:10.1007/s11434-010-4047-x
Pyranylidene carbene complexes bearing a pyran ring carbene ligand represent a unique kind of organometallic species due to their special structures and reactivity. In this review, we present a comprehensive summary on the synthesis and reactivity of pyranylidene carbene complexes during the past four decades.
Co-reporter:Shaoguang Zhang;Wen-Xiong Zhang Dr. Dr.
Chemistry - A European Journal 2010 Volume 16( Issue 28) pp:8419-8426
Publication Date(Web):
DOI:10.1002/chem.201000708
Abstract
An efficient multicomponent synthesis of 5-azaindoles and dihydropyrrolo[3,2-c]azepines was achieved by zirconocene-mediated coupling of silicon-tethered diynes, nitriles, and isocyanides. The synthesis, structures, and intramolecular cyclization of mono- and bis(iminoacyl)Zr intermediates were investigated to elucidate the reaction process. Upon hydrolysis, the isolated mono(iminoacyl)Zr intermediates underwent intramolecular cyclization to afford tetrasubstituted 5-azaindoles, whereas intramolecular cyclization of bis(iminoacyl)Zr intermediates led to the formation of dihydropyrrolo[3,2-c]azepines. The structure of a bis(iminoacyl)Zr intermediate, formed through insertion of two molecules of CyNC into the ZrC bond, and structures of two dihydropyrrolo[3,2-c]azepines were characterized by single-crystal X-ray structural analysis.
Co-reporter:Lantao Liu, Wen-Xiong Zhang, Qian Luo, Heng Li and Zhenfeng Xi
Organometallics 2010 Volume 29(Issue 1) pp:278-281
Publication Date(Web):December 1, 2009
DOI:10.1021/om900872u
The X-ray crystallographic structures of a trimeric all-alkyl-substituted 1,4-dilithio-1,3-butadiene with a Li6 pseudooctahedron and a dimeric Me3Si-substituted 1,4-dilithio-1,3-butadiene with a Li4 tetrahedron are reported. The reactions of theses compounds with CO were investigated.
Co-reporter:Zitao Wang ; Yang Wang ; Wen-Xiong Zhang ; Zhaomin Hou
Journal of the American Chemical Society 2009 Volume 131(Issue 42) pp:15108-15109
Publication Date(Web):October 2, 2009
DOI:10.1021/ja9069419
An organolithium-promoted multicomponent reaction (MCR) involving readily available terminal alkynes, elemental sulfur, and carbodiimides has been achieved for the first time. This MCR offers an atom-economic route to 2,3-dihydropyrimidinthiones which are difficult to access by other means via an interesting and useful C═N double bond cleavage and an sp3 C−H bond functionalization of carbodiimides.
Co-reporter:Fei Zhao, Chao Wang, Lantao Liu, Wen-Xiong Zhang and Zhenfeng Xi
Chemical Communications 2009 (Issue 43) pp:6569-6571
Publication Date(Web):11 Sep 2009
DOI:10.1039/B914619A
Mediated by AlCl3, substituted spiro[4.5]deca-tetraenes underwent novel and selective skeletal rearrangement to generate indene derivatives in high to excellent yields.
Co-reporter:Chao Wang, Liang Deng, Jun Yan, Hailin Wang, Qian Luo, Wen-Xiong Zhang and Zhenfeng Xi
Chemical Communications 2009 (Issue 29) pp:4414-4416
Publication Date(Web):11 Jun 2009
DOI:10.1039/B908803B
Selective cleavage of both active and inert C–C bonds in substituted allylcyclopropanes was realized via a novel ligand-controlled approach using zirconocene(II)-1-butene or zirconocene(II)-benzyne complexes.
Co-reporter:Hui-Jun Zhang, Tianhao Meng, Bernard Demerseman, Christian Bruneau and Zhenfeng Xi
Organic Letters 2009 Volume 11(Issue 19) pp:4458-4461
Publication Date(Web):September 2, 2009
DOI:10.1021/ol901840c
By treatment with LiAlH4, 1,4-dicyano-1,4-bis(trimethylsilyl)-1,3-dienes underwent a novel hydride-induced nucleophilic intermolecular 1,4-addition to the α,β-unsaturated nitrile moiety, followed by an immediate nucleophilic intramolecular 1,2-addition to the remaining CN group, to afford multiply functionalized cyclopentadienes in high isolated yields. These multifunctional (−CN, −NH2, −SiMe3, −H) cyclopentadienes are of a rich and interesting reaction chemistry as preliminarily demonstrated by their reaction with ArNCO involving both the substituent and the ring.
Co-reporter:Hui-Jun Zhang;Bernard Demerseman;Christian Bruneau
Advanced Synthesis & Catalysis 2009 Volume 351( Issue 16) pp:2724-2728
Publication Date(Web):
DOI:10.1002/adsc.200900498
Abstract
The allylation of 1,3-dicarbonyl compounds and malononitrile with aliphatic allylic substrates is achieved under mild conditions in the presence of new ruthenium catalysts. The ruthenium complex [Ru(C5Me5)(2-quinolinecarboxylato)(CH2CHCH-n-Pr)] [BF4] as a precatalyst, allows the synthesis of mono-allylated branched derivatives. On the other hand, the parent complex [Ru(C5Me5)(MeCN)3] [PF6] as a precatalyst, straightforwardly favours the bis-allylation of the procarbonucleophiles leading to bis-allylated bis-linear products. The involvement of the two precatalysts provides a sequential synthesis of unsymmetrical mixed linear-branched bis-allylated derivatives.
Co-reporter:Qifeng Wang, Lantao Liu, Chen Lin, Hui Sun, Wen-Xiong Zhang and Zhenfeng Xi
Dalton Transactions 2009 (Issue 47) pp:10433-10435
Publication Date(Web):08 Oct 2009
DOI:10.1039/B918679B
Hydride-induced cleavage of the C–O bond in the pyran ring followed by ring closure of unsaturated acylmetalates and reductive elimination was achieved.
Co-reporter:Wen-Xiong Zhang Dr.;Shaoguang Zhang;Xiaohua Sun Dr.;Masayoshi Nishiura Dr.;Zhaomin Hou Dr. Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 39) pp:7227-7231
Publication Date(Web):
DOI:10.1002/anie.200903329
Co-reporter:Lantao Liu Dr.;Wen-Xiong Zhang Dr.;Chao Wang Dr.;Congyang Wang Dr. Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 43) pp:8111-8114
Publication Date(Web):
DOI:10.1002/anie.200904298
Co-reporter:Junhui Liu, Shaoguang Zhang, Wen-Xiong Zhang and Zhenfeng Xi
Organometallics 2009 Volume 28(Issue 2) pp:413-417
Publication Date(Web):December 29, 2008
DOI:10.1021/om800841s
Well-defined π-conjugated systems containing two or three silacyclobutene units have been synthesized in high yields via regioselective coupling of bis(phenylethynyl)silanes with dialkynylbenzene or trialkynylbenzene in the presence of Cp2ZrBun2 (Negishi reagent). The UV−vis absorption and fluorescence spectra of these new π-conjugated systems have demonstrated that the increase in silacyclobutene units per molecule from two or three brings about an increase of the extinction coefficient.
Co-reporter:Wen-Xiong Zhang, Dongzhen Li, Zitao Wang and Zhenfeng Xi
Organometallics 2009 Volume 28(Issue 3) pp:882-887
Publication Date(Web):January 14, 2009
DOI:10.1021/om801035t
The commercially readily available alkyl aluminums, such as AlMe3, AlEt3, and AlEt2Cl, can serve as excellent catalyst precursors for the catalytic addition of amines to carbodiimides, yielding quantitatively their corresponding trisubstituted guanidines. Aromatic C−X (X = F, Cl, Br, and I) bonds, nitro NO2, and terminal alkyne units can survive the reaction conditions. An aluminum guanidinate species has been characterized by single-crystal X-ray structural analysis and has been confirmed to be a true catalyst species.
Co-reporter:Hui-Jun Zhang, Bernard Demerseman, Loïc Toupet, Zhenfeng Xi and Christian Bruneau
Organometallics 2009 Volume 28(Issue 17) pp:5173-5182
Publication Date(Web):July 10, 2009
DOI:10.1021/om900437m
The trans addition of HSiEt3 to propargylic ethyl carbonates catalyzed by the ruthenium complex [Ru(C5Me5)(MeCN)3][PF6], 1, leads to H2C═C(SiEt3)CH(R)OCO2Et (R = H, Me, Prn, or Ph) derivatives as allylic substrates for subsequent ruthenium-catalyzed nucleophilic allylic substitution reactions. Ruthenium catalysts based on a [Ru(C5Me5)(2-quinolinecarboxylato)] or [Ru(C5Me5)(2,2′-bipyridine)]+ fragment are more efficient than 1 for access to various H2C═C(SiEt3)CH(R)Nu-functionalized vinylsilanes (R = H, Me; NuH = secondary amine, phenol, dimethylmalonate, methanol). New [Ru(C5Me5)(2-quinolinecarboxylato)(η3-allyl)]+ cationic ruthenium(IV) intermediates are characterized, and experiments using a deuterium-labeled allylic carbonate provide evidence for a regioselectivity arising from the presence of the 2-quinolinecarboxylato (N,O)-chelate.
Co-reporter:Shaoguang Zhang;Xiaohua Sun Dr.;Wen-Xiong Zhang Dr. Dr.
Chemistry - A European Journal 2009 Volume 15( Issue 46) pp:12608-12617
Publication Date(Web):
DOI:10.1002/chem.200902411
Abstract
A zirconocene-mediated one-pot multicomponent synthesis process leading to the synthesis of pyrrolo[3,2-c]pyridine (5-azaindoles) and pyrrole derivatives has been developed starting from a silicon-tethered diyne and three or two different or identical organonitriles. Pyrrolo[3,2-c]pyridine and pyrrole derivatives with diverse structures and substitution patterns have been highly selectively prepared by this protocol. A wide variety of organonitriles and silicon-tethered diynes, either aliphatic or aromatic with both electron-withdrawing and -donating groups, have been used to afford 5-azaindoles and/or pyrroles in good-to-excellent isolated yields. A key intermediate formed in the Cp2ZrII-mediated reaction of one (PhC≡C)2SiMe2, two molecules of iPrCN and one p-tolylCN was characterised by X-ray single-crystal structural analysis, which has allowed us to gain a good understanding of the reaction process.
Co-reporter:Qian Luo, Li Gu, Chao Wang, Junhui Liu, Wenxiong Zhang, Zhenfeng Xi
Tetrahedron Letters 2009 50(26) pp: 3213-3215
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.01.152
Co-reporter:Lantao Liu Dr.;Wen-Xiong Zhang Dr.;Chao Wang Dr.;Congyang Wang Dr. Dr.
Angewandte Chemie 2009 Volume 121( Issue 43) pp:8255-8258
Publication Date(Web):
DOI:10.1002/ange.200904298
Co-reporter:Qian Luo, Chao Wang, Wen-Xiong Zhang and Zhenfeng Xi
Chemical Communications 2008 (Issue 13) pp:1593-1595
Publication Date(Web):25 Feb 2008
DOI:10.1039/B719007G
Mediated by CuCl, 1,4-dilithio-1,3-dienes reacted with carbon monoxide (CO) to generate multi-substituted cyclopentadienones and/or their head-to-head dimers, via tandem CO insertion and intra/intermolecular cycloaddition of organocopper compounds.
Co-reporter:Hui-Jun Zhang;Bernard Demerseman;Loïc Toupet;Christian Bruneau
Advanced Synthesis & Catalysis 2008 Volume 350( Issue 10) pp:1601-1609
Publication Date(Web):
DOI:10.1002/adsc.200800135
Abstract
Stable [ruthenium(R-substituted-tetramethylcyclopentadiene)(2-quinolinecarboxylato)(1-R′-substituted-allyl) hexafluorophosphate (R=Me, R′=H, Me, n-Pr, Ph; R=t-Bu, R′=Me) and [ruthenium(pentamethylcyclopentadiene)(2-quinolinecarboxylato)(1-n-propylallyl)] tetrafluoroborate (4′a), as allylruthenium(IV) complexes, have been synthesized in one step, starting from [ruthenium(R-substituted-tetramethylcyclopentadiene)tris(acetonitrile) hexafluorophosphate or tetrafluoroborate complexes, quinaldic acid, and allylic alcohols. Single stereoisomers are obtained and the X-ray single crystal structure determinations of 3b (R=t-Bu, R′=Me) and 4′a allow one to specify the preferred arrangement. Complexes 3a (R=R′=Me) and 3b are involved as precatalysts favoring the formation of branched products in regioselective nucleophilic allylic substitution reactions, starting from ethyl 2-(E)-hexen-1-yl carbonate and chlorohexene as unsymmetrical aliphatic allylic substrates. Phenols, dimethyl malonate, and primary (aniline) and secondary (pyrrolidine, piperidine) amines have been used as nucleophiles under mild basic conditions. For the first time, the regioselectivity in favor of the branched product obtained from purely aliphatic allylic substrates is close to the high regioselectivity previously reached starting from cinnamyl-type substrates in the presence of ruthenium catalysts.
Co-reporter:Hui-Jun Zhang;Bernard Demerseman;Christian Bruneau
European Journal of Inorganic Chemistry 2008 Volume 2008( Issue 20) pp:3212-3217
Publication Date(Web):
DOI:10.1002/ejic.200800295
Abstract
[Ru(η5-C5Me4R)(MeCN)3][PF6] (R = CH2tBu, iPr, tBu, and CF3; 2–5) complexes were synthesized in two steps starting from the appropriate cyclopentadienes and RuCl3·3H2O. The fully substituted ruthenocenes [Ru(C5Me5)(C5Me4R*)], [Ru(C5Me5)(C5nPr4R*)] {R* = (1R,5S)-6,6-dimethylbicyclo[3.1.1]hept-2-en-2-yl} and [Ru(C5Me5)(C5nPr5)] were obtained by treating [Ru(C5Me5)Cl]4 with the corresponding cyclopentadienyllithium salts. Complexes 2–5 were evaluated as catalyst precursors for nucleophilic allylic substitution reactions, and the results were compared to those obtained with the [Ru(C5Me5)(MeCN)3][PF6] (1) precatalyst. The etherification of p-methoxyphenol with the typical aliphatic chlorohexene allylic substrate shows that the introduction of the bulky tert-butyl and trifluoromethyl groups into the tetramethylcyclopentadienyl ring results in a valuable enhancement in regioselectivity in favour of the branched allyl aryl ether.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2008)
Co-reporter:Nan Yu;Masayoshi Nishiura Dr.;Xiaofang Li Dr. Dr.;Zhaomin Hou Dr.
Chemistry – An Asian Journal 2008 Volume 3( Issue 8-9) pp:1406-1414
Publication Date(Web):
DOI:10.1002/asia.200800145
Abstract
The one-pot salt-metathesis reaction of ScCl3, cyclopentadienyl lithium salts, and allylmagnesium chlorides afforded with ease the corresponding base-free half-sandwich scandium di(η3-allyl) complexes [(C5Me4SiMe3)Sc(C3H5)2] (1 a), [(C5Me5)Sc(C3H5)2] (1 b), and [(C5Me5)Sc(2-MeC3H4)2] (1 c) in high yields. Reaction of 1 a with 1 equivalent of [PhNMe2H][B(C6F5)4] in toluene gave rapidly the N,N-dimethylaniline-coordinated cationic mono(η3-allyl) complex [(C5Me4SiMe3)Sc(η3-C3H5)(η6-PhNMe2)][B(C6F5)4] (2). The similar reaction of 1 a with [Ph3C][B(C6F5)4] yielded the analogous toluene-separated ion pair [(C5Me4SiMe3)Sc(η3-C3H5)(η6-PhMe)][B(C6F5)4] (3). When [PhNMe2H][BPh4] was treated with 1 a, the contact ion pair [(C5Me4SiMe3)Sc(η3-C3H5)( μ,η6-Ph)BPh3] (4) was obtained. Recrystallization of 2, 3, and 4 in THF yielded the corresponding thf-separated ion pair complexes [(C5Me4SiMe3)Sc(η3-C3H5)(thf)2][B(C6F5)4] (5) and [(C5Me4SiMe3)Sc(η3-C3H5)(thf)2][BPh4] (6). The N,N-dimethylaniline-coordinated cationic scandium allyl complex 2 and the toluene-coordinated analogue 3 showed high activity (activity: 3>2) toward the polymerization and copolymerization of isoprene and norbornene to afford random copolymers with a broad range of isoprene content (33–86 mol %). The tight ion pair 4 and the thf-coordinated complexes 5 and 6 showed no activity under the same conditions. These results offer unprecedented insight into the structure–activity relationship of a cationic metal polymerization-catalyst system.
Co-reporter:Qifeng Wang ; Wen-Xiong Zhang
Organometallics 2008 Volume 27(Issue 15) pp:3627-3629
Publication Date(Web):July 9, 2008
DOI:10.1021/om800565r
This paper reports a conceptually new and concise synthetic method leading to unprecedented, fully alkyl substituted pyranylidene carbene complexes 2 from M(CO)6 (M = Cr, Mo, W) and the readily available 1-lithio-1,3-butadiene 1, bearing a leaving group, via an intramolecular trapping of metallaacylate intermediates. These carbenes have been demonstrated to have interesting and unique reactivity.
Co-reporter:Nan Yu;Congyang Wang Dr.;Fei Zhao;Lantao Liu;Wen-Xiong Zhang Dr. Dr.
Chemistry - A European Journal 2008 Volume 14( Issue 18) pp:5670-5679
Publication Date(Web):
DOI:10.1002/chem.200701742
Abstract
Addition cyclization of 1,2,3,4-tetrasubstituted 1,4-dilithio-1,3-dienes (Type I) with four equivalents of various aromatic nitriles in the presence of hexamethylphosphoramide (HMPA) gives exclusively fully substituted pyridines in moderate to good yields. Similarly, trisubstituted pyridines can be prepared by the reaction of 2,3-dialkyl- or diaryl-substituted 1,4-dilithio-1,3-dienes (Type II) with nitriles. However, five- or six-membered-ring fused 2,3-disubstituted 1,4-dilithio-1,3-dienes (Type III) reacted with various aromatic and aliphatic nitriles without α-hydrogen atoms to afford tricyclic Δ1-bipyrrolines in high yields. The reaction of six-membered-ring fused 2,3-disubstituted 1,4-dilithio-1,3-diene (Type III) with 2-cyanopyridine afforded the corresponding pyridine, and no tricyclic Δ1-bipyrroline was observed. Seven-membered-ring fused dilithiodienes reacted with PhCN or trimethylacetonitrile to afford the corresponding pyridines in good yield. When 1,2,3,4-tetrasubstituted dilithio reagents (Type I) were treated with Me3SiCN, a tandem silylation/intramolecular substitution process readily occurred to yield siloles, whereas the reaction of 2,3-disubstituted dilithio reagents (Types II and III) with Me3SiCN gave rise to (Z,Z)-dienylsilanes with high stereoselectivity. These results revealed that the formation of tricyclic Δ1-bipyrrolines, pyridines, siloles, and (Z,Z)-dienylsilanes are strongly dependent on the substitution patterns of the dilithio butadienes and the nature of the nitriles employed.
Co-reporter:Congyang Wang and Zhenfeng Xi
Chemical Society Reviews 2007 vol. 36(Issue 9) pp:1395-1406
Publication Date(Web):28 Mar 2007
DOI:10.1039/B608694M
Transition metal-mediated or -catalyzed carbon–carbon bond or carbon–heteroatom bond forming reactions are among the most powerful tools in organic synthesis. In addition, Lewis acid-mediated or -catalyzed organic transformations are widely used. In this context, an effective combination of these two powerful protocols or an efficient cooperation of transition metals with Lewis acids should open a new era in synthetic chemistry. This tutorial review summarizes representative synthetic methodologies developed in the past decades employing this co-operative effect.
Co-reporter:Chao Wang and Zhenfeng Xi
Chemical Communications 2007 (Issue 48) pp:5119-5133
Publication Date(Web):19 Oct 2007
DOI:10.1039/B709839A
Substituted cyclooctatetraenes are a class of interesting and important compounds both theoretically and synthetically. Since Reppe first discovered the Ni-catalyzed tetramerization of ethyne affording cyclooctatetraene in 1948, transition metal mediated synthesis of this type of compounds has become a primary methodology. In this Feature Article, based on our own recent results and other groups' related reports, we describe major achievements on transition metal mediated or catalyzed synthetic methods for substituted cyclooctatetraenes, with focus on reaction patterns, mechanisms, and structural diversity of products.
Co-reporter:Qiaoshu Hu, Chao Wang, Dongzhen Li and Zhenfeng Xi
Organic & Biomolecular Chemistry 2007 vol. 5(Issue 13) pp:2114-2118
Publication Date(Web):31 May 2007
DOI:10.1039/B705110G
Stable enols were synthesized from the reaction of (1Z,3Z)-1,4-dilithio-1,3-dienes with acid chlorides and structurally characterized by single-crystal X-ray analysis. These stable enols were formed by a novel de-aromatization/Michael addition/re-aromatization domino process.
Co-reporter:Chao Wang;Zhi-Hui Wang;Guo-Liang Mao;Zhen-Feng Xi
European Journal of Organic Chemistry 2007 Volume 2007(Issue 8) pp:1267-1273
Publication Date(Web):8 JAN 2007
DOI:10.1002/ejoc.200600504
Multiply substituted cyclopentadienes were formed through reactions between phenyl-substituted 1,4-dilithio-1,3-dienes such as 1b and 1c and two molecules of acyl chlorides. The phenyl substituents on the skeletons of the dilithiobutadiene derivatives played an essential role in the reaction pattern. Interestingly, when anhydrides were used, normal alkyl-substituted 1,4-dilithio-1,3-dienes such as 1a could also react similarly to the phenyl-substituted 1b and 1c, affording analogous types of multiply substituted cyclopentadienes in excellent isolated yields. Esters were found to afford mixtures of products when treated with 1a–c. When 1,4-dilithio-1,3-dienes were treated with DMF, multiply substituted 2,4-diene-1,6-dials and/or 2,5-dihydrofuran derivatives were obtained in good yields, depending on the substitution patterns of the butadienyl skeletons.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2007)
Co-reporter:Congyang Wang Dr.;Chao Wang;Qifeng Wang;Zhihui Wang Dr.;Hui Sun;Xiangyu Guo Dr.
Chemistry - A European Journal 2007 Volume 13(Issue 22) pp:
Publication Date(Web):11 MAY 2007
DOI:10.1002/chem.200700028
Stereodefined multisubstituted 1-cyano- and 1,4-dicyano-1,3-butadiene derivatives were obtained in excellent yields of the isolated product from their corresponding monohalo- and dihalobutadienes and CuCN. This reaction proceeded with high stereoselectivity and retention of the stereochemistry of the starting halobutadienes. A study of the utility of the thus-obtained 1-cyano- and 1,4-dicyano-1,3-butadiene derivatives was demonstrated by their reactions with organolithium reagents. 2H-Pyrrole or iminocyclopentadiene derivatives were formed in high yields from 1-cyano-4-halo-1,3-butadienes and organolithium reagents. When 1,4-dicyano-1,3-butadienes were treated with organolithium reagents followed by trapping with electrophiles, a tandem process took place to afford 2H-pyrrolyl nitriles in excellent yields. Reduction of 1,4-dicyano-1,3-butadiene derivatives with LiAlH4 showed novel reaction patterns relative to normal nitriles.
Co-reporter:Junhui Liu, Wen-Xiong Zhang, Xiangyu Guo, Zhaomin Hou and Zhenfeng Xi
Organometallics 2007 Volume 26(Issue 27) pp:6812-6820
Publication Date(Web):December 5, 2007
DOI:10.1021/om701002k
2,5-Bis(alkynylsilyl)-1-zirconacyclopentadienes 3 were formed highly regio- and chemoselectively in excellent yield by the homocoupling reaction of 2 equiv of bis(alkynyl)silanes 1 mediated by a low-valent zirconocene species (Negishi reagent) generated in situ from Cp2ZrBu2. Single-crystal X-ray structural analysis of 2,5-bis(phenylalkynyldimethylsilyl)-1-zirconacyclopentadiene (3a) revealed a sandwich-type conformation. Hydrolysis or halogenation of these zirconacyclopentadienes 3 afforded multisubstituted stereodefined Si-bridged conjugated systems. Skeletal rearrangement of these 2,5-bis(alkynylsilyl)-1-zirconacyclopentadienes with aromatic substituents afforded zirconacyclohexadiene−silacyclobutene fused ring compounds 4. Using these zirconacyclopentadienes 3 as starting reactive organometallic reagents, interesting cyclic compounds such as bis(alkynylsilyl)benzene derivatives and bis(alkynylsilyl)thiophene derivatives could be prepared in high yield. Further applications of these alkynylsilyl-substituted compounds for more complicated products and functional materials can be expected.
Co-reporter:Jiang Lu, Guoliang Mao, Wenxiong Zhang and Zhenfeng Xi
Chemical Communications 2005 (Issue 38) pp:4848-4850
Publication Date(Web):05 Sep 2005
DOI:10.1039/B509142J
Multiply-substituted iminocyclopentadienes were formed from Lewis acid-promoted reactions of zirconacyclopentadienes and isocyanates via a one-pot three-component coupling process; the CO double bond of the RNCO moiety in the isocyanate was cleaved, and the isocyanates behaved formally as a one-carbon unit with Lewis acid-dependent and substituent-dependent reactions being realized.
Co-reporter:Tao Yu;Xiaohua Sun;Congyang Wang;Liang Deng Dr.
Chemistry - A European Journal 2005 Volume 11(Issue 6) pp:
Publication Date(Web):26 JAN 2005
DOI:10.1002/chem.200400697
Bis(alkynyl)silanes react with low valent zirconocene species to afford zirconacyclobutene intermediates. These in situ generated reactive organometallic intermediates can react with alkynes, ketones, aldehydes, and isocyanates by means of a novel skeletal rearrangement. When a zirconacyclobutene intermediate was treated with an alkyne, an α-alkynylsilyl zirconacyclopentadiene was formed. Addition of dimethyl acetylenedicarboxylate (DMAD) and CuCl resulted in one-pot formation of an alkynylsilyl–benzene derivative from three different alkynes. At a higher temperature, the α-alkynylsilylzirconacyclopentadiene was transformed by means of an intramolecular skeletal rearrangement to a zirconacyclohexadiene–silacyclobutene fused-ring compound, which reacted with DMAD in the presence of CuCl affording the same alkynylsilyl–benzene derivative. When treated with a ketone, an aldehdye, or an isocyanate, the zirconocyclobutene intermediate also underwent the above-mentioned skeletal rearrangement, generating zirconocene-mediated cross-coupling products.
Co-reporter:Zhenfeng Xi
European Journal of Organic Chemistry 2004 Volume 2004(Issue 13) pp:
Publication Date(Web):5 MAY 2004
DOI:10.1002/ejoc.200400033
The organolithium compounds, 1,4-dilithio-1,3-diene and 1-lithio-1,3-diene derivatives, show unprecedented reaction patterns with organic substrates. The 1,4-dilithio-1,3-dienes react with aldehydes or ketones via deoxygenation of the C=O moieties to afford cyclopentadiene derivatives; pyridine derivatives are obtained in excellent yields when these organolithium reagents are treated with nitriles. In addition to novel reaction patterns, the experimental results have demonstrated that both the mono- and the dilithio reagents are synthetically useful building blocks. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2004)
Co-reporter:Hongyun Fang;Guotao Li;Guoliang Mao Dr.
Chemistry - A European Journal 2004 Volume 10(Issue 14) pp:
Publication Date(Web):26 MAY 2004
DOI:10.1002/chem.200400025
1,4-Dilithiobutadiene derivatives 1, 1,4-bis(bromomagnesio)butadiene derivatives 2 and metallacyclic (1,3-butadiene-1,4-diyl)magnesium reagents 3 were prepared and their reactions with ketones, aldehydes, and PhNO were investigated. Multiply substituted cyclopentadienes and N-Ph pyrroles were formed by unprecedented reaction conditions. The carbonyl group of aldehydes and ketones was deoxygenated during the reaction and behaved formally as a one-carbon unit; the NO moiety of PhNO was cleaved to afford N-Ph pyrrole derivatives. Furthermore, different reactivities among these three types of reagents 1, 2 and 3 were revealed. The 1,4-dilithium reagents 1 readily reacted with both aldehydes and ketones; the 1,4-dimagnesium reagents 2 reacted with aldehydes, but not ketones; the metallacyclopentadiene reagents of magnesium 3 showed higher reactivity and did react with ketones.
Co-reporter:Changjia Zhao, Tao Yu and Zhenfeng Xi
Chemical Communications 2002 (Issue 2) pp:142-143
Publication Date(Web):09 Jan 2002
DOI:10.1039/B109761J
Zirconacyclopentenes generated in situ from alkynes and
ethylene undergo selective reaction with aldehydes to afford homoallylketones
in yields exceeding 70% in the presence of AlCl3.
Co-reporter:Changjia Zhao;Pixu Li;Xiaoyu Cao Dr.
Chemistry - A European Journal 2002 Volume 8(Issue 18) pp:
Publication Date(Web):16 SEP 2002
DOI:10.1002/1521-3765(20020916)8:18<4292::AID-CHEM4292>3.0.CO;2-G
One-pot procedures for the preparation of highly substituted indenes, tetrahydroindenes, and cyclopentadienes have been developed by using a combination of zirconocene-mediated CC-bond-forming reactions with Lewis acid mediated activation of carbonyl groups. The carbonyl groups of aldehydes were deoxygenated in the reaction and behaved formally as a one-carbon unit. A variety of Lewis acids were checked and showed different reactivities in this reaction.
Co-reporter:Changjia Zhao;Pixu Li;Xiaoyu Cao Dr.
Chemistry - A European Journal 2002 Volume 8(Issue 20) pp:
Publication Date(Web):14 OCT 2002
DOI:10.1002/1521-3765(20021018)8:20<4573::AID-CHEM4573>3.0.CO;2-L
Co-reporter:Zhenfeng Xi Dr.;Qiuling Song;Jinglong Chen;Hairong Guan;Pixu Li
Angewandte Chemie 2001 Volume 113(Issue 10) pp:
Publication Date(Web):15 MAY 2001
DOI:10.1002/1521-3757(20010518)113:10<1967::AID-ANGE1967>3.0.CO;2-S
Die Desoxygenierung der C=O-Funktion von Aldehyden und Ketonen mit Dialkenyllithiumverbindungen führt, unter Bildung einer neuen C(sp3)-C(sp2)-Bindung, zu hochsubstituierten Derivaten des Cyclopentadiens [Gl. (1)].
Co-reporter:Zhenfeng Xi Dr.;Pixu Li
Angewandte Chemie 2000 Volume 112(Issue 16) pp:
Publication Date(Web):11 AUG 2000
DOI:10.1002/1521-3757(20000818)112:16<3057::AID-ANGE3057>3.0.CO;2-Y
Co-reporter:Shaoguang Zhang ; Wen-Xiong Zhang ; Jing Zhao
Journal of the American Chemical Society () pp:
Publication Date(Web):
DOI:10.1021/ja1074932
The t-BuCN-stabilized zirconacyclopropene−azasilacyclopentadiene complexes 2 are generated in situ in high yields from the Si-tethered diynes, Cp2Zr(II) species, and 2 equiv of t-BuCN via a coordination-induced Zr−C/Si−C bond cleavage and reorganization. Complexes 2 have been demonstrated to be synthetically very useful. A variety of novel Zr/Si organo-bimetallic compounds and Si/N heterocyclic compounds, such as azasilacyclopentadienes, azasilacyclohexadienes, and allenylazazirconacycles, are obtained in high yields when complexes 2 are treated with ketones, carbodiimides, alkynes, elemental sulfur (S8), acid chlorides, or nitriles. In this chemistry, t-BuCN behaves as both an initiator and a brake/release handle to initiate and control the reaction process.
Co-reporter:Shaoguang Zhang, Ming Zhan, Qifeng Wang, Chao Wang, Wen-Xiong Zhang and Zhenfeng Xi
Inorganic Chemistry Frontiers 2014 - vol. 1(Issue 2) pp:NaN134-134
Publication Date(Web):2014/01/28
DOI:10.1039/C3QO00019B
Co2(CO)8-mediated cyclodimerization of 1,2,3,4-tetrasubstituted 1,4-dilithio-1,3-butadienes readily afforded octa-substituted semibullvalenes. The X-ray crystal structure of 1,2,5,6-tetraethyl-3,4,7,8-tetraphenyl semibullvalene was determined to show an unsymmetrical, localized structure, which is in sharp contrast with the C2 symmetrical structure of its tetramethyl analogue. In addition, this Co2(CO)8-mediated cyclodimerization was found to be dependent on the substitution pattern of 1,4-dilithio-1,3-butadienes. The 1,4-diphenyl dilithio compound gave its corresponding cyclooctatetraene derivative, while the dilithio reagent without substituents at its 1,4-positions afforded a cyclopentadienone [4 + 2] dimer.
Co-reporter:Yun Liang, Weizhi Geng, Junnian Wei, Kunbing Ouyang and Zhenfeng Xi
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 8) pp:NaN1542-1542
Publication Date(Web):2012/01/05
DOI:10.1039/C2OB06941E
The first transition-metal-catalyzed activation of silyl C(sp3)–H bond was realized and synthetically applied. A variety of organic skeletons substituted with SiMe3 groups could undergo the Pd-catalyzed intramolecular coupling reaction, resulting in an unprecedented synthetic method for yielding six-membered silacycles. It was found that the adjacent Si atom played an essential role for the activation of the C(sp3)–H bond of the SiMe3 group; no activation reaction of the C(sp3)–H bond of the CMe3 group took place under the same reaction conditions.
Co-reporter:Qifeng Wang, Lantao Liu, Chen Lin, Hui Sun, Wen-Xiong Zhang and Zhenfeng Xi
Dalton Transactions 2009(Issue 47) pp:NaN10435-10435
Publication Date(Web):2009/10/08
DOI:10.1039/B918679B
Hydride-induced cleavage of the C–O bond in the pyran ring followed by ring closure of unsaturated acylmetalates and reductive elimination was achieved.
Co-reporter:Congyang Wang and Zhenfeng Xi
Chemical Society Reviews 2007 - vol. 36(Issue 9) pp:NaN1406-1406
Publication Date(Web):2007/03/28
DOI:10.1039/B608694M
Transition metal-mediated or -catalyzed carbon–carbon bond or carbon–heteroatom bond forming reactions are among the most powerful tools in organic synthesis. In addition, Lewis acid-mediated or -catalyzed organic transformations are widely used. In this context, an effective combination of these two powerful protocols or an efficient cooperation of transition metals with Lewis acids should open a new era in synthetic chemistry. This tutorial review summarizes representative synthetic methodologies developed in the past decades employing this co-operative effect.
Co-reporter:Qian Luo, Chao Wang, Yuexing Li, Kunbing Ouyang, Li Gu, Masanobu Uchiyama and Zhenfeng Xi
Chemical Science (2010-Present) 2011 - vol. 2(Issue 11) pp:NaN2274-2274
Publication Date(Web):2011/08/26
DOI:10.1039/C1SC00356A
An efficient and specific cleavage of the endo-C(sp2)-Si bond of silole rings was developed. The stable silole ring was effectively opened in an unexpected regioselectivity by using an AcOH/ROH system, in which the AcOH behaved as a catalyst and the ROH as a nucleophile. Depending on the nature of the substituents at the 2- and 5-positions of the silole ring, the cooperative effect of the AcOH/ROH system exclusively cleaved one of the two endo-C-Si bonds to afford silylbutadiene derivatives.
Co-reporter:Fei Zhao, Chao Wang, Lantao Liu, Wen-Xiong Zhang and Zhenfeng Xi
Chemical Communications 2009(Issue 43) pp:NaN6571-6571
Publication Date(Web):2009/09/11
DOI:10.1039/B914619A
Mediated by AlCl3, substituted spiro[4.5]deca-tetraenes underwent novel and selective skeletal rearrangement to generate indene derivatives in high to excellent yields.
Co-reporter:Chao Wang, Liang Deng, Jun Yan, Hailin Wang, Qian Luo, Wen-Xiong Zhang and Zhenfeng Xi
Chemical Communications 2009(Issue 29) pp:NaN4416-4416
Publication Date(Web):2009/06/11
DOI:10.1039/B908803B
Selective cleavage of both active and inert C–C bonds in substituted allylcyclopropanes was realized via a novel ligand-controlled approach using zirconocene(II)-1-butene or zirconocene(II)-benzyne complexes.
Co-reporter:Hui-jun Zhang, Junnian Wei, Fei Zhao, Yun Liang, Zitao Wang and Zhenfeng Xi
Chemical Communications 2010 - vol. 46(Issue 39) pp:NaN7441-7441
Publication Date(Web):2010/09/06
DOI:10.1039/C0CC02380A
Dibenzo[a,c]cyclooctatetraenes were selectively generated in good yields from the palladium-catalyzed [4+4] cyclic homocoupling of borylvinyl iodobenzene derivatives, which were prepared via the selective Suzuki–Miyaura cross-coupling of the gem-diboryl reagents with iodo compounds.
Co-reporter:Fei Zhao, Shaoguang Zhang and Zhenfeng Xi
Chemical Communications 2011 - vol. 47(Issue 15) pp:NaN4357-4357
Publication Date(Web):2011/02/21
DOI:10.1039/C0CC05665K
Silyl-substituted 1,3-butadienes are useful building blocks and are readily applied in several types of reactions such as Diels–Alder reaction, ene reaction and allylation. They can also participate in different tandem reactions such as Diels–Alder/allylation, ene/allylation, ene/allylation/Diels–Alder reaction, ene/allylation/ene reaction and ene/allylation/Diels–Alder/allylation reaction. This feature article reviews the synthesis of silyl-substituted 1,3-butadienes, and their applications in the reaction types mentioned above, involving a tandem Diels–Alder/ene/allylation process. This article also introduces some reactions of alkenylsilanes and allylsilanes for comparison and discussion about the tandem reaction. The tandem reactions described in this article are a powerful tool to construct complicated multicyclic compounds with high selectivity and high efficiency.
Co-reporter:Junnian Wei, Yongliang Zhang, Wen-Xiong Zhang and Zhenfeng Xi
Inorganic Chemistry Frontiers 2014 - vol. 1(Issue 8) pp:NaN987-987
Publication Date(Web):2014/08/08
DOI:10.1039/C4QO00191E
Selective I/Mg exchange reactions of 1,4-diiodo-1,3-dienes and o-iodo-2-(2-iodovinyl)benzenes were achieved viaiPrMgCl·LiCl. Various 1-iodo-4-MgCl-1,3-dienes and 1-iodovinyl phenylmagnesium chlorides were thus efficiently generated and synthetically applied to afford new C–C bonds via reactions of the alkenyl or aryl C–MgCl bonds with different electrophiles. Useful conjugated compounds including polysubstituted benzenes, naphthalenes and phosphine compounds could be synthesized readily via further applications of the remaining alkenyl C–I bonds and subsequent cyclization reactions.
Co-reporter:Wei Hao, Han Wang, Patrick J. Walsh and Zhenfeng Xi
Inorganic Chemistry Frontiers 2015 - vol. 2(Issue 9) pp:NaN1084-1084
Publication Date(Web):2015/07/14
DOI:10.1039/C5QO00197H
The combination of Pd(π-allyl)Cp and PPh3 was found to generate an efficient catalyst for the formation of indole-containing alkyl iodides from ortho-amino iodobenzenes and both aromatic and aliphatic alkynes. A unique feature of this reaction is a palladium-promoted C(sp3)–I bond formation via reductive elimination. This catalytic process was found to be very sensitive to the size and equivalents of phosphine ligands and the nature of the base.
Co-reporter:Shaoguang Zhang, Ming Zhan, Qian Luo, Wen-Xiong Zhang and Zhenfeng Xi
Chemical Communications 2013 - vol. 49(Issue 55) pp:NaN6148-6148
Publication Date(Web):2013/05/20
DOI:10.1039/C3CC43061H
2,6-Diazasemibullvalenes (NSBVs) readily reacted with oxygen under mild conditions, affording Δ1-bipyrrolinones via C–N bond cleavage and C–H bond oxidation. Pyrrolino[3,2-b]pyrrolinone derivatives were efficiently generated when NSBVs were treated with N-oxides in the presence of Lewis acids. Δ1-Bipyrrolinones thus obtained could be readily transformed into other heterocycles.
Co-reporter:Qian Luo, Chao Wang, Wen-Xiong Zhang and Zhenfeng Xi
Chemical Communications 2008(Issue 13) pp:
Publication Date(Web):
DOI:10.1039/B719007G
Co-reporter:Chao Wang and Zhenfeng Xi
Chemical Communications 2007(Issue 48) pp:NaN5133-5133
Publication Date(Web):2007/10/19
DOI:10.1039/B709839A
Substituted cyclooctatetraenes are a class of interesting and important compounds both theoretically and synthetically. Since Reppe first discovered the Ni-catalyzed tetramerization of ethyne affording cyclooctatetraene in 1948, transition metal mediated synthesis of this type of compounds has become a primary methodology. In this Feature Article, based on our own recent results and other groups' related reports, we describe major achievements on transition metal mediated or catalyzed synthetic methods for substituted cyclooctatetraenes, with focus on reaction patterns, mechanisms, and structural diversity of products.
Co-reporter:Wen-Xiong Zhang
Inorganic Chemistry Frontiers 2014 - vol. 1(Issue 9) pp:
Publication Date(Web):2014/10/07
DOI:10.1039/C4QO00231H
Metal-mediated organic reactions have become one of the great frontiers of organic synthesis. These processes usually involve multiple transient or unobservable reactive intermediates. The isolation and study of these reactive organometallic intermediates would not only lead to a better understanding of their reactivity, but also guide the discovery of new reactions, culminating in the advancement of novel synthetic methods. Herein we provide a perspective of a research strategy which focuses on organometallic intermediate-based organic synthesis. Specifically, we highlight the applications of our organo-di-lithio reagents to the synthesis of a diverse range of reactive intermediates and small molecules. A series of organo-di-lithio reagents has been synthesized, isolated, and applied in organic and organometallic synthesis. These isolated dilithio reagents can be transformed into a variety of organo-di-metallic or metallacyclic compounds via transmetallation, which in turn provides access to organic and organometallic compounds with interesting and useful properties. Selected examples by other research groups are also briefly mentioned, in which reactive organometallic intermediates are isolated for a better understanding of reaction mechanisms, and in some cases are further applied to synthesis.
Co-reporter:Qiaoshu Hu, Chao Wang, Dongzhen Li and Zhenfeng Xi
Organic & Biomolecular Chemistry 2007 - vol. 5(Issue 13) pp:NaN2118-2118
Publication Date(Web):2007/05/31
DOI:10.1039/B705110G
Stable enols were synthesized from the reaction of (1Z,3Z)-1,4-dilithio-1,3-dienes with acid chlorides and structurally characterized by single-crystal X-ray analysis. These stable enols were formed by a novel de-aromatization/Michael addition/re-aromatization domino process.





















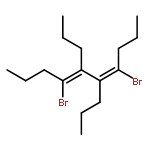



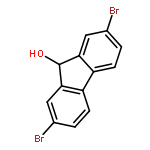

![Tricyclo[4.2.0.02,5]octa-3,7-diene, 1,2,3,4,5,6,7,8-octapropyl-](http://img.cochemist.com/ccimg/643800/643744-52-9.png)
![Tricyclo[4.2.0.02,5]octa-3,7-diene, 1,2,3,4,5,6,7,8-octapropyl-](http://img.cochemist.com/ccimg/643800/643744-52-9_b.png)
![Benzenemethanamine, N-[[2,6-bis(1-methylethyl)phenyl]carbonimidoyl]-](http://img.cochemist.com/ccimg/491900/491869-07-9.png)
![Benzenemethanamine, N-[[2,6-bis(1-methylethyl)phenyl]carbonimidoyl]-](http://img.cochemist.com/ccimg/491900/491869-07-9_b.png)


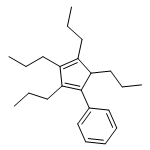






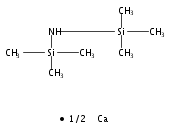








![Silane, trimethyl[[9-(trimethylsilyl)-9H-fluoren-9-yl]oxy]-](http://img.cochemist.com/ccimg/61000/60991-98-2.png)
![Silane, trimethyl[[9-(trimethylsilyl)-9H-fluoren-9-yl]oxy]-](http://img.cochemist.com/ccimg/61000/60991-98-2_b.png)

![Silane, trimethyl[[9-(2-propenyl)-9H-fluoren-9-yl]oxy]-](http://img.cochemist.com/ccimg/51600/51519-04-1.png)
![Silane, trimethyl[[9-(2-propenyl)-9H-fluoren-9-yl]oxy]-](http://img.cochemist.com/ccimg/51600/51519-04-1_b.png)













![4,7,13,16,21,24-Hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane](http://img.cochemist.com/ccimg/24000/23978-09-8.png)
![4,7,13,16,21,24-Hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane](http://img.cochemist.com/ccimg/24000/23978-09-8_b.png)





![Cyclopropa[cd]pentalene, 2a,2b,4a,4b-tetrahydro-](/data/chemimg/945100/6909-37-1.png)
![Cyclopropa[cd]pentalene, 2a,2b,4a,4b-tetrahydro-](/data/chemimg/945100/6909-37-1_b.png)






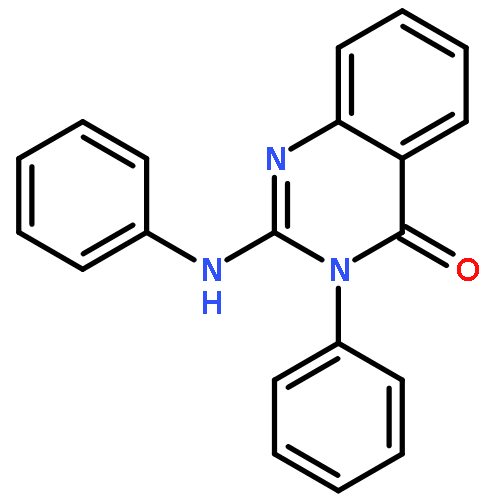






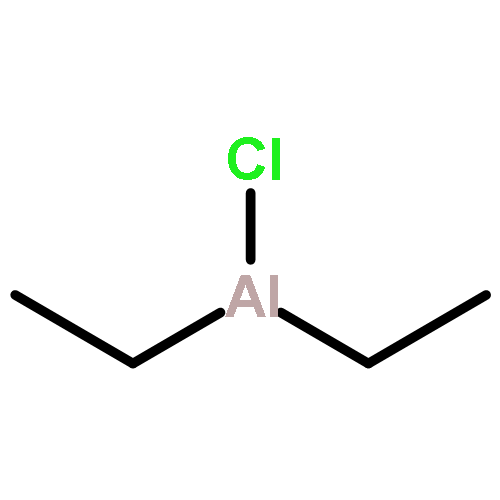
![Phosphine, diphenyl[2-(2,3,4,5-tetraethyl-2,4-cyclopentadien-1-yl)phenyl]-](/data/chemimg/3726300/1415808-61-5.png)
![Phosphine, diphenyl[2-(2,3,4,5-tetraethyl-2,4-cyclopentadien-1-yl)phenyl]-](/data/chemimg/3726300/1415808-61-5_b.png)


![Lithium, μ-[1,1'-biphenyl]-2,2'-diyldi-](/data/chemimg/1753500/16291-32-0.png)
![Lithium, μ-[1,1'-biphenyl]-2,2'-diyldi-](/data/chemimg/1753500/16291-32-0_b.png)








8:18<4292::AID-CHEM4292>3.0.CO;2-G/asset/image_n/ntrans.gif?v=1&t=iwlqqxvv&s=859c7c4367ea4d677d7a3cbd01748026dfdfbd55){kind=link}