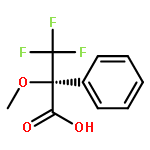
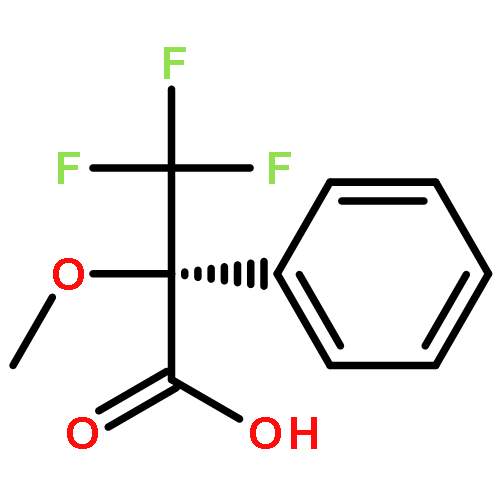
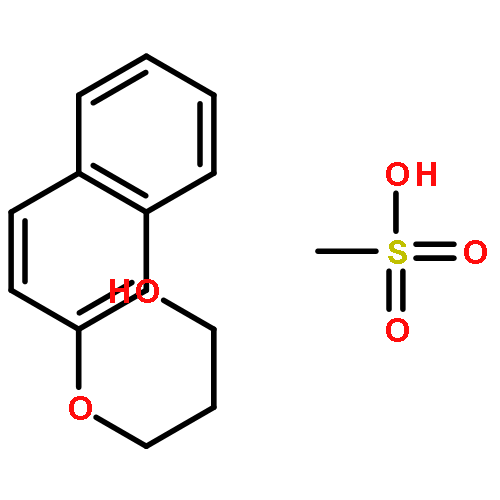
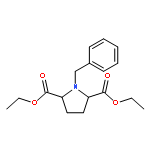
Co-reporter:Philip A. Waghorn, Mark R. Jackson, Veronique Gouverneur, Katherine A. Vallis
European Journal of Medicinal Chemistry 2017 Volume 125() pp:117-129
Publication Date(Web):5 January 2017
DOI:10.1016/j.ejmech.2016.09.028
•123I-radiolabeled small molecule inhibitors of telomerase were synthesized.•Synchronous telomerase inhibition and radionuclide therapy was achieved.•123I-MST-312 caused cytotoxicity in telomerase positive cancer cells.The expression of telomerase in approximately 85% of cancers and its absence in the majority of normal cells makes it an attractive target for cancer therapy. However the lag period between initiation of telomerase inhibition and growth arrest makes direct inhibition alone an insufficient method of treatment. However, telomerase inhibition has been shown to enhance cancer cell radiosensitivity. To investigate the strategy of simultaneously inhibiting telomerase while delivering targeted radionuclide therapy to cancer cells, 123I-radiolabeled inhibitors of telomerase were synthesized and their effects on cancer cell survival studied. An 123I-labeled analogue of the telomerase inhibitor MST-312 inhibited telomerase with an IC50 of 1.58 μM (MST-312 IC50: 0.23 μM). Clonogenic assays showed a dose dependant effect of 123I-MST-312 on cell survival in a telomerase positive cell line, MDA-MB-435.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Raul Pereira;Lukas Pfeifer;Jean Fournier;Véronique Gouverneur;Ján Cvengroš
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 3) pp:628-633
Publication Date(Web):2017/01/18
DOI:10.1039/C6OB02359B
The typically planar amide when incorporated into bicyclic systems can undergo a significant distortion from planarity resulting in physical properties and reactivity that deviate from classical amide behaviour. Herein, we report a succinct protocol that utilises potassium permanganate to selectively α-oxygenate the benzylic position of ethano-Tröger's base derivatives to yield a new class of twisted bisamides. Additionally, we report the first synthesis of an ethano-Tröger's base derivative bearing substituents in the positions ortho to the nitrogen atoms.
Co-reporter:Paolo Ricci;Tanatorn Khotavivattana;Lukas Pfeifer;Maurice Médebielle;John Richard Morphy;Véronique Gouverneur
Chemical Science (2010-Present) 2017 vol. 8(Issue 2) pp:1195-1199
Publication Date(Web):2017/01/30
DOI:10.1039/C6SC02790C
Alkenes substituted with a thiourea undergo C–CF3 followed by intramolecular C–S bond formation with the Togni reagent and trifluoroacetic acid (TFA) at room temperature; thiols and thioamides are not suitable S-sources for this reaction. This anti-addition process involves a CF3 radical, and affords CF3-substituted thiazolines and thiazines for medicinal applications. A metal or photoredox catalyst is not required as the thiourea acts as a reductant, as well as serving as an S-source capable of adding to a C-centered radical. Mechanistic work comparing the reactivity of thiourea, urea, thioamide and thiol in the context of alkene trifluoromethylation demonstrates that in this series, the thiourea is unique for its ability to release CF3 radical from the Togni reagent, and to orchestrate trifluoromethylation followed by S-cyclization with both activated and unactivated alkenes.
Co-reporter:Sean Preshlock, Matthew Tredwell, and Véronique Gouverneur
Chemical Reviews 2016 Volume 116(Issue 2) pp:719
Publication Date(Web):January 11, 2016
DOI:10.1021/acs.chemrev.5b00493
Diverse radiochemistry is an essential component of nuclear medicine; this includes imaging techniques such as positron emission tomography (PET). As such, PET can track diseases at an early stage of development, help patient care planning through personalized medicine and support drug discovery programs. Fluorine-18 is the most frequently used radioisotope in PET radiopharmaceuticals for both clinical and preclinical research. Its physical and nuclear characteristics (97% β+ decay, 109.8 min half-life, 635 keV positron energy) and high specific activity make it an attractive nuclide for labeling and molecular imaging. Arenes and heteroarenes are privileged candidates for 18F-incorporation as they are metabolically robust and therefore widely used by medicinal chemists and radiochemists alike. For many years, the range of (hetero)arenes amenable to 18F-fluorination was limited by the lack of chemically diverse precursors, and of radiochemical methods allowing 18F-incorporation in high selectivity and efficiency (radiochemical yield and purity, specific activity, and radio-scalability). The appearance of late-stage fluorination reactions catalyzed by transition metal or small organic molecules (organocatalysis) has encouraged much research on the use of these activation manifolds for 18F-fluorination. In this piece, we review all of the reactions known to date to install the 18F substituent and other key 18F-motifs (e.g., CF3, CHF2, OCF3, SCF3, OCHF2) of medicinal relevance onto (hetero)arenes. The field has changed significantly in the past five years, and the current trend suggests that the radiochemical space available for PET applications will expand rapidly in the near future.
Co-reporter:Lukas Pfeifer, Keary M. Engle, George W. Pidgeon, Hazel A. Sparkes, Amber L. Thompson, John M. Brown, and Véronique Gouverneur
Journal of the American Chemical Society 2016 Volume 138(Issue 40) pp:13314-13325
Publication Date(Web):September 8, 2016
DOI:10.1021/jacs.6b07501
Hydrogen bonding with fluoride is a key interaction encountered when analyzing the mode of action of 5′-fluoro-5′-deoxyadenosine synthase, the only known enzyme capable of catalyzing the formation of a C–F bond from F–. Further understanding of the effect of hydrogen bonding on the structure and reactivity of complexed fluoride is therefore important for catalysis and numerous other applications, such as anion supramolecular chemistry. Herein we disclose a detailed study examining the structure of 18 novel urea–fluoride complexes in the solid state, by X-ray and neutron diffraction, and in solution phase and explore the reactivity of these complexes as a fluoride source in SN2 chemistry. Experimental data show that the structure, coordination strength, and reactivity of the urea–fluoride complexes are tunable by modifying substituents on the urea receptor. Hammett analysis of aryl groups on the urea indicates that fluoride binding is dependent on σp and σm parameters with stronger binding being observed for electron-deficient urea ligands. For the first time, defined urea–fluoride complexes are used as fluoride-binding reagents for the nucleophilic substitution of a model alkyl bromide. The reaction is slower in comparison with known alcohol–fluoride complexes, but SN2 is largely favored over E2, at a ratio surpassing all hydrogen-bonded complexes documented in the literature for the model alkyl bromide employed. Increased second-order rate constants at higher dilution support the hypothesis that the reactive species is a 1:1 urea–fluoride complex of type [UF]− (U = urea) resulting from partial dissociation of the parent compound [U2F]−. The dissociation processes can be quantified through a combination of UV and NMR assays, including DOSY and HOESY analyses that illuminate the complexation state and H-bonding in solution.
Co-reporter:Faye Buckingham and Véronique Gouverneur
Chemical Science 2016 vol. 7(Issue 3) pp:1645-1652
Publication Date(Web):17 Dec 2015
DOI:10.1039/C5SC04229A
Positron emission tomography (PET) is becoming more frequently used by medicinal chemists to facilitate the selection of the most promising lead compounds for further evaluation. For PET, this entails the preparation of 11C- or 18F-labeled drugs or radioligands. With the importance of chirality and fluorine substitution in drug development, chemists can be faced with the challenge of preparing enantiopure molecules featuring the 18F-tag on a stereogenic carbon. Asymmetric 18F-fluorination is an emerging field of research that provides an alternative to resolution or conventional SN2-based radiochemistry. To date, both transition metal complexes and organomediators have been successfully employed for 18F-incorporation at a stereogenic carbon.
Co-reporter:Sean Preshlock, Samuel Calderwood, Stefan Verhoog, Matthew Tredwell, Mickael Huiban, Antje Hienzsch, Stefan Gruber, Thomas C. Wilson, Nicholas J. Taylor, Thomas Cailly, Michael Schedler, Thomas Lee Collier, Jan Passchier, René Smits, Jan Mollitor, Alexander Hoepping, Marco Mueller, Christophe Genicot, Joël Mercier and Véronique Gouverneur
Chemical Communications 2016 vol. 52(Issue 54) pp:8361-8364
Publication Date(Web):23 May 2016
DOI:10.1039/C6CC03295H
[18F]FMTEB, [18F]FPEB, [18F]flumazenil, [18F]DAA1106, [18F]MFBG, [18F]FDOPA, [18F]FMT and [18F]FDA are prepared from the corresponding arylboronic esters and [18F]KF/K222 in the presence of Cu(OTf)2py4. The method was successfully applied using three radiosynthetic platforms, and up to 26 GBq of non-carrier added starting activity of 18F-fluoride.
Co-reporter:Raul Pereira, Jamie Wolstenhulme, Graham Sandford, Timothy D. W. Claridge, Véronique Gouverneur and Ján Cvengroš
Chemical Communications 2016 vol. 52(Issue 8) pp:1606-1609
Publication Date(Web):25 Nov 2015
DOI:10.1039/C5CC08375C
Methylation of 2,8-dimethyl-6H,12H-5,11-ethanodibenzo[b,f][1,5]-diazocine (ethano-Tröger's base) with methyl iodide followed by ion metathesis and fluorination with N-fluoro-2,3,4,5,6-pentachloropyridinium triflate affords a new electrophilic N–F reagent, that is more reactive than Selectfluor. 2D 19F–15N HMQC experiments provide 1JNF coupling constants which are diagnostic for the N–F functional group.
Co-reporter:Stephen Hyde;Dr. Janis Veliks;Dr. Benoît Liégault;Dr. David Grassi;Dr. Marc Taillefer; Véronique Gouverneur
Angewandte Chemie International Edition 2016 Volume 55( Issue 11) pp:3785-3789
Publication Date(Web):
DOI:10.1002/anie.201511954
Abstract
Copper-catalyzed Si−H, B−H, P−H, S−H, and N−H insertion reactions of 2,2,2-trifluoro-1-diazoethane and 1-aryl 2,2,2-trifluorodiazoethanes generated a large number of new fluorine-containing chemical entities for medicinal chemists. With selected Si−H and B−H insertion reactions, we demonstrate successful extension to asymmetric catalysis.
Co-reporter:Stephen Hyde;Dr. Janis Veliks;Dr. Benoît Liégault;Dr. David Grassi;Dr. Marc Taillefer; Véronique Gouverneur
Angewandte Chemie 2016 Volume 128( Issue 11) pp:3849-3853
Publication Date(Web):
DOI:10.1002/ange.201511954
Abstract
Copper-catalyzed Si−H, B−H, P−H, S−H, and N−H insertion reactions of 2,2,2-trifluoro-1-diazoethane and 1-aryl 2,2,2-trifluorodiazoethanes generated a large number of new fluorine-containing chemical entities for medicinal chemists. With selected Si−H and B−H insertion reactions, we demonstrate successful extension to asymmetric catalysis.
Co-reporter:Veronique Gouverneur and Konrad Seppelt
Chemical Reviews 2015 Volume 115(Issue 2) pp:563
Publication Date(Web):January 28, 2015
DOI:10.1021/cr500686k
Co-reporter:Keary M. Engle, Lukas Pfeifer, George W. Pidgeon, Guy T. Giuffredi, Amber L. Thompson, Robert S. Paton, John M. Brown and Véronique Gouverneur
Chemical Science 2015 vol. 6(Issue 9) pp:5293-5302
Publication Date(Web):22 Jun 2015
DOI:10.1039/C5SC01812A
The nucleophilic reactivity of fluoride ion is altered in the presence of hydrogen-bond donors, including alcohols. Relatively little is known about the coordination involved; to rectify this, the X-ray structures of fourteen novel fluoride–alcohol complexes with tetrabutylammonium as the counterion have been determined. The coordination number varies from two to four depending on the steric bulk of the alcohol and is closely linked to trends in reactivity. This diversity in coordination stoichiometry is unprecedented but significant, as it implies differences in the ability of the fluoride–alcohol complexes to dissociate in solution with release of a more active and/or selective fluoride source.
Co-reporter:Tanatorn Khotavivattana;Stefan Verhoog;Dr. Matthew Tredwell;Lukas Pfeifer;Samuel Calderwood;Dr. Katherine Wheelhouse;Dr. Thomas LeeCollier; Véronique Gouverneur
Angewandte Chemie International Edition 2015 Volume 54( Issue 34) pp:9991-9995
Publication Date(Web):
DOI:10.1002/anie.201504665
Abstract
We report that halogenophilic silver(I) triflate permits halogen exchange (halex) nucleophilic 18F-fluorination of aryl-OCHFCl, -OCF2Br and -SCF2Br precursors under mild conditions. This AgI-mediated process allows for the first time access to a range of 18F-labeled aryl-OCHF2, -OCF3 and -SCF3 derivatives, inclusive of [18F]riluzole. The 18F-labeling of these medicinally important motifs expands the radiochemical space available for PET applications.
Co-reporter:Faye Buckingham;Dr. Anna K. Kirjavainen;Dr. Sarita Forsback;Anna Krzyczmonik;Thomas Keller;Dr. Ian M. Newington;Dr. Matthias Glaser;Dr. Sajinder K. Luthra; Olof Solin; Véronique Gouverneur
Angewandte Chemie International Edition 2015 Volume 54( Issue 45) pp:13366-13369
Publication Date(Web):
DOI:10.1002/anie.201506035
Abstract
The first organomediated asymmetric 18F fluorination has been accomplished using a chiral imidazolidinone and [18F]N-fluorobenzenesulfonimide. The method provides access to enantioenriched 18F-labeled α-fluoroaldehydes (>90 % ee), which are versatile chiral 18F synthons for the synthesis of radiotracers. The utility of this process is demonstrated with the synthesis of the PET (positron emission tomography) tracer (2S,4S)-4-[18F]fluoroglutamic acid.
Co-reporter:Tanatorn Khotavivattana;Stefan Verhoog;Dr. Matthew Tredwell;Lukas Pfeifer;Samuel Calderwood;Dr. Katherine Wheelhouse;Dr. Thomas LeeCollier; Véronique Gouverneur
Angewandte Chemie 2015 Volume 127( Issue 34) pp:10129-10133
Publication Date(Web):
DOI:10.1002/ange.201504665
Abstract
We report that halogenophilic silver(I) triflate permits halogen exchange (halex) nucleophilic 18F-fluorination of aryl-OCHFCl, -OCF2Br and -SCF2Br precursors under mild conditions. This AgI-mediated process allows for the first time access to a range of 18F-labeled aryl-OCHF2, -OCF3 and -SCF3 derivatives, inclusive of [18F]riluzole. The 18F-labeling of these medicinally important motifs expands the radiochemical space available for PET applications.
Co-reporter:Faye Buckingham;Dr. Anna K. Kirjavainen;Dr. Sarita Forsback;Anna Krzyczmonik;Thomas Keller;Dr. Ian M. Newington;Dr. Matthias Glaser;Dr. Sajinder K. Luthra; Olof Solin; Véronique Gouverneur
Angewandte Chemie 2015 Volume 127( Issue 45) pp:13564-13567
Publication Date(Web):
DOI:10.1002/ange.201506035
Abstract
The first organomediated asymmetric 18F fluorination has been accomplished using a chiral imidazolidinone and [18F]N-fluorobenzenesulfonimide. The method provides access to enantioenriched 18F-labeled α-fluoroaldehydes (>90 % ee), which are versatile chiral 18F synthons for the synthesis of radiotracers. The utility of this process is demonstrated with the synthesis of the PET (positron emission tomography) tracer (2S,4S)-4-[18F]fluoroglutamic acid.
Co-reporter:Jamie R. Wolstenhulme and Véronique Gouverneur
Accounts of Chemical Research 2014 Volume 47(Issue 12) pp:3560
Publication Date(Web):November 7, 2014
DOI:10.1021/ar500282z
The vicinal fluorofunctionalization of alkenes is an attractive transformation that converts feedstock olefins into valuable cyclic fluorinated molecules for application in the pharmaceutical, agrochemical, medical, and material sectors. The challenges associated with asymmetric fluorocyclizations induced by F+ reagents are distinct from other types of halocyclizations. Processes initiated by the addition of an F+ reagent onto an alkene do not involve the reversible formation of bridged fluoronium ions but generate acyclic β-fluorocationic intermediates. This mechanistic feature implies that fluorocyclizations are not stereospecific. A discontinuity exists between the importance of this class of fluorocyclization and the activation modes currently available to implement successful catalysis. Progress toward fluorocyclization has been achieved by investing in neutral and cationic [NF] reagent development. The body of work on asymmetric fluorination using chiral cationic [NF]+ reagents prepared by fluorine transfer from the dicationic [NF]2+ reagent Selectfluor to quinuclidines, inspired the development of asymmetric F+-induced fluorocyclizations catalyzed by cinchona alkaloids; for catalysis, the use of N-fluorobenzenesulfonimide, which is less reactive than Selectfluor, ensures that the achiral F+ source remains unreactive toward the alkene. These organocatalyzed enantioselective fluorocyclizations can be applied to indoles to install the fluorine on a quaternary benzylic stereogenic carbon center and to afford fluorinated analogues of natural products featuring the hexahydropyrrolo[2,3-b]indole or the tetrahydro-2H-furo[2,3-b]indole skeleton. In an alternative approach, the poor solubility of dicationic Selectfluor bis(tetrafluoroborate) in nonpolar solvent was exploited with anionic phase transfer catalysis as the operating activation mode. Exchange of the tetrafluoroborate ions of Selectfluor with bulky lipophilic chiral anions (e.g., TRIP and derivatives) brings into solution the resulting chiral Selectfluor reagent, now capable of asymmetric fluorocyclization. This strategy is best applied to a subset of substrates bearing a nucleophilic pendent group (benzamide is best) capable of hydrogen bonding for association with the chiral phosphate catalyst. These contributions focused on fluoroheterocyclization involving either O- or N-nucleophiles. As for other halocyclizations, alkenes armed with π C-nucleophiles represent the most demanding class of substrates for asymmetric F+-induced electrophilic fluorination–cyclization. Successful implementation required the design of new chiral Selectfluor reagents featuring stereogenicity on the DABCO core. These reagents, accessible from chiral vicinal diamines, allowed the synthesis of unusual chiral fluorine-containing tetracyclic compounds, some composed of carbon, hydrogen, and fluorine exclusively. The challenges associated with F+-induced fluorocarbocyclizations prompted methodologists to consider chemistry where the Csp3–F bond formation event follows a catalyst-controlled cyclization. An exciting development built on in the area of transition metal π-cyclization of polyenes leading to cationic metal–alkyl intermediates. When intercepted by oxidative fluorodemetalation with a F+ source, the resulting products are complex polycyclic structures emerging from an overall catalytic cascade fluorocarbocyclization. Complementing F+-based reactions, examples of fluorocyclizations with fluoride in the presence of an oxidant were reported. Despite some exciting developments, the field of asymmetric fluorocyclizations is in its infancy and undoubtedly requires new activation modes, catalysts, as well as F+ and F– reagents to progress into general retrosynthetic approach toward enantioenriched fluorocycles. Numerous opportunities emerge, not least the use of a latent fluorine source as a means to minimize background fluorination.
Co-reporter:Dr. Enrico Emer;Lukas Pfeifer;Dr. John M. Brown ; Véronique Gouverneur
Angewandte Chemie 2014 Volume 126( Issue 16) pp:4265-4269
Publication Date(Web):
DOI:10.1002/ange.201310056
Abstract
This paper describes the hydrofluorination of alkenes through sequential H− and F+ addition under palladium catalysis. The reaction is cis specific, thus providing access to benzylic fluorides. The mechanism of this reaction involves an ionic pathway and is distinct from known hydrofluorinations involving radical intermediates. The first catalytic enantioselective hydrofluorination is also disclosed.
Co-reporter:Dr. Matthew Tredwell;Dr. Sean M. Preshlock;Nicholas J. Taylor;Dr. Stefan Gruber;Dr. Mickael Huiban;Dr. Jan Passchier;Dr. Joël Mercier;Dr. Christophe Génicot; Véronique Gouverneur
Angewandte Chemie 2014 Volume 126( Issue 30) pp:7885-7889
Publication Date(Web):
DOI:10.1002/ange.201404436
Abstract
Molecules labeled with fluorine-18 are used as radiotracers for positron emission tomography. An important challenge is the labeling of arenes not amenable to aromatic nucleophilic substitution (SNAr) with [18F]F−. In the ideal case, the 18F fluorination of these substrates would be performed through reaction of [18F]KF with shelf-stable readily available precursors using a broadly applicable method suitable for automation. Herein, we describe the realization of these requirements with the production of 18F arenes from pinacol-derived aryl boronic esters (arylBPin) upon treatment with [18F]KF/K222 and [Cu(OTf)2(py)4] (OTf=trifluoromethanesulfonate, py=pyridine). This method tolerates electron-poor and electron-rich arenes and various functional groups, and allows access to 6-[18F]fluoro-L-DOPA, 6-[18F]fluoro-m-tyrosine, and the translocator protein (TSPO) PET ligand [18F]DAA1106.
Co-reporter:Dr. Matthew Tredwell;Dr. Sean M. Preshlock;Nicholas J. Taylor;Dr. Stefan Gruber;Dr. Mickael Huiban;Dr. Jan Passchier;Dr. Joël Mercier;Dr. Christophe Génicot; Véronique Gouverneur
Angewandte Chemie 2014 Volume 126( Issue 30) pp:
Publication Date(Web):
DOI:10.1002/ange.201405437
Co-reporter:Dr. Enrico Emer;Lukas Pfeifer;Dr. John M. Brown ; Véronique Gouverneur
Angewandte Chemie International Edition 2014 Volume 53( Issue 16) pp:4181-4185
Publication Date(Web):
DOI:10.1002/anie.201310056
Abstract
This paper describes the hydrofluorination of alkenes through sequential H− and F+ addition under palladium catalysis. The reaction is cis specific, thus providing access to benzylic fluorides. The mechanism of this reaction involves an ionic pathway and is distinct from known hydrofluorinations involving radical intermediates. The first catalytic enantioselective hydrofluorination is also disclosed.
Co-reporter:Dr. Matthew Tredwell;Dr. Sean M. Preshlock;Nicholas J. Taylor;Dr. Stefan Gruber;Dr. Mickael Huiban;Dr. Jan Passchier;Dr. Joël Mercier;Dr. Christophe Génicot; Véronique Gouverneur
Angewandte Chemie International Edition 2014 Volume 53( Issue 30) pp:7751-7755
Publication Date(Web):
DOI:10.1002/anie.201404436
Abstract
Molecules labeled with fluorine-18 are used as radiotracers for positron emission tomography. An important challenge is the labeling of arenes not amenable to aromatic nucleophilic substitution (SNAr) with [18F]F−. In the ideal case, the 18F fluorination of these substrates would be performed through reaction of [18F]KF with shelf-stable readily available precursors using a broadly applicable method suitable for automation. Herein, we describe the realization of these requirements with the production of 18F arenes from pinacol-derived aryl boronic esters (arylBPin) upon treatment with [18F]KF/K222 and [Cu(OTf)2(py)4] (OTf=trifluoromethanesulfonate, py=pyridine). This method tolerates electron-poor and electron-rich arenes and various functional groups, and allows access to 6-[18F]fluoro-L-DOPA, 6-[18F]fluoro-m-tyrosine, and the translocator protein (TSPO) PET ligand [18F]DAA1106.
Co-reporter:Dr. Matthew Tredwell;Dr. Sean M. Preshlock;Nicholas J. Taylor;Dr. Stefan Gruber;Dr. Mickael Huiban;Dr. Jan Passchier;Dr. Joël Mercier;Dr. Christophe Génicot; Véronique Gouverneur
Angewandte Chemie International Edition 2014 Volume 53( Issue 30) pp:
Publication Date(Web):
DOI:10.1002/anie.201405437
Co-reporter:Satoshi Mizuta ; Stefan Verhoog ; Keary M. Engle ; Tanatorn Khotavivattana ; Miriam O’Duill ; Katherine Wheelhouse ; Gerasimos Rassias ; Maurice Médebielle ;Véronique Gouverneur
Journal of the American Chemical Society 2013 Volume 135(Issue 7) pp:2505-2508
Publication Date(Web):February 1, 2013
DOI:10.1021/ja401022x
A visible-light-mediated hydrotrifluoromethylation of unactivated alkenes that uses the Umemoto reagent as the CF3 source and MeOH as the reductant is disclosed. This effective transformation operates at room temperature in the presence of 5 mol % Ru(bpy)3Cl2; the process is characterized by its operational simplicity and functional group tolerance.
Co-reporter:Elena Benedetto, Matthew Tredwell, Charlotte Hollingworth, Tanatorn Khotavivattana, John M. Brown and Véronique Gouverneur
Chemical Science 2013 vol. 4(Issue 1) pp:89-96
Publication Date(Web):24 Oct 2012
DOI:10.1039/C2SC21789A
This paper describes a new catalytic method for the regio- and stereocontrolled fluorination of allylic carbonates. This transformation uses TBAF·4tBuOH as the fluoride source and [Ir(COD)Cl]2 as the catalyst; the most commonly used [Ir(COD)Cl]2/phosphoramidite system is ineffective. Synthetically, this reaction is characterized by a high degree of structural conservation in going from substrates to the products. The fluorination of (E)-allylic carbonates leading to linear (E)-allylic fluorides (l:b > 20:1, E:Z > 20:1) is unprecedented and a unique feature of fluoride as the nucleophile. The first examples of transition metal catalyzed fluorination affording (Z)-allyl fluorides (Z:E ratio >20:1) are disclosed along with the successful fluorination of branched, linear (E)- and (Z)-allyl carbonates with [18F] fluoride in the presence of [Ir(COD)Cl]2. 18O-Labeling of the reactant reveals internal return during the allylic ionization step, and pathways for effective intra- and intermolecular isotope exchange.
Co-reporter:Satoshi Mizuta, Keary M. Engle, Stefan Verhoog, Oscar Galicia-López, Miriam O’Duill, Maurice Médebielle, Katherine Wheelhouse, Gerasimos Rassias, Amber L. Thompson, and Véronique Gouverneur
Organic Letters 2013 Volume 15(Issue 6) pp:1250-1253
Publication Date(Web):March 6, 2013
DOI:10.1021/ol400184t
A new catalytic method to access allylic secondary CF3 products is described. These reactions use the visible light excited Ru(bpy)3Cl2·6H2O catalyst and the Togni or Umemoto reagent as the CF3 source. The photoredox catalytic manifold delivers enantioenriched allylic trifluoromethylated products not accessible under Cu(I) catalysis.
Co-reporter:Ida S. R. Stenhagen, Anna K. Kirjavainen, Sarita J. Forsback, Charlotte G. Jørgensen, Edward G. Robins, Sajinder K. Luthra, Olof Solin and Véronique Gouverneur
Chemical Communications 2013 vol. 49(Issue 14) pp:1386-1388
Publication Date(Web):03 Jan 2013
DOI:10.1039/C2CC38646A
The Ag-mediated electrophilic [18F]fluorination of an arylboronic ester is reported. This new radiochemical transformation uses [18F]selectfluor bis(triflate) in acetone. The process gave 6-[18F]fluoro-L-DOPA with a RCY of 19 ± 12% and a specific activity of 2.6 ± 0.3 GBq μmol−1.
Co-reporter:Satoshi Mizuta, Ida S. R. Stenhagen, Miriam O’Duill, Jamie Wolstenhulme, Anna K. Kirjavainen, Sarita J. Forsback, Matthew Tredwell, Graham Sandford, Peter R. Moore, Mickael Huiban, Sajinder K. Luthra, Jan Passchier, Olof Solin, and Véronique Gouverneur
Organic Letters 2013 Volume 15(Issue 11) pp:2648-2651
Publication Date(Web):May 20, 2013
DOI:10.1021/ol4009377
Treatment of readily available α,α-difluoro- and α-fluoroarylacetic acids with Selectfluor under Ag(I) catalysis led to decarboxylative fluorination. This operationally simple reaction gave access to tri- and difluoromethylarenes applying a late-stage fluorination strategy. Translation to [18F]labeling is demonstrated using [18F]Selectfluor bis(triflate), a reagent affording [18F]tri- and [18F]difluoromethylarenes not within reach with [18F]F2.
Co-reporter:Jamie R. Wolstenhulme;Jessica Rosenqvist;Dr. Oscar Lozano;John Ilupeju;Nathalie Wurz;Keary M. Engle;George W. Pidgeon;Dr. Peter R. Moore; Graham Sford; Véronique Gouverneur
Angewandte Chemie 2013 Volume 125( Issue 37) pp:9978-9982
Publication Date(Web):
DOI:10.1002/ange.201304845
Co-reporter:Dr. Guy T. Giuffredi; Véronique Gouverneur;Dr. Bruno Bernet
Angewandte Chemie 2013 Volume 125( Issue 40) pp:10718-10722
Publication Date(Web):
DOI:10.1002/ange.201303766
Co-reporter:Jamie R. Wolstenhulme;Jessica Rosenqvist;Dr. Oscar Lozano;John Ilupeju;Nathalie Wurz;Keary M. Engle;George W. Pidgeon;Dr. Peter R. Moore; Graham Sford; Véronique Gouverneur
Angewandte Chemie International Edition 2013 Volume 52( Issue 37) pp:9796-9800
Publication Date(Web):
DOI:10.1002/anie.201304845
Co-reporter:Dr. Guy T. Giuffredi; Véronique Gouverneur;Dr. Bruno Bernet
Angewandte Chemie International Edition 2013 Volume 52( Issue 40) pp:10524-10528
Publication Date(Web):
DOI:10.1002/anie.201303766
Co-reporter:George Blessley, Patrick Holden, Matthew Walker, John M. Brown, and Véronique Gouverneur
Organic Letters 2012 Volume 14(Issue 11) pp:2754-2757
Publication Date(Web):May 18, 2012
DOI:10.1021/ol300977f
Benzylic fluorides are suitable substrates for Pd(0)-catalyzed Tsuji–Trost substitution using carbon, nitrogen, oxygen, and sulfur nucleophiles and for cross-coupling with phenylboronic acid. For the bifunctional substrate 4-chlorobenzyl fluoride, fine-tuning of the reaction conditions allows for the regioselective displacement of either the chlorine or fluorine substituent. The leaving group ability of fluoride vs other groups displaced in substitution is CF3CO2 ≈ p-NO2C6H4CO2 ≈ OCO2CH3 > F > CH3CO2, a ranking similar to allylic fluorides under Pd catalysis.
Co-reporter:Charlotte Hollingworth and Véronique Gouverneur
Chemical Communications 2012 vol. 48(Issue 24) pp:2929-2942
Publication Date(Web):12 Dec 2011
DOI:10.1039/C2CC16158C
Transition metal catalyzed transformations using fluorinating reagents have been developed extensively for the preparation of synthetically valuable fluorinated targets. This is a topic of critical importance to facilitate laboratory and industrial chemical synthesis of fluorine containing pharmaceuticals and agrochemicals. Translation to 18F-radiochemistry is also emerging as a vibrant research field because functional imaging based on Positron Emission Tomography (PET) is increasingly used for both diagnosis and pharmaceutical development. This review summarizes how fluoride sources have been used for the catalytic nucleophilic fluorination of various substrates inclusive of aryl triflates, alkynes, allylic halides, allylic esters, allylic trichloroacetimidates, benzylic halides, tertiary alkyl halides and epoxides. Until recently, progress in this field of research has been slow in part because of the challenges associated with the dual reactivity profile of fluoride (nucleophile or base). Despite these difficulties, some remarkable breakthroughs have emerged. This includes the demonstration that Pd(0)/Pd(II)-catalyzed nucleophilic fluorination to access fluoroarenes from aryl triflates is feasible, and the first examples of Tsuji–Trost allylic alkylation with fluoride using either allyl chlorides or allyl precursors bearing O-leaving groups. More recently, allylic fluorides were also made accessible under iridium catalysis. Another reaction, which has been greatly improved based on careful mechanistic work, is the catalytic asymmetric hydrofluorination of meso epoxides. Notably, each individual transition metal catalyzed nucleophilic fluorination reported to date employs a different F-reagent, an observation indicating that this area of research will benefit from a larger pool of nucleophilic fluoride sources. In this context, a striking recent development is the successful design, synthesis and applications of a fluoride-derived electrophilic late stage fluorination reagent. This new class of reagents could greatly benefit preclinical and clinical PET imaging.
Co-reporter:Lorraine E. Combettes, Oscar Lozano, Véronique Gouverneur
Journal of Fluorine Chemistry 2012 Volume 143() pp:167-176
Publication Date(Web):November 2012
DOI:10.1016/j.jfluchem.2012.05.013
The intramolecular fluoroamination of homoallylic amines activated by a triisopropylsilyl or p-tolyldiisopropylsilyl group was successfully performed in the presence of Selectfluor® in acetonitrile leading to anti or syn 3-fluoropyrrolidines. The stereochemical outcome of these fluorocyclizations is dictated by the geometry of the alkene precursor. In comparison with oxygen nucleophile, the use of N-tosyl or N-Boc nucleophiles benefits from superior control over stereoselectivity but suffers from competitive fluorodesilylation.The fluoroamination of homoallylic amines activated by a triisopropylsilyl or p-tolyldiisopropylsilyl group is a suitable route to access to anti or syn 3-fluoropyrrolidines. The stereochemical outcome of these fluorocyclizations is dictated by the geometry of the alkene precursor.
Co-reporter:Lorraine E. Combettes;Philip Clausen-Thue;Dr. Michael A. King;Dr. Barbara Odell;Dr. Amber L. Thompson; Véronique Gouverneur;Dr. Tim D. W. Claridge
Chemistry - A European Journal 2012 Volume 18( Issue 41) pp:13133-13141
Publication Date(Web):
DOI:10.1002/chem.201201577
Abstract
A series of 3-fluoropyrrolidines have been studied to investigate the influence of the stereoelectronic fluorine gauche effect on ring conformations in the solid state by single-crystal X-ray analysis and in solution phase by NMR spectroscopy. As part of these studies 1D 19F–1H heteronuclear NOE (HOESY) experiments have been optimised for applications to small molecules and are described in detail. These have been employed to estimate 19F–1H internuclear distances and were combined with vicinal 3J(F,H) and 3J(H,H) scalar coupling constants to analyse the ring conformations. Where possible the derived solution-phase structural data have been compared with those of the crystalline state. The results demonstrate the influence of the gauche effect in stabilising Cγ-exo conformations of the fluorinated pyrrolidines. It was further shown that when steric interactions were also present, this conformational bias was diminished and the contribution of the alternative Cγ-endo conformation was seen to increase in solution at lower sample temperatures.
Co-reporter:Lorraine E. Combettes;Dr. Marie Schuler;Rakesh Patel;Baltasar Bonillo;Dr. Barbara Odell;Dr. Amber L. Thompson;Dr. Tim D. W. Claridge; Véronique Gouverneur
Chemistry - A European Journal 2012 Volume 18( Issue 41) pp:13126-13132
Publication Date(Web):
DOI:10.1002/chem.201201576
Abstract
Various 3-fluoropyrrolidines and 4-fluoropyrrolidin-2-ones were prepared by 5-exo-trig iodocyclisation from allylic fluorides bearing a pending nitrogen nucleophile. These bench-stable precursors were made accessible upon electrophilic fluorination of the corresponding allylsilanes. The presence of the allylic fluorine substituent induces syn-stereocontrol upon iodocyclisation with diastereomeric ratios ranging from 10:1 to > 20:1 for all N-tosyl-3-fluoropent-4-en-1-amines and amides. The sense and level of stereocontrol is strikingly similar to the corresponding iodocyclisation of structurally related allylic fluorides bearing pending oxygen nucleophiles. These results suggest that the syn selectivity observed upon ring closure involves I2–π complexes with the fluorine positioned inside.
Co-reporter:Lorraine E. Combettes;Philip Clausen-Thue;Dr. Michael A. King;Dr. Barbara Odell;Dr. Amber L. Thompson; Véronique Gouverneur;Dr. Tim D. W. Claridge
Chemistry - A European Journal 2012 Volume 18( Issue 41) pp:
Publication Date(Web):
DOI:10.1002/chem.201290181
Co-reporter:Dr. Matthew Tredwell; Véronique Gouverneur
Angewandte Chemie International Edition 2012 Volume 51( Issue 46) pp:11426-11437
Publication Date(Web):
DOI:10.1002/anie.201204687
Abstract
Molecular imaging has witnessed an upsurge in growth, with positron emission tomography leading the way. This trend has encouraged numerous synthetic chemists to enter the field of 18F-radiochemistry and provide generic solutions to address the well-recognized challenges of late-stage fluorination. This Minireview focuses on recent developments in the 18F-labeling of aromatic substrates.
Co-reporter:Dr. Satoshi Mizuta;Oscar Galicia-López;Keary M. Engle;Stefan Verhoog;Dr. Katherine Wheelhouse;Dr. Gerasimos Rassias; Véronique Gouverneur
Chemistry - A European Journal 2012 Volume 18( Issue 28) pp:8583-8587
Publication Date(Web):
DOI:10.1002/chem.201201707
Co-reporter:Elena Benedetto, Massaba Keita, Matthew Tredwell, Charlotte Hollingworth, John M. Brown, and Véronique Gouverneur
Organometallics 2012 Volume 31(Issue 4) pp:1408-1416
Publication Date(Web):January 20, 2012
DOI:10.1021/om201029m
Allyl fluorides are reactive toward Pt-catalyzed alkylation with malonate and likewise toward N- and O-nucleophiles under mild conditions. The reactivity of fluoride as a leaving group equals or exceeds that of the esters and carbonates commonly employed in allylic alkylation. The order of leaving-group ability with Pt catalysts was found to be F ≥ OCO2Me ≫ OBz ≥ OAc. This discouraged the application of platinum catalysts for the reverse reaction, fluorination of allylic substrates. Fluoride displacement involves predominant or complete retention of configuration in all the observed cases, and this was confirmed as a general feature of Pt catalysis, the stereochemical integrity being as high or higher as in Pd catalysis for the examples chosen.
Co-reporter:Dr. Zhanghua Gao;Dr. Yee Hwee Lim;Dr. Matthew Tredwell;Dr. Lei Li;Stefan Verhoog;Dr. Matthew Hopkinson;Wojciech Kaluza;Dr. Thomas Lee Collier;Dr. Jan Passchier;Dr. Mickael Huiban; Véronique Gouverneur
Angewandte Chemie International Edition 2012 Volume 51( Issue 27) pp:6733-6737
Publication Date(Web):
DOI:10.1002/anie.201201502
Co-reporter:Dr. Zhanghua Gao;Dr. Yee Hwee Lim;Dr. Matthew Tredwell;Dr. Lei Li;Stefan Verhoog;Dr. Matthew Hopkinson;Wojciech Kaluza;Dr. Thomas Lee Collier;Dr. Jan Passchier;Dr. Mickael Huiban; Véronique Gouverneur
Angewandte Chemie 2012 Volume 124( Issue 27) pp:6837-6841
Publication Date(Web):
DOI:10.1002/ange.201201502
Co-reporter:Lei Li, Matthew N. Hopkinson, Rodrigue Leuma Yona, Romain Bejot, Antony D. Gee and Véronique Gouverneur
Chemical Science 2011 vol. 2(Issue 1) pp:123-131
Publication Date(Web):01 Oct 2010
DOI:10.1039/C0SC00362J
The availability of radiolabelled probes is important for in vivo studies by positron emission tomography (PET). Among the frontier challenges in 18F-radiochemistry are the interconnected goals of increasing synthetic efficiency and diversity in the construction of 18F-labelled radiotracers. 18F-Radioretrosynthetic strategies implemented to date are typically linear sequences of transformations designed with the aim of introducing the 18F-label ideally in the last step or at least as late as possible. Here we report that convergent 18F-radiochemistry allows for the rapid assembly of functionalised 18F-radiotracers from readily accessible 18F-labelled prosthetic groups. Using multicomponent reactions for proof of concept, Ugi-4CR, Passerini-3CR, Biginelli-3CR and Groebke-3CR were performed successfully using 18F-benzaldehydes and these highly convergent reactions delivered, in high radiochemical yield (RCY), structurally complex 18F-radiotracers with the label positioned on an aryl motif not responsive to direct nucleophilic fluorination. These data establish an unprecedented connection between radiochemistry for PET and the field of multicomponent chemistry and demonstrate that convergent retroradiosynthesis is a powerful strategy expanding dramatically the scope of 18F-prosthetic group radiochemistry. The preparation of 18F-labelled prosthetic groups from [18F]fluoride ion is commonly performed in many labelling laboratories, so the concept of convergent 18F-radiosynthesis can easily be applied immediately.
Co-reporter:Laurence Carroll, Sophie Boldon, Romain Bejot, Jane E. Moore, Jérôme Declerck and Véronique Gouverneur
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 1) pp:136-140
Publication Date(Web):22 Nov 2010
DOI:10.1039/C0OB00564A
The Staudinger ligation of phosphine-substituted thioesters with 18F-fluoroethylazide has been successfully applied to access 18F-labelled molecules in radiochemical yields superior to 95%; the first fluorous variant of a Staudinger radio-ligation has been validated.
Co-reporter:Guy T. Giuffredi;Bruno Bernet;Véronique Gouverneur
European Journal of Organic Chemistry 2011 Volume 2011( Issue 20-21) pp:3825-3836
Publication Date(Web):
DOI:10.1002/ejoc.201100565
Abstract
A range of racemic 4-deoxy-4,4-difluorinated carbohydrates were prepared by using a diversity-oriented de novo synthesis starting with three commercially available two-carbon building blocks. A common gem-difluorinated glycal was prepared in 35 % yield in seven steps, from which five different 4-deoxy-4,4-difluoro- and 4-deoxy-2,4,4-trifluorohexopyranosides were accessed using well-established functional group manipulations.
Co-reporter:Guy T. Giuffredi, Laura E. Jennings, Bruno Bernet, Véronique Gouverneur
Journal of Fluorine Chemistry 2011 Volume 132(Issue 10) pp:772-778
Publication Date(Web):October 2011
DOI:10.1016/j.jfluchem.2011.05.017
The title compounds were prepared by two independent syntheses using inexpensive commercially available starting materials. 4-Deoxy-4-fluoro-α-d-talopyranoside served as a precursor to 4-deoxy-4-fluoro-α-d-idopyranoside, allowing for inversion of configuration at C-3 via a three-step protocol. The synthesis of 2,4-dideoxy-2,4-difluoro-α-d-talopyranoside is based on two nucleophilic fluorination events at C-2 then at C-4 using TBAF·3H2O and TBAF·4tBuOH as a fluoride source. All compounds are prepared as pure stereoisomers and are therefore suitable probes for OH⋯F H-bonding studies by 1H NMR spectroscopy.Graphical abstract4-Deoxy-4-fluoro-α-d-idopyranosides and 2,4-dideoxy-2,4-difluoro-α-d-talopyranosides were prepared in two independent syntheses, in good yields from commercially available starting materials.Highlights► Novel methyl 4-deoxy-4-fluoro-6-O-trityl-α-d-idopyranoside was accessed in 3 steps in 25% overall yield from a readily available precursor. ► Fluorination of 2-O-Tf-α-d-glucopyranoside with TBAF·3H2O in tBuOH gave 2-fluoro-α-d-mannopyranoside in a vastly improved yield of 77%. ► The novel methyl 2,4-dideoxy-2,4-difluoro-6-O-pivaloyl-α-d-talopyranoside was synthesized in 7 steps with an overall yield of 22%. ► The target compounds presented in this paper can be regarded as ideal probes for the study of intramolecular OH⋯F H-bonds by 1H NMR spectroscopy.
Co-reporter:Matthew N. Hopkinson; Antony D. Gee; Véronique Gouverneur
Chemistry - A European Journal 2011 Volume 17( Issue 30) pp:8248-8262
Publication Date(Web):
DOI:10.1002/chem.201100736
Abstract
When reacted in the presence of external oxidants, gold complexes are capable of catalyzing oxidative homo- and cross-coupling reactions involving the formation of new CC bonds. Over the last few years, several cascade processes have been reported in which coupling is preceded by a gold-mediated aryl CH functionalization or nucleophilic addition. These reactions combine the unique reactivity of gold with oxidative coupling, enabling the construction of CC bonds between coupling partners that are not easily accessed using alternative catalysts. In this Concept paper, the development of gold-catalyzed oxidative coupling reactions is discussed focusing on CC bond-forming reactions of broad synthetic appeal.
Co-reporter:Dr. Oscar Lozano;George Blessley;Dr. Teresa MartinezdelCampo;Dr. Amber L. Thompson;Guy T. Giuffredi;Dr. Michela Bettati;Dr. Matthew Walker;Dr. Richard Borman; Véronique Gouverneur
Angewandte Chemie 2011 Volume 123( Issue 35) pp:8255-8259
Publication Date(Web):
DOI:10.1002/ange.201103151
Co-reporter:Dr. Oscar Lozano;George Blessley;Dr. Teresa MartinezdelCampo;Dr. Amber L. Thompson;Guy T. Giuffredi;Dr. Michela Bettati;Dr. Matthew Walker;Dr. Richard Borman; Véronique Gouverneur
Angewandte Chemie International Edition 2011 Volume 50( Issue 35) pp:8105-8109
Publication Date(Web):
DOI:10.1002/anie.201103151
Co-reporter:Charlotte Hollingworth;Dr. Amaruka Hazari;Matthew N. Hopkinson;Dr. Matthew Tredwell;Elena Benedetto;Dr. Mickael Huiban; Antony D. Gee;Dr. John M. Brown; Véronique Gouverneur
Angewandte Chemie International Edition 2011 Volume 50( Issue 11) pp:2613-2617
Publication Date(Web):
DOI:10.1002/anie.201007307
Co-reporter:Matthew N. Hopkinson, Jonathan E. Ross, Guy T. Giuffredi, Antony D. Gee, and Véronique Gouverneur
Organic Letters 2010 Volume 12(Issue 21) pp:4904-4907
Publication Date(Web):October 11, 2010
DOI:10.1021/ol102061k
A gold(I)-catalyzed cascade cyclization−oxidative cross-coupling process has been applied to prepare β-alkynyl-γ-butenolides directly from allenoates and various terminal alkynes. Following an initial gold-catalyzed C−O bond forming allenoate cyclization, a mechanism based on a AuI/AuIII redox cycle has been proposed with Selectfluor acting as the external oxidant.
Co-reporter:James Stephen Harvey and Véronique Gouverneur
Chemical Communications 2010 vol. 46(Issue 40) pp:7477-7485
Publication Date(Web):13 Sep 2010
DOI:10.1039/C0CC01939A
Catalytic enantioselective strategies have become synthetically useful to access P-stereogenic phosphines. To date, enantioselective desymmetrisations and dynamic kinetic resolutions dominate the field. Desymmetrisation strategies do not necessarily require the formation of a P-carbon or P-heteroatom bond. This approach has been validated with variable levels of success using organocatalysed asymmetric deprotonation (chiral diamine) or methylation (phase transfer catalysis), enzyme-mediated esterification, rhodium catalysed [2+2+2] cycloadditions and more recently molybdenum-based ring closing metathesis. The dynamic kinetic resolution of racemic P-templates relying on a P–C bond-forming event has been the object of extensive investigations, which have culminated with the arylation and alkylation (benzylation) of equilibrating diastereomeric palladium, platinum or ruthenium phosphido complexes. Although all these routes allow access to a myriad of highly interesting P-stereogenic compounds, the level of enantiocontrol is substrate- and reactant-dependent. Pleasingly, ee’s up to 98% were obtained on selected systems.
Co-reporter:Laurence Carroll, Romain Bejot, Rebekka Hueting, Robert King, Paul Bonnitcha, Simon Bayly, Martin Christlieb, Jonathan R. Dilworth, Antony D. Gee, Jérôme Declerck and Véronique Gouverneur
Chemical Communications 2010 vol. 46(Issue 23) pp:4052-4054
Publication Date(Web):20 Apr 2010
DOI:10.1039/B926980K
The synthesis of three pairs of orthogonally labelled fluorinated Cu bis(thiosemicarbazonato) complexes is presented. These are the first examples of 18F-labelled Cu(II)-complexes designed to serve as new hypoxia selective PET tracers and as mechanistic probes to study the mode of action of this class of markers. In vitro evaluation revealed that the fluorinated Cu-complex derived from amide coupling is suitable for in vivo work.
Co-reporter:James Stephen Harvey, Guy T. Giuffredi and Véronique Gouverneur
Organic Letters 2010 Volume 12(Issue 6) pp:1236-1239
Publication Date(Web):February 25, 2010
DOI:10.1021/ol100098c
A range of P-containing ene-diynes suitable for desymmetrization was prepared via a two-step process starting from phosphorus oxychloride. In the presence of the Hoveyda−Grubbs II catalyst, these substrates underwent diastereoselective enyne ring-closing metathesis leading to various synthetically useful P-stereogenic heterocycles featuring an exocyclic alkynyl group. These products are amenable to further functional manipulation.
Co-reporter:Laurent Bonnac, Sarah E. Lee, Guy T. Giuffredi, Lucy M. Elphick, Alexandra A. Anderson, Emma S. Child, David J. Mann and Véronique Gouverneur
Organic & Biomolecular Chemistry 2010 vol. 8(Issue 6) pp:1445-1454
Publication Date(Web):25 Jan 2010
DOI:10.1039/B922442D
Enantioenriched tetrafluorinated aryl-C-nucleosides were synthesised in four steps from 1-benzyloxy-4-bromo-3,3,4,4-tetrafluorobutan-2-ol. The presence of the tetrafluorinated ethylene group is compatible with O-phosphorylation of the primary alcohol, as demonstrated by the successful preparation of the tetrafluorinated naphthyl-C-nucleotide.
Co-reporter:Matthew N. Hopkinson;Antony D. Gee;Véronique Gouverneur
Israel Journal of Chemistry 2010 Volume 50( Issue 5-6) pp:675-690
Publication Date(Web):
DOI:10.1002/ijch.201000078
Abstract
Performing gold-catalyzed organic transformations in the presence of fluorinating reagents can lead to both fluorinated and non-fluorinated products. Gold(I) complexes can activate alkynes towards nucleophilic attack by fluoride leading to fluoroalkenes under mild conditions. Fluorinated products can also be prepared upon performing gold-catalyzed transformations in the presence of electrophilic sources of fluorine. In most cases, however, the combination of gold and electrophilic fluorinating reagents does not lead to fluorination but delivers products of oxidative homo- or cross-coupling. In these processes the “F+” source is likely acting as a sacrificial two-electron external oxidant performing the key oxidation of gold(I) to gold(III) in the redox cycle. Oxidative coupling is an emerging field of gold catalysis which, when combined with the well-established reactivity of gold as a soft π-acid, holds promise as a mild and efficient method for the construction of complex organic molecules.
Co-reporter:Harriet Teare;Dr. EdwardG. Robins;Anna Kirjavainen;Sarita Forsback; Graham Sford; Olof Solin;Dr. SajinderK. Luthra; Véronique Gouverneur
Angewandte Chemie International Edition 2010 Volume 49( Issue 38) pp:6821-6824
Publication Date(Web):
DOI:10.1002/anie.201002310
Co-reporter:Harriet Teare;Dr. EdwardG. Robins;Anna Kirjavainen;Sarita Forsback; Graham Sford; Olof Solin;Dr. SajinderK. Luthra; Véronique Gouverneur
Angewandte Chemie 2010 Volume 122( Issue 38) pp:6973-6976
Publication Date(Web):
DOI:10.1002/ange.201002310
Co-reporter:MatthewN. Hopkinson;Arnaud Tessier Dr.;Andrew Salisbury;GuyT. Giuffredi;LorraineE. Combettes;AntonyD. Gee ;Véronique Gouverneur
Chemistry - A European Journal 2010 Volume 16( Issue 16) pp:4739-4743
Publication Date(Web):
DOI:10.1002/chem.201000322
Co-reporter:Guy T. Giuffredi, Sophie Purser, Marcin Sawicki, Amber L. Thompson, Véronique Gouverneur
Tetrahedron: Asymmetry 2010 Volume 21(Issue 1) pp:123-127
Publication Date(Web):29 January 2010
DOI:10.1016/j.tetasy.2009.10.016
Co-reporter:Sarah E. Lee, Lucy M. Elphick, Alexandra A. Anderson, Laurent Bonnac, Emma S. Child, David J. Mann, Véronique Gouverneur
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 14) pp:3804-3807
Publication Date(Web):15 July 2009
DOI:10.1016/j.bmcl.2009.04.028
We hereby present a simple yet novel chemical synthesis of a family of γ-modified ATPs bearing functional groups on the γ-phosphate that are amenable to further derivatization by highly selective chemical manipulations (e.g., click chemistry, Staudinger ligations). A preliminary screen of these compounds as phosphate donors with a typical wild type protein kinase (cdk2) and one of its known substrates p27kip1 is also presented.
Co-reporter:Guy T. Giuffredi, Sophie Purser, Marcin Sawicki, Amber L. Thompson, Véronique Gouverneur
Tetrahedron: Asymmetry 2009 Volume 20(6–8) pp:910-920
Publication Date(Web):7 May 2009
DOI:10.1016/j.tetasy.2009.03.001
A highly efficient anti-SE2′ electrophilic fluorination of enantioenriched allylsilanes a subsequent dihydroxylation of the resulting allylic fluorides were used as key steps for the synthesis of three fluorinated carbohydrate analogues, 1,5-di-O-benzyl-2-deoxy-2-fluoro-d-glucitol, 2,6-di-O-benzyl-5-deoxy-5-fluoro-l-glucitol and 1,5-di-O-benzyl-2-deoxy-2-fluoro-d-mannitol. A new catalytic asymmetric route to 1-benzyloxy-4-trimethylsilyl-but-3-yn-2-ol, a common precursor to two advanced allylsilanes, is also described featuring a Noyori asymmetric transfer hydrogenation reaction.(2S,3E)-1-(Benzyloxy)-4-(trimethylsilyl)but-3-en-2-yl (benzyloxy)acetateC23H30O4SiEe = 93%[α]D21=+21.3 (c 1.1, CH2Cl2)Source of chirality: asymmetric synthesisAbsolute configuration: (S)(2R,3R,4E)-2,6-Bis(benzyloxy)-3-(trimethylsilyl)hex-4-enoic acidC23H30O4SiEe = 93%[α]D21=+15.0 (c 1.0, CH2Cl2)Source of chirality: asymmetric synthesisAbsolute configuration: (2R,3R)(2R,3R,4E)-2,6-Bis(benzyloxy)-3-(trimethylsilyl)hex-4-en-1-olC23H32O3SiEe = 93%[α]D21=-12.6 (c 1.0, CH2Cl2)Source of chirality: asymmetric synthesisAbsolute configuration: (2R,3R)(2S,3E,5R)-2,6-Bis(benzyloxy)-5-fluorohex-3-en-1-olC20H23FO3Ee = 93%[α]D21=-32.2 (c 1.0, CH2Cl2)Source of chirality: asymmetric synthesisAbsolute configuration: (2S,5R)1,5-di-O-Benzyl-2-deoxy-2-fluoro-d-glucitolC20H25FO5Ee = 93%[α]D25=+2.6 (c 0.4, MeOH)Source of chirality: asymmetric synthesisAbsolute configuration: (2S,3S,4S,5R)2,6-Di-O-benzyl-5-deoxy-5-fluoro-l-glucitolC20H25FO5Ee = 93%[α]D25=-2.0 (c 1.3, MeOH)Source of chirality: asymmetric synthesisAbsolute configuration: (2R,3R,4R,5S)(2S,3Z)-1-(Benzyloxy)-4-(trimethylsilyl)but-3-en-2-yl (benzyloxy)acetateC23H30O4SiEe = 93%[α]D21=+21.3 (c 1.1, CH2Cl2)Source of chirality: asymmetric synthesisAbsolute configuration: (S)(2R,3S,4E)-2,6-Bis(benzyloxy)-3-(trimethylsilyl)hex-4-enoic acidC23H30O4SiEe = 93%[α]D21=-11.3 (c 1.0, CH2Cl2)Source of chirality: asymmetric synthesisAbsolute configuration: (2R,3S)(2R,3S,4E)-2,6-Bis(benzyloxy)-3-(trimethylsilyl)hex-4-en-1-olC23H32O3SiEe = 93%[α]D21=-22.0 (c 1.0, CH2Cl2)Source of chirality: asymmetric synthesisAbsolute configuration: (2R,3S)(2S,3E,5S)-2,6-Bis(benzyloxy)-5-fluorohex-3-en-1-olC20H23FO3Ee = 93%[α]D21=-38.4 (c 1.1, CH2Cl2)Source of chirality: asymmetric synthesisAbsolute configuration: (2S,5S)1,5-di-O-Benzyl-2-deoxy-2-fluoro-d-mannitolC20H25FO5Ee = 93%[α]D21=+20.4 (c 0.5, CH2Cl2)Source of chirality: asymmetric synthesisAbsolute configuration: (2S,3S,4S,5R)(2S)-1-(Benzyloxy)-4-(trimethylsilyl)but-3-yn-2-yl (2S)-3,3,3-trifluoro-2-methoxy-2-phenylpropanoateC24H27F3O4SiEe = 93%[α]D25=+40.2 (c 1.0, CH2Cl2)Source of chirality: asymmetric synthesisAbsolute configuration: (2S,2′S)(2S,3E)-1-(Benzyloxy)-4-(trimethylsilyl)but-3-en-2-yl (2S)-3,3,3-trifluoro-2-methoxy-2-phenylpropanoateC24H29F3O4SiEe = 93%[α]D25=+46.3 (c 1.0, CH2Cl2)Source of chirality: asymmetric synthesisAbsolute configuration: (2S,2′S)(2S,3Z)-1-(Benzyloxy)-4-(trimethylsilyl)but-3-en-2-yl (2S)-3,3,3-trifluoro-2-methoxy-2-phenylpropanoateC24H29F3O4SiEe = 93%[α]D25=+45.8 (c 1.1, CH2Cl2)Source of chirality: asymmetric synthesisAbsolute configuration: (2S,2′S)(2R,3R,4E)-2,6-Bis(benzyloxy)-3-(trimethylsilyl)hex-4-en-1-yl (2S)-3,3,3-trifluoro-2-methoxy-2-phenylpropanoateC33H39F3O5SiEe = 93%[α]D25=+24.3 (c 1.0, CH2Cl2)Source of chirality: asymmetric synthesisAbsolute configuration: (2R,3R,2′S)(2R,3S,4E)-2,6-Bis(benzyloxy)-3-(trimethylsilyl)hex-4-en-1-yl (2S)-3,3,3-trifluoro-2-methoxy-2-phenylpropanoateC33H39F3O5SiEe = 93%[α]D25=+11.0 (c 0.7, CH2Cl2)Source of chirality: asymmetric synthesisAbsolute configuration: (2R,3S,2′S)(2S,3E,5R)-2,6-Bis(benzyloxy)-5-fluorohex-3-en-1-yl (2S)-3,3,3-trifluoro-2-methoxy-2-phenylpropanoateC30H30F4O5Ee = 93%[α]D25=+16.0 (c 0.7, CH2Cl2)Source of chirality: asymmetric synthesisAbsolute configuration: (2S,5R,2′S)(2S,3E,5S)-2,6-Bis(benzyloxy)-5-fluorohex-3-en-1-yl (2S)-3,3,3-trifluoro-2-methoxy-2-phenylpropanoateC30H30F4O5Ee = 93%[α]D25=+12.9 (c 0.9, CH2Cl2)Source of chirality: asymmetric synthesisAbsolute configuration: (2S,5S,2′S)
Co-reporter:Sophie Boldon, Jane E. Moore, Véronique Gouverneur
Journal of Fluorine Chemistry 2009 130(12) pp: 1151-1156
Publication Date(Web):
DOI:10.1016/j.jfluchem.2009.05.017
Co-reporter:Yu-hong Lam, Steven J. Stanway, Véronique Gouverneur
Tetrahedron 2009 65(48) pp: 9905-9933
Publication Date(Web):
DOI:10.1016/j.tet.2009.08.005
Co-reporter:JohnM. Brown Dr. ;Véronique Gouverneur
Angewandte Chemie International Edition 2009 Volume 48( Issue 46) pp:
Publication Date(Web):
DOI:10.1002/anie.200902121
Co-reporter:Romain Bejot Dr.;Thomas Fowler;Laurence Carroll;Sophie Boldon;JaneE. Moore Dr.;Jérôme Declerck Dr.;Véronique Gouverneur
Angewandte Chemie 2009 Volume 121( Issue 3) pp:
Publication Date(Web):
DOI:10.1002/ange.200890336
Co-reporter:Romain Bejot Dr.;Thomas Fowler;Laurence Carroll;Sophie Boldon;JaneE. Moore Dr.;Jérôme Declerck Dr.;Véronique Gouverneur
Angewandte Chemie 2009 Volume 121( Issue 3) pp:594-597
Publication Date(Web):
DOI:10.1002/ange.200803897
Co-reporter:Amaruka Hazari;Véronique Gouverneur ;JohnM. Brown Dr.
Angewandte Chemie 2009 Volume 121( Issue 7) pp:1322-1325
Publication Date(Web):
DOI:10.1002/ange.200804310
Co-reporter:SusanC. Wilkinson;Oscar Lozano Dr.;Marie Schuler Dr.;MariaC. Pacheco Dr.;Roger Salmon;Véronique Gouverneur
Angewandte Chemie 2009 Volume 121( Issue 38) pp:7217-7220
Publication Date(Web):
DOI:10.1002/ange.200901795
Co-reporter:Romain Bejot Dr.;Thomas Fowler;Laurence Carroll;Sophie Boldon;JaneE. Moore Dr.;Jérôme Declerck Dr.;Véronique Gouverneur
Angewandte Chemie International Edition 2009 Volume 48( Issue 3) pp:586-589
Publication Date(Web):
DOI:10.1002/anie.200803897
Co-reporter:Romain Bejot Dr.;Thomas Fowler;Laurence Carroll;Sophie Boldon;JaneE. Moore Dr.;Jérôme Declerck Dr.;Véronique Gouverneur
Angewandte Chemie International Edition 2009 Volume 48( Issue 3) pp:
Publication Date(Web):
DOI:10.1002/anie.200890287
Co-reporter:JamesStephen Harvey;StevenJ. Malcolmson;KatherineS. Dunne Dr.;SimonJ. Meek Dr.;AmberL. Thompson Dr.;RichardR. Schrock ;AmirH. Hoveyda ;Véronique Gouverneur
Angewandte Chemie International Edition 2009 Volume 48( Issue 4) pp:762-766
Publication Date(Web):
DOI:10.1002/anie.200805066
Co-reporter:Amaruka Hazari;Véronique Gouverneur ;JohnM. Brown Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 7) pp:1296-1299
Publication Date(Web):
DOI:10.1002/anie.200804310
Co-reporter:SusanC. Wilkinson;Oscar Lozano Dr.;Marie Schuler Dr.;MariaC. Pacheco Dr.;Roger Salmon;Véronique Gouverneur
Angewandte Chemie International Edition 2009 Volume 48( Issue 38) pp:7083-7086
Publication Date(Web):
DOI:10.1002/anie.200901795
Co-reporter:M. Carmen Pacheco, Sophie Purser and Véronique Gouverneur
Chemical Reviews 2008 Volume 108(Issue 6) pp:1943
Publication Date(Web):June 11, 2008
DOI:10.1021/cr068410e
Co-reporter:Sophie Purser, Peter R. Moore, Steve Swallow and Véronique Gouverneur
Chemical Society Reviews 2008 vol. 37(Issue 2) pp:320-330
Publication Date(Web):13 Dec 2007
DOI:10.1039/B610213C
It has become evident that fluorinated compounds have a remarkable record in medicinal chemistry and will play a continuing role in providing lead compounds for therapeutic applications. This tutorial review provides a sampling of renowned fluorinated drugs and their mode of action with a discussion clarifying the role and impact of fluorine substitution on drug potency.
Co-reporter:Sophie Boldon, Jane E. Moore and Véronique Gouverneur
Chemical Communications 2008 (Issue 31) pp:3622-3624
Publication Date(Web):04 Jun 2008
DOI:10.1039/B804484H
A novel fluorous tagging–detagging strategy has been developed featuring a fluorination as the detagging process; fluorous allylsilanes were prepared by cross-metathesis and subsequently subjected to electrophilic fluorodesilylation; Selectfluor was used as the detagging reagent; the resulting allylic fluorides were successfully purified by fluorous solid phase extraction.
Co-reporter:Laurence Carroll, Samantha McCullough, Tom Rees, Timothy D. W. Claridge and Véronique Gouverneur
Organic & Biomolecular Chemistry 2008 vol. 6(Issue 10) pp:1731-1733
Publication Date(Web):03 Apr 2008
DOI:10.1039/B803888K
The electrophilic fluorodesilylation of enantioenriched allenylsilanes proceeds with efficient transfer of chirality. The silylation–fluorination of propargylic alcohols occurs with overall retention of stereochemistry, a result consistent with a stereospecific anti SE2′ mechanism for the fluorination step.
Co-reporter:Marie Schuler Dr.;Franck Silva Dr.;Carla Bobbio Dr.;Arnaud Tessier Dr. ;Véronique Gouverneur Dr.
Angewandte Chemie 2008 Volume 120( Issue 41) pp:8045-8048
Publication Date(Web):
DOI:10.1002/ange.200802162
Co-reporter:Marie Schuler Dr.;Franck Silva Dr.;Carla Bobbio Dr.;Arnaud Tessier Dr. ;Véronique Gouverneur Dr.
Angewandte Chemie 2008 Volume 120( Issue 41) pp:
Publication Date(Web):
DOI:10.1002/ange.200890256
Co-reporter:Marie Schuler Dr.;Franck Silva Dr.;Carla Bobbio Dr.;Arnaud Tessier Dr. ;Véronique Gouverneur Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 41) pp:7927-7930
Publication Date(Web):
DOI:10.1002/anie.200802162
Co-reporter:Marie Schuler Dr.;Franck Silva Dr.;Carla Bobbio Dr.;Arnaud Tessier Dr. ;Véronique Gouverneur Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 41) pp:
Publication Date(Web):
DOI:10.1002/anie.200890202
Co-reporter:Harriet Teare, Edward G. Robins, Erik Årstad, Sajinder K. Luthra and Véronique Gouverneur
Chemical Communications 2007 (Issue 23) pp:2330-2332
Publication Date(Web):14 Mar 2007
DOI:10.1039/B701177F
A novel [18F]NF reagent and two novel radiochemical transformations have been developed: [18F]NFSi has been prepared from sodium dibenzenesulfonimide and reacted in the presence of silyl enol ethers and allylsilanes to deliver labelled fluorinated ketones and allylic fluorides respectively; the radiosynthesis of the fluorinated A ring of vitamin D3 has also been completed with success.
Co-reporter:Yu-hong Lam;Carla Bobbio Dr.;Ian R. Cooper Dr.;Véronique Gouverneur Dr.
Angewandte Chemie 2007 Volume 119(Issue 27) pp:
Publication Date(Web):5 JUN 2007
DOI:10.1002/ange.200701365
Eine „umgekehrte“ Strategie aus Cycloaddition und Fluorierung liefert enantiomerenangereicherte fluorierte Verbindungen mit bis zu fünf stereogenen Zentren, von denen eines fluoriert ist. Teil dieses hoch konvergenten Prozesses ist eine milde Fluorierung, die mit hoher Ausbeute gelingt und hinsichtlich Richtung und Ausmaß der Stereokontrolle getestet wurde.
Co-reporter:Yu-hong Lam;Carla Bobbio Dr.;Ian R. Cooper Dr.;Véronique Gouverneur Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 27) pp:
Publication Date(Web):5 JUN 2007
DOI:10.1002/anie.200701365
A “reverse” cycloaddition–fluorination strategy has been validated to access enantioenriched fluorinated compounds featuring up to five stereogenic centers, one of which is fluorinated. This highly convergent process features a mild and high-yielding fluorination, for which the sense and level of stereocontrol have been probed.
Co-reporter:Matthew Tredwell Dr.;JenniferA.R. Luft;Marie Schuler Dr.;Kenny Tenza Dr.;KendallN. Houk ;Véronique Gouverneur Dr.
Angewandte Chemie 2007 Volume 120( Issue 2) pp:363-366
Publication Date(Web):
DOI:10.1002/ange.200703465
Co-reporter:Matthew Tredwell Dr.;JenniferA.R. Luft;Marie Schuler Dr.;Kenny Tenza Dr.;KendallN. Houk ;Véronique Gouverneur Dr.
Angewandte Chemie International Edition 2007 Volume 47( Issue 2) pp:357-360
Publication Date(Web):
DOI:10.1002/anie.200703465
Co-reporter:Laurence Carroll, Mª Carmen Pacheco, Ludivine Garcia and Véronique Gouverneur
Chemical Communications 2006 (Issue 39) pp:4113-4115
Publication Date(Web):23 Aug 2006
DOI:10.1039/B610013A
Allenylsilanes reacted at room temperature in acetonitrile with Selectfluor, an electrophilic fluorinating reagent, to give secondary propargylic fluorides in moderate to good yields; mechanistically, a side-product resulting from a 1,2-silyl shift testifies to the presence of a cationic intermediate.
Co-reporter:Carla Bobbio and Véronique Gouverneur
Organic & Biomolecular Chemistry 2006 vol. 4(Issue 11) pp:2065-2075
Publication Date(Web):03 May 2006
DOI:10.1039/B603163C
The appearance of structurally diverse fluorinating reagents displaying a large spectrum of reactivity has been critical to the development of the catalytic asymmetric fluorination processes known to date. In this article, we discuss how this area of research emerged and which strategies have allowed for the successful development of both nucleophilic and electrophilic catalytic enantioselective fluorinations. We also present the fundamental understanding of catalytic activity and enantioselectivity for the most efficient processes and highlight the first synthetic application with the preparation of a complex fluorinated target.
Co-reporter:Matthew Tredwell and Véronique Gouverneur
Organic & Biomolecular Chemistry 2006 vol. 4(Issue 1) pp:26-32
Publication Date(Web):23 Nov 2005
DOI:10.1039/B513399H
The fluorination of organosilanes with the silyl groups directly attached or adjacent to an aryl or alkenyl group has been only very recently examined despite the fact that the corresponding fluorinated products are synthetically useful building blocks. In these reactions, the silyl group enhances the reactivity of the π-nucleophile and controls the sense of regiochemistry upon addition of the electrophilic source of fluorine. These reactions take advantage of the β effect of the silicon–carbon bond and recent results from the literature revealed that this chemistry allows for the preparation of a variety of novel fluorinated building blocks including enantioenriched derivatives.
Co-reporter:Maud Reiter Dr.;Hazel Turner;Véronique Gouverneur Dr.
Chemistry - A European Journal 2006 Volume 12(Issue 27) pp:
Publication Date(Web):3 JUL 2006
DOI:10.1002/chem.200600415
Structurally diverse β-hydroxyenones are shown to undergo nonoxidative 6-endo-trig ring closure to form highly substituted tetrahydropyranones. Amberlyst-15, Al(ClO4)3⋅9 H2O and [Pd(MeCN)4](BF4)2 were found to be suitable catalysts for these intramolecular conjugate additions, preventing side reactions, such as dehydration or retroaldolisation. The use of [Pd(MeCN)4](BF4)2 is particularly effective, as this palladium-mediated reaction is under kinetic control and generates tri- and tetrasubstituted tetrahydropyranones with high levels of diastereocontrol. In the presence of the Lewis acid Al(ClO4)3⋅9 H2O, the reaction proceeded with a similar level of diastereocontrol; however, in contrast to [Pd(MeCN)4](BF4)2, this catalyst can promote enolisation. The palladium-mediated reaction was also found to be compatible with an enantioenriched β-hydroxyenone substrate, giving no loss of enantiopurity upon ring closure. The most distinctive synthetic development to emerge from this new chemistry is the possibility to access tri- and tetrasubstituted 2,6-anti-tetrahydropyranones from anti-aldol precursors. These compounds are particularly difficult to access by using alternative methodologies. Two modes of activation were envisaged for the ring closure, involving metal coordination to either the CC or CO functional groups. Experimental results suggest that CO coordination was the preferred mode of activation for reactions performed in the presence of Al(ClO4)3⋅9 H2O or [Pd(MeCN)4](BF4)2.
Co-reporter:Véronique Gouverneur Dr.;Maud Reiter
Chemistry - A European Journal 2005 Volume 11(Issue 20) pp:
Publication Date(Web):8 JUL 2005
DOI:10.1002/chem.200500406
Very little information is available on hetero-Diels–Alderases for the assembly of heterocyclic products despite the synthetic value of these [4+2] cycloadditions. Hetero-Diels–Alderase antibodies raised against a bicyclic transition state analogue have been generated for the cycloaddition of ethylglyoxylate with an all-carbon diene. More recently, a conceptually novel biocatalytic approach to hetero-Diels–Alder (HDA) adducts derived from carbonyl dienophiles has been developed mirroring a stepwise aldol Michael mechanism instead of a concerted pathway. In this approach, the two key steps are an antibody-mediated kinetic resolution of β-hydroxyenones and a subsequent ring-closure process. An attractive feature of this methodology is the possibility to convert the enantioenriched aldol intermediates into tetrahydropyranones or dihydropyranones. This bioorganic route is best applied for the preparation of enantioenriched HDA adducts derived from poorly electrophilic acceptors, therefore complementing existing catalytic routes to these adducts based on the use of small organocatalysts or chiral Lewis acids.
Co-reporter:Sébastien Thibaudeau, Robert Fuller and Véronique Gouverneur
Organic & Biomolecular Chemistry 2004 vol. 2(Issue 8) pp:1110-1112
Publication Date(Web):11 Mar 2004
DOI:10.1039/B402097A
Ru-based catalysts can be used in E-selective cross metathesis (CM) reactions to synthesise various functionalised internal allylic monofluorides.
Co-reporter:Catherine A. Slinn, Alison J. Redgrave, S. Lucy Hind, Chris Edlin, Steven P. Nolan and Veronique Gouverneur
Organic & Biomolecular Chemistry 2003 vol. 1(Issue 21) pp:3820-3825
Publication Date(Web):10 Sep 2003
DOI:10.1039/B306940K
Ru– and Mo-based catalysts can be used in ring closing metathesis (RCM) reactions to synthesise cyclic phosphines protected as their borane complexes. The compatibility of the Schrock Mo-catalyst and the N-heterocyclic carbene Ru-catalysts with this class of substrates is particularly noteworthy as asymmetric RCM (ARCM) is now emerging as a new tool for the preparation of homochiral phosphines. One of the key results is that the Mo-catalyst allows the ring closure of the unprotected diallylphenylphosphine with 95% conversion.
Co-reporter:Benjamin Greedy;Jean-Marc Paris Dr.;Thierry Vidal Dr.;Véronique Gouverneur Dr.
Angewandte Chemie International Edition 2003 Volume 42(Issue 28) pp:
Publication Date(Web):16 JUL 2003
DOI:10.1002/anie.200351405
EnantiopureN-fluorocinchona alkaloids promoted the electrophilic fluorodesilylation of allyl silanes in a conceptually new approach to the regio- and enantioselective formation of allylic fluorides (see scheme). Excellent conversions were observed in these reactions, which afforded the desired allylic fluorides with up to 96 % ee.
Co-reporter:Benjamin Greedy;Jean-Marc Paris Dr.;Thierry Vidal Dr.;Véronique Gouverneur Dr.
Angewandte Chemie 2003 Volume 115(Issue 28) pp:
Publication Date(Web):16 JUL 2003
DOI:10.1002/ange.200351405
EnantiomerenreineN-Fluor-Cinchona-Alkaloide katalysieren die elektrophile Fluordesilylierung von Allylsilanen. Dieses neue Konzept ermöglicht die regio- und enantioselektive Synthese von Allylfluoriden (siehe Schema). Bei hervorragenden Ausbeuten konnten Enantiomerenüberschüsse bis zu 96 % ee erzielt werden.
Co-reporter:Anne Briot;Murielle Bujard;Véronique Gouverneur;Charles Mioskowski
European Journal of Organic Chemistry 2002 Volume 2002(Issue 1) pp:
Publication Date(Web):10 DEC 2001
DOI:10.1002/1099-0690(20021)2002:1<139::AID-EJOC139>3.0.CO;2-S
The synthesis of the first two haptens designed to elicit catalytic hydrolytic antibodies for the dynamic kinetic resolution of racemic 4-substituted 4H-oxazolin-5-ones is reported. A cyclic phosphinate and a 2,5-dihydro-1H-pyrrolium derivative were chosen as “first generation” haptens. The cyclic phosphinate was designed to mimic the higher energy transition state along the reaction coordinate for the hydrolytic process, while the dihydro-1H-pyrrolium group of the second hapten was selected to generate hydrolytic antibodies that might use a “bait and switch” mechanism. These two haptens were prepared successfully by use of a ring-closing metathesis reaction as the key step. This study is the first pilot investigation towards an antibody-promoted dynamic kinetic resolution and should allow the development of a new biocatalytic route to enantiomerically pure natural and unnatural amino acids.
Co-reporter:Virginie Maggiotti;Marina Resmini Dr.;Véronique Gouverneur Dr.
Angewandte Chemie International Edition 2002 Volume 41(Issue 6) pp:
Publication Date(Web):15 MAR 2002
DOI:10.1002/1521-3773(20020315)41:6<1012::AID-ANIE1012>3.0.CO;2-I
Direct asymmetric aldol reactions at the less substituted carbon atom of unmodified unsymmetrical ketones are achieved in the presence of the commercially available aldolase I antibody 84G3 [Eq. (1)]. All the reactions proceeded with enantiomeric excesses greater than 94 %. Both enantiomers are accessible by using either an aldol or a retro-aldol reaction.
Co-reporter:Véronique Gouverneur Dr.;Benjamin Greedy
Chemistry - A European Journal 2002 Volume 8(Issue 4) pp:
Publication Date(Web):13 FEB 2002
DOI:10.1002/1521-3765(20020215)8:4<766::AID-CHEM766>3.0.CO;2-U
Recently, with the appearance of electrophilic sources of fluorine including the commercially available N−F reagents, the concept of electrophilic fluorodesilylation has emerged as a new strategy to prepare a variety of fluorine containing compounds. This paper highlights how this concept has been applied to the preparation of a series of fluorinated molecules including fluoroaromatic compounds, fluoroalkenes, difluoroamides, difluoroalcohols, difluoroethers and α-fluorinated carbonyl derivatives.
Co-reporter:Virginie Maggiotti;Marina Resmini Dr.;Véronique Gouverneur Dr.
Angewandte Chemie 2002 Volume 114(Issue 6) pp:
Publication Date(Web):15 MAR 2002
DOI:10.1002/1521-3757(20020315)114:6<1054::AID-ANGE1054>3.0.CO;2-H
Direkte asymmetrische Aldolreaktionen am weniger substituierten Kohlenstoffatom unmodifizierter unsymmetrischer Ketone gelingen in Gegenwart des kommerziell erhältlichen Aldolase-I-Antikörpers 84G3 [Gl. (1)]. Bei allen Reaktionen war der Enantiomerenüberschuss größer als 94 %. Indem entweder die Aldol- oder die Retro-Aldolreaktion genutzt wird, sind beide Enantiomere zugänglich.
Co-reporter:V. Maggiotti, J.-B. Wong, R. Razet, A.R. Cowley, V. Gouverneur
Tetrahedron: Asymmetry 2002 Volume 13(Issue 16) pp:1789-1798
Publication Date(Web):27 August 2002
DOI:10.1016/S0957-4166(02)00410-X
Compounds 1–7 are the products formed by aldol condensation of p-nitrobenzaldehyde with a series of unsymmetrical ketones, the reaction occurring at the less substituted carbon. The asymmetric synthesis of 1–7 using the Evans's asymmetric aldol methodology is described in detail. These syntheses were completed to allow us to assign the absolute configuration of products 1–3, obtained in a single step in the presence of the aldolase I antibody 84G3.Graphic(4S)-1-Chloro-4-hydroxy-4-(4-nitrophenyl)butan-2-oneE.e. >99%[α]D25=−30.4 (c 0.5, DCM)Source of chirality: chiral starting materialAbsolute configuration: 4S(3′S,4S)-4-Benzyl-3-[3′-hydroxy-3′-(4-nitrophenyl)propionyl]oxazolidin-2-oneE.e. >99%[α]D25=+29.5 (c 1, DCM)Source of chirality: (S)-4-benzyloxazolidin-2-oneAbsolute configuration: 3′S,4S(3S)-3-(tert-Butyldimethylsilanyloxy)-3-(4-nitrophenyl)propionic acidE.e. >99%[α]D25=−49.8 (c 1, DCM)Source of chirality: chiral starting materialAbsolute configuration: 3S(1S)-1-Hydroxy-1-(4-nitrophenyl)heptan-3-oneE.e. >99%[α]D25=−55.3 (c 1, DCM)Source of chirality: chiral starting materialAbsolute configuration: 1S(4S)-4-Hydroxy-1-methoxy-4-(4-nitrophenyl)butan-2-oneE.e. >99%[α]D25=−37.5 (c 0.5, DCM)Source of chirality: chiral starting materialAbsolute configuration: 4S(4S)-4-Hydroxy-1-methylsulfanyl-4-(4-nitrophenyl)butan-2-oneE.e. >99%[α]D25=−32.2 (c 1, DCM)Source of chirality: chiral starting materialAbsolute configuration: 4S(4S)-1,4-Dihydroxy-4-(4-nitrophenyl)butan-2-oneE.e. >99%[α]D25=−38.7 (c 0.5, DCM)Source of chirality: chiral starting materialAbsolute configuration: 4S(1S)-1-Hydroxy-5-methoxy-1-(4-nitrophenyl)pentan-3-oneE.e. >99%[α]D25=−42.6 (c 1, DCM)Source of chirality: chiral starting materialAbsolute configuration: 1S(1S)-1-Hydroxy-5-methylsulfanyl-1-(4-nitrophenyl)pentan-3-oneE.e. >99%[α]D25=−47.2 (c 1, DCM)Source of chirality: chiral starting materialAbsolute configuration: 1S
Co-reporter:Benjamin Greedy and Veronique Gouverneur
Chemical Communications 2001 (Issue 3) pp:233-234
Publication Date(Web):16 Jan 2001
DOI:10.1039/B009179K
A range of alkenyltrimethylsilanes are converted to alkenyl
fluorides by reaction with one equivalent of Selectfluor™
(1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane
bis(tetrafluoroborate)), or difluoromethyl-substituted alcohols, ethers or
amides using an excess of Selectfluor™ in the presence of
various nucleophiles.
Co-reporter:Marc Schuman Dr.;Michael Trevitt;Andrew Redd;Véronique Gouverneur Dr.
Angewandte Chemie 2000 Volume 112(Issue 14) pp:
Publication Date(Web):11 JUL 2000
DOI:10.1002/1521-3757(20000717)112:14<2604::AID-ANGE2604>3.0.CO;2-#
Co-reporter:Raul Pereira, Lukas Pfeifer, Jean Fournier, Véronique Gouverneur and Ján Cvengroš
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 3) pp:NaN633-633
Publication Date(Web):2016/12/06
DOI:10.1039/C6OB02359B
The typically planar amide when incorporated into bicyclic systems can undergo a significant distortion from planarity resulting in physical properties and reactivity that deviate from classical amide behaviour. Herein, we report a succinct protocol that utilises potassium permanganate to selectively α-oxygenate the benzylic position of ethano-Tröger's base derivatives to yield a new class of twisted bisamides. Additionally, we report the first synthesis of an ethano-Tröger's base derivative bearing substituents in the positions ortho to the nitrogen atoms.
Co-reporter:Thomas C. Wilson, Greg McSweeney, Sean Preshlock, Stefan Verhoog, Matthew Tredwell, Thomas Cailly and Véronique Gouverneur
Chemical Communications 2016 - vol. 52(Issue 90) pp:NaN13280-13280
Publication Date(Web):2016/10/12
DOI:10.1039/C6CC07417K
A general method for the copper mediated nucleophilic 123I-iodination of (hetero)aryl boronic esters and acids has been developed. The broad substrate scope of this radiosynthetic approach allows access to [123I]DPA-713, [123I]IMPY, [123I]MIBG and [123I]IPEB that are four commonly used SPECT radiotracers. Our results infer that aryl boronic reagents can now be employed as common precursors for both fluorine-18 and iodine-123 radiolabelling.
Co-reporter:Ida S. R. Stenhagen, Anna K. Kirjavainen, Sarita J. Forsback, Charlotte G. Jørgensen, Edward G. Robins, Sajinder K. Luthra, Olof Solin and Véronique Gouverneur
Chemical Communications 2013 - vol. 49(Issue 14) pp:NaN1388-1388
Publication Date(Web):2013/01/03
DOI:10.1039/C2CC38646A
The Ag-mediated electrophilic [18F]fluorination of an arylboronic ester is reported. This new radiochemical transformation uses [18F]selectfluor bis(triflate) in acetone. The process gave 6-[18F]fluoro-L-DOPA with a RCY of 19 ± 12% and a specific activity of 2.6 ± 0.3 GBq μmol−1.
Co-reporter:Charlotte Hollingworth and Véronique Gouverneur
Chemical Communications 2012 - vol. 48(Issue 24) pp:NaN2942-2942
Publication Date(Web):2011/12/12
DOI:10.1039/C2CC16158C
Transition metal catalyzed transformations using fluorinating reagents have been developed extensively for the preparation of synthetically valuable fluorinated targets. This is a topic of critical importance to facilitate laboratory and industrial chemical synthesis of fluorine containing pharmaceuticals and agrochemicals. Translation to 18F-radiochemistry is also emerging as a vibrant research field because functional imaging based on Positron Emission Tomography (PET) is increasingly used for both diagnosis and pharmaceutical development. This review summarizes how fluoride sources have been used for the catalytic nucleophilic fluorination of various substrates inclusive of aryl triflates, alkynes, allylic halides, allylic esters, allylic trichloroacetimidates, benzylic halides, tertiary alkyl halides and epoxides. Until recently, progress in this field of research has been slow in part because of the challenges associated with the dual reactivity profile of fluoride (nucleophile or base). Despite these difficulties, some remarkable breakthroughs have emerged. This includes the demonstration that Pd(0)/Pd(II)-catalyzed nucleophilic fluorination to access fluoroarenes from aryl triflates is feasible, and the first examples of Tsuji–Trost allylic alkylation with fluoride using either allyl chlorides or allyl precursors bearing O-leaving groups. More recently, allylic fluorides were also made accessible under iridium catalysis. Another reaction, which has been greatly improved based on careful mechanistic work, is the catalytic asymmetric hydrofluorination of meso epoxides. Notably, each individual transition metal catalyzed nucleophilic fluorination reported to date employs a different F-reagent, an observation indicating that this area of research will benefit from a larger pool of nucleophilic fluoride sources. In this context, a striking recent development is the successful design, synthesis and applications of a fluoride-derived electrophilic late stage fluorination reagent. This new class of reagents could greatly benefit preclinical and clinical PET imaging.
Co-reporter:James Stephen Harvey and Véronique Gouverneur
Chemical Communications 2010 - vol. 46(Issue 40) pp:NaN7485-7485
Publication Date(Web):2010/09/13
DOI:10.1039/C0CC01939A
Catalytic enantioselective strategies have become synthetically useful to access P-stereogenic phosphines. To date, enantioselective desymmetrisations and dynamic kinetic resolutions dominate the field. Desymmetrisation strategies do not necessarily require the formation of a P-carbon or P-heteroatom bond. This approach has been validated with variable levels of success using organocatalysed asymmetric deprotonation (chiral diamine) or methylation (phase transfer catalysis), enzyme-mediated esterification, rhodium catalysed [2+2+2] cycloadditions and more recently molybdenum-based ring closing metathesis. The dynamic kinetic resolution of racemic P-templates relying on a P–C bond-forming event has been the object of extensive investigations, which have culminated with the arylation and alkylation (benzylation) of equilibrating diastereomeric palladium, platinum or ruthenium phosphido complexes. Although all these routes allow access to a myriad of highly interesting P-stereogenic compounds, the level of enantiocontrol is substrate- and reactant-dependent. Pleasingly, ee’s up to 98% were obtained on selected systems.
Co-reporter:Harriet Teare, Edward G. Robins, Erik Årstad, Sajinder K. Luthra and Véronique Gouverneur
Chemical Communications 2007(Issue 23) pp:NaN2332-2332
Publication Date(Web):2007/03/14
DOI:10.1039/B701177F
A novel [18F]NF reagent and two novel radiochemical transformations have been developed: [18F]NFSi has been prepared from sodium dibenzenesulfonimide and reacted in the presence of silyl enol ethers and allylsilanes to deliver labelled fluorinated ketones and allylic fluorides respectively; the radiosynthesis of the fluorinated A ring of vitamin D3 has also been completed with success.
Co-reporter:Lei Li, Matthew N. Hopkinson, Rodrigue Leuma Yona, Romain Bejot, Antony D. Gee and Véronique Gouverneur
Chemical Science (2010-Present) 2011 - vol. 2(Issue 1) pp:NaN131-131
Publication Date(Web):2010/10/01
DOI:10.1039/C0SC00362J
The availability of radiolabelled probes is important for in vivo studies by positron emission tomography (PET). Among the frontier challenges in 18F-radiochemistry are the interconnected goals of increasing synthetic efficiency and diversity in the construction of 18F-labelled radiotracers. 18F-Radioretrosynthetic strategies implemented to date are typically linear sequences of transformations designed with the aim of introducing the 18F-label ideally in the last step or at least as late as possible. Here we report that convergent 18F-radiochemistry allows for the rapid assembly of functionalised 18F-radiotracers from readily accessible 18F-labelled prosthetic groups. Using multicomponent reactions for proof of concept, Ugi-4CR, Passerini-3CR, Biginelli-3CR and Groebke-3CR were performed successfully using 18F-benzaldehydes and these highly convergent reactions delivered, in high radiochemical yield (RCY), structurally complex 18F-radiotracers with the label positioned on an aryl motif not responsive to direct nucleophilic fluorination. These data establish an unprecedented connection between radiochemistry for PET and the field of multicomponent chemistry and demonstrate that convergent retroradiosynthesis is a powerful strategy expanding dramatically the scope of 18F-prosthetic group radiochemistry. The preparation of 18F-labelled prosthetic groups from [18F]fluoride ion is commonly performed in many labelling laboratories, so the concept of convergent 18F-radiosynthesis can easily be applied immediately.
Co-reporter:Laurence Carroll, Samantha McCullough, Tom Rees, Timothy D. W. Claridge and Véronique Gouverneur
Organic & Biomolecular Chemistry 2008 - vol. 6(Issue 10) pp:NaN1733-1733
Publication Date(Web):2008/04/03
DOI:10.1039/B803888K
The electrophilic fluorodesilylation of enantioenriched allenylsilanes proceeds with efficient transfer of chirality. The silylation–fluorination of propargylic alcohols occurs with overall retention of stereochemistry, a result consistent with a stereospecific anti SE2′ mechanism for the fluorination step.
Co-reporter:Laurent Bonnac, Sarah E. Lee, Guy T. Giuffredi, Lucy M. Elphick, Alexandra A. Anderson, Emma S. Child, David J. Mann and Véronique Gouverneur
Organic & Biomolecular Chemistry 2010 - vol. 8(Issue 6) pp:NaN1454-1454
Publication Date(Web):2010/01/25
DOI:10.1039/B922442D
Enantioenriched tetrafluorinated aryl-C-nucleosides were synthesised in four steps from 1-benzyloxy-4-bromo-3,3,4,4-tetrafluorobutan-2-ol. The presence of the tetrafluorinated ethylene group is compatible with O-phosphorylation of the primary alcohol, as demonstrated by the successful preparation of the tetrafluorinated naphthyl-C-nucleotide.
Co-reporter:Laurence Carroll, Sophie Boldon, Romain Bejot, Jane E. Moore, Jérôme Declerck and Véronique Gouverneur
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 1) pp:NaN140-140
Publication Date(Web):2010/11/22
DOI:10.1039/C0OB00564A
The Staudinger ligation of phosphine-substituted thioesters with 18F-fluoroethylazide has been successfully applied to access 18F-labelled molecules in radiochemical yields superior to 95%; the first fluorous variant of a Staudinger radio-ligation has been validated.
Co-reporter:Sophie Purser, Peter R. Moore, Steve Swallow and Véronique Gouverneur
Chemical Society Reviews 2008 - vol. 37(Issue 2) pp:NaN330-330
Publication Date(Web):2007/12/13
DOI:10.1039/B610213C
It has become evident that fluorinated compounds have a remarkable record in medicinal chemistry and will play a continuing role in providing lead compounds for therapeutic applications. This tutorial review provides a sampling of renowned fluorinated drugs and their mode of action with a discussion clarifying the role and impact of fluorine substitution on drug potency.
Co-reporter:Keary M. Engle, Lukas Pfeifer, George W. Pidgeon, Guy T. Giuffredi, Amber L. Thompson, Robert S. Paton, John M. Brown and Véronique Gouverneur
Chemical Science (2010-Present) 2015 - vol. 6(Issue 9) pp:NaN5302-5302
Publication Date(Web):2015/06/22
DOI:10.1039/C5SC01812A
The nucleophilic reactivity of fluoride ion is altered in the presence of hydrogen-bond donors, including alcohols. Relatively little is known about the coordination involved; to rectify this, the X-ray structures of fourteen novel fluoride–alcohol complexes with tetrabutylammonium as the counterion have been determined. The coordination number varies from two to four depending on the steric bulk of the alcohol and is closely linked to trends in reactivity. This diversity in coordination stoichiometry is unprecedented but significant, as it implies differences in the ability of the fluoride–alcohol complexes to dissociate in solution with release of a more active and/or selective fluoride source.
Co-reporter:Elena Benedetto, Matthew Tredwell, Charlotte Hollingworth, Tanatorn Khotavivattana, John M. Brown and Véronique Gouverneur
Chemical Science (2010-Present) 2013 - vol. 4(Issue 1) pp:NaN96-96
Publication Date(Web):2012/10/24
DOI:10.1039/C2SC21789A
This paper describes a new catalytic method for the regio- and stereocontrolled fluorination of allylic carbonates. This transformation uses TBAF·4tBuOH as the fluoride source and [Ir(COD)Cl]2 as the catalyst; the most commonly used [Ir(COD)Cl]2/phosphoramidite system is ineffective. Synthetically, this reaction is characterized by a high degree of structural conservation in going from substrates to the products. The fluorination of (E)-allylic carbonates leading to linear (E)-allylic fluorides (l:b > 20:1, E:Z > 20:1) is unprecedented and a unique feature of fluoride as the nucleophile. The first examples of transition metal catalyzed fluorination affording (Z)-allyl fluorides (Z:E ratio >20:1) are disclosed along with the successful fluorination of branched, linear (E)- and (Z)-allyl carbonates with [18F] fluoride in the presence of [Ir(COD)Cl]2. 18O-Labeling of the reactant reveals internal return during the allylic ionization step, and pathways for effective intra- and intermolecular isotope exchange.
Co-reporter:Faye Buckingham and Véronique Gouverneur
Chemical Science (2010-Present) 2016 - vol. 7(Issue 3) pp:NaN1652-1652
Publication Date(Web):2015/12/17
DOI:10.1039/C5SC04229A
Positron emission tomography (PET) is becoming more frequently used by medicinal chemists to facilitate the selection of the most promising lead compounds for further evaluation. For PET, this entails the preparation of 11C- or 18F-labeled drugs or radioligands. With the importance of chirality and fluorine substitution in drug development, chemists can be faced with the challenge of preparing enantiopure molecules featuring the 18F-tag on a stereogenic carbon. Asymmetric 18F-fluorination is an emerging field of research that provides an alternative to resolution or conventional SN2-based radiochemistry. To date, both transition metal complexes and organomediators have been successfully employed for 18F-incorporation at a stereogenic carbon.
Co-reporter:Sophie Boldon, Jane E. Moore and Véronique Gouverneur
Chemical Communications 2008(Issue 31) pp:NaN3624-3624
Publication Date(Web):2008/06/04
DOI:10.1039/B804484H
A novel fluorous tagging–detagging strategy has been developed featuring a fluorination as the detagging process; fluorous allylsilanes were prepared by cross-metathesis and subsequently subjected to electrophilic fluorodesilylation; Selectfluor was used as the detagging reagent; the resulting allylic fluorides were successfully purified by fluorous solid phase extraction.
Co-reporter:Laurence Carroll, Romain Bejot, Rebekka Hueting, Robert King, Paul Bonnitcha, Simon Bayly, Martin Christlieb, Jonathan R. Dilworth, Antony D. Gee, Jérôme Declerck and Véronique Gouverneur
Chemical Communications 2010 - vol. 46(Issue 23) pp:NaN4054-4054
Publication Date(Web):2010/04/20
DOI:10.1039/B926980K
The synthesis of three pairs of orthogonally labelled fluorinated Cu bis(thiosemicarbazonato) complexes is presented. These are the first examples of 18F-labelled Cu(II)-complexes designed to serve as new hypoxia selective PET tracers and as mechanistic probes to study the mode of action of this class of markers. In vitro evaluation revealed that the fluorinated Cu-complex derived from amide coupling is suitable for in vivo work.
Co-reporter:Sean Preshlock, Samuel Calderwood, Stefan Verhoog, Matthew Tredwell, Mickael Huiban, Antje Hienzsch, Stefan Gruber, Thomas C. Wilson, Nicholas J. Taylor, Thomas Cailly, Michael Schedler, Thomas Lee Collier, Jan Passchier, René Smits, Jan Mollitor, Alexander Hoepping, Marco Mueller, Christophe Genicot, Joël Mercier and Véronique Gouverneur
Chemical Communications 2016 - vol. 52(Issue 54) pp:NaN8364-8364
Publication Date(Web):2016/05/23
DOI:10.1039/C6CC03295H
[18F]FMTEB, [18F]FPEB, [18F]flumazenil, [18F]DAA1106, [18F]MFBG, [18F]FDOPA, [18F]FMT and [18F]FDA are prepared from the corresponding arylboronic esters and [18F]KF/K222 in the presence of Cu(OTf)2py4. The method was successfully applied using three radiosynthetic platforms, and up to 26 GBq of non-carrier added starting activity of 18F-fluoride.
Co-reporter:Raul Pereira, Jamie Wolstenhulme, Graham Sandford, Timothy D. W. Claridge, Véronique Gouverneur and Ján Cvengroš
Chemical Communications 2016 - vol. 52(Issue 8) pp:NaN1609-1609
Publication Date(Web):2015/11/25
DOI:10.1039/C5CC08375C
Methylation of 2,8-dimethyl-6H,12H-5,11-ethanodibenzo[b,f][1,5]-diazocine (ethano-Tröger's base) with methyl iodide followed by ion metathesis and fluorination with N-fluoro-2,3,4,5,6-pentachloropyridinium triflate affords a new electrophilic N–F reagent, that is more reactive than Selectfluor. 2D 19F–15N HMQC experiments provide 1JNF coupling constants which are diagnostic for the N–F functional group.
Co-reporter:Paolo Ricci, Tanatorn Khotavivattana, Lukas Pfeifer, Maurice Médebielle, John Richard Morphy and Véronique Gouverneur
Chemical Science (2010-Present) 2017 - vol. 8(Issue 2) pp:NaN1199-1199
Publication Date(Web):2016/09/30
DOI:10.1039/C6SC02790C
Alkenes substituted with a thiourea undergo C–CF3 followed by intramolecular C–S bond formation with the Togni reagent and trifluoroacetic acid (TFA) at room temperature; thiols and thioamides are not suitable S-sources for this reaction. This anti-addition process involves a CF3 radical, and affords CF3-substituted thiazolines and thiazines for medicinal applications. A metal or photoredox catalyst is not required as the thiourea acts as a reductant, as well as serving as an S-source capable of adding to a C-centered radical. Mechanistic work comparing the reactivity of thiourea, urea, thioamide and thiol in the context of alkene trifluoromethylation demonstrates that in this series, the thiourea is unique for its ability to release CF3 radical from the Togni reagent, and to orchestrate trifluoromethylation followed by S-cyclization with both activated and unactivated alkenes.