Co-reporter:Shashank Kulkarni; Spyros P. Nikas; Rishi Sharma; Shan Jiang; Carol A. Paronis; Michael Z. Leonard; Bin Zhang; Chandrashekhar Honrao; Srikrishnan Mallipeddi; Jimit Girish Raghav; Othman Benchama; Torbjörn U. C. Järbe; Jack Bergman;Alexandros Makriyannis
Journal of Medicinal Chemistry 2016 Volume 59(Issue 14) pp:6903-6919
Publication Date(Web):July 1, 2016
DOI:10.1021/acs.jmedchem.6b00717
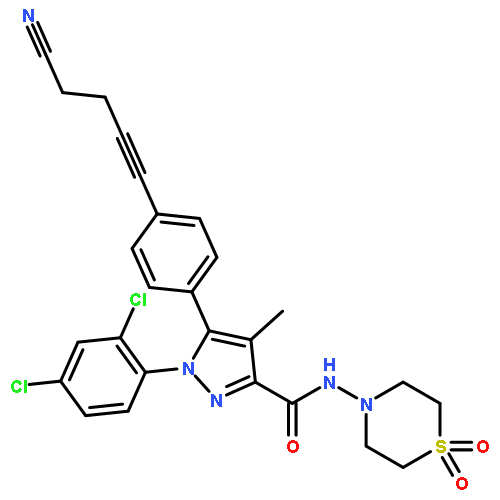
In pursuit of safer controlled-deactivation cannabinoids with high potency and short duration of action, we report the design, synthesis, and pharmacological evaluation of novel C9- and C11-hydroxy-substituted hexahydrocannabinol (HHC) and tetrahydrocannabinol (THC) analogues in which a seven atom long side chain, with or without 1′-substituents, carries a metabolically labile 2′,3′-ester group. Importantly, in vivo studies validated our controlled deactivation approach in rodents and non-human primates. The lead molecule identified here, namely, butyl-2-[(6aR,9R,10aR)-1-hydroxy-9-(hydroxymethyl)-6,6-dimethyl-6a,7,8,9,10,10a-hexahydro-6H-benzo[c]chromen-3-yl]-2-methylpropanoate (AM7499), was found to exhibit remarkably high in vitro and in vivo potency with shorter duration of action than the currently existing classical cannabinoid agonists.
Co-reporter:Rishi Sharma, Spyros P. Nikas, Jason Jianxin Guo, Srikrishnan Mallipeddi, JodiAnne T. Wood, and Alexandros Makriyannis
ACS Medicinal Chemistry Letters 2014 Volume 5(Issue 4) pp:400-404
Publication Date(Web):January 14, 2014
DOI:10.1021/ml4005304
As a part of our controlled-deactivation ligand development project, we recently disclosed a series of (−)-Δ8-tetrahydrocannabinols (THCs) with a metabolically labile ester group at the 2′-position of the side chain. Now, we have replaced the C-ring in the classical THC structure with a hydrolyzable seven-membered lactone. One of the synthesized analogues binds with high affinity to the CB1 receptor (Ki = 4.6 nM) and exhibits much lower affinities for the mCB2 and the hCB2. Also, in vitro functional characterization found the compound to be an agonist at rCB1. Consistent with our rational design, the lead cannabinergic lactone identified here is susceptible to metabolic inactivation by plasma esterases, while the respective acid metabolite is inactive at CB receptors. These results are highlighted with molecular modeling of the two regiosomeric lactones.Keywords: Baeyer−Villiger rearrangement; Cannabinoids; lactones;
Co-reporter:Rishi Sharma ; Spyros P. Nikas ; Carol A. Paronis ; JodiAnne T. Wood ; Aneetha Halikhedkar ; Jason Jianxin Guo ; Ganesh A. Thakur ; Shashank Kulkarni ; Othman Benchama ; Jimit Girish Raghav ; Roger S. Gifford ; Torbjörn U. C. Järbe ; Jack Bergman ;Alexandros Makriyannis
Journal of Medicinal Chemistry 2013 Volume 56(Issue 24) pp:10142-10157
Publication Date(Web):November 28, 2013
DOI:10.1021/jm4016075
We report an approach for obtaining novel cannabinoid analogues with controllable deactivation and improved druggability. Our design involves the incorporation of a metabolically labile ester group at the 2′-position on a series of (−)-Δ8-THC analogues. We have sought to introduce benzylic substituents α to the ester group which affect the half-lives of deactivation through enzymatic activity while enhancing the affinities and efficacies of individual ligands for the CB1 and CB2 receptors. The 1′-(S)-methyl, 1′-gem-dimethyl, and 1′-cyclobutyl analogues exhibit remarkably high affinities for both CB receptors. The novel ligands are susceptible to enzymatic hydrolysis by plasma esterases in a controllable manner, while their metabolites are inactive at the CB receptors. In further in vitro and in vivo experiments key analogues were shown to be potent CB1 receptor agonists and to exhibit CB1-mediated hypothermic and analgesic effects.
Co-reporter:Shakiru O. Alapafuja ; Spyros P. Nikas ; Indu T. Bharathan ; Vidyanand G. Shukla ; Mahmoud L. Nasr ; Anna L. Bowman ; Nikolai Zvonok ; Jing Li ; Xiaomeng Shi ; John R. Engen ;Alexandros Makriyannis
Journal of Medicinal Chemistry 2012 Volume 55(Issue 22) pp:10074-10089
Publication Date(Web):October 19, 2012
DOI:10.1021/jm301205j
Sulfonyl fluorides are known to inhibit esterases. Early work from our laboratory has identified hexadecyl sulfonylfluoride (AM374) as a potent in vitro and in vivo inhibitor of fatty acid amide hydrolase (FAAH). We now report on later generation sulfonyl fluoride analogs that exhibit potent and selective inhibition of FAAH. Using recombinant rat and human FAAH, we show that 5-(4-hydroxyphenyl)pentanesulfonyl fluoride (AM3506) has similar inhibitory activity for both the rat and the human enzyme, while rapid dilution assays and mass spectrometry analysis suggest that the compound is a covalent modifier for FAAH and inhibits its action in an irreversible manner. Our SAR results are highlighted by molecular docking of key analogs.
Co-reporter:Spyros P. Nikas, Marsha D'Souza, Alexandros Makriyannis
Tetrahedron 2012 68(31) pp: 6329-6337
Publication Date(Web):
DOI:10.1016/j.tet.2012.05.010
Co-reporter:Spyros P. Nikas ; Shakiru O. Alapafuja ; Ioannis Papanastasiou ; Carol A. Paronis ; Vidyanand G. Shukla ; Demetris P. Papahatjis ; Anna L. Bowman ; Aneetha Halikhedkar ; Xiuwen Han ;Alexandros Makriyannis
Journal of Medicinal Chemistry 2010 Volume 53(Issue 19) pp:6996-7010
Publication Date(Web):September 9, 2010
DOI:10.1021/jm100641g
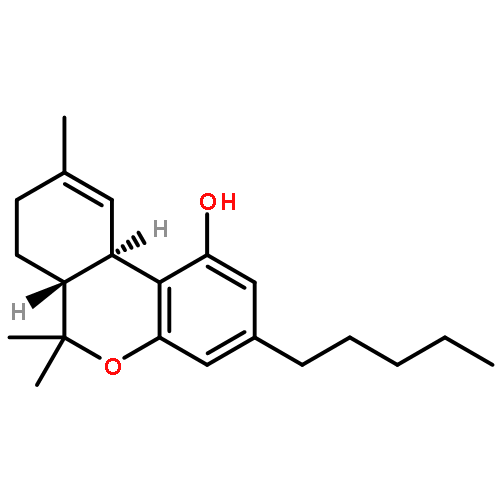
In pursuit of a more detailed understanding of the structural requirements for the key side chain cannabinoid pharmacophore, we have extended our SAR to cover a variety of conformationally modified side chains within the 9-keto and 9-hydroxyl tricyclic structures. Of the compounds described here, those with a seven-atom long side chain substituted with a cyclopentyl ring at C1′ position have very high affinities for both CB1 and CB2 (0.97 nM < Ki < 5.25 nM), with no preference for either of the two receptors. However, presence of the smaller cyclobutyl group at the C1′ position leads to an optimal affinity and selectivity interaction with CB1. Thus, two of the C1′-cyclobutyl analogues, namely, (6aR,10aR)-3-(1-hexyl-cyclobut-1-yl)-6,6a,7,8,10,10a-hexahydro-1-hydroxy-6,6-dimethyl-9H-dibenzo[b,d]pyran-9-one and (6aR,9R,10aR)-3-(1-hexyl-cyclobut-1-yl)-6a,7,8,9,10,10a-hexahydro-6,6-dimethyl-6H-dibenzo[b,d]pyran-1,9 diol (7e-β, AM2389), exhibited remarkably high affinities (0.84 and 0.16 nM, respectively) and significant selectivities (16- and 26-fold, respectively) for CB1. Compound 7e-β was found to exhibit exceptionally high in vitro and in vivo potency with a relatively long duration of action.

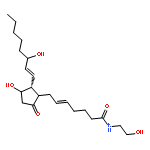
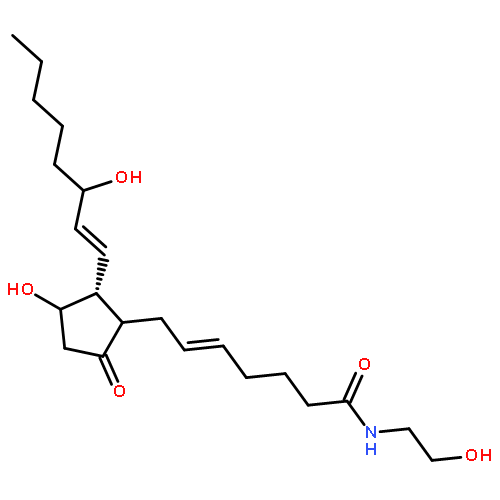
![2H-Cyclopenta[b]furan-4-carboxaldehyde,5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]hexahydro-2-oxo-, [3aR-(3aa,4a,5b,6aa)]- (9CI)](http://img.cochemist.com/ccimg/64100/64091-14-1.png)
![2H-Cyclopenta[b]furan-4-carboxaldehyde,5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]hexahydro-2-oxo-, [3aR-(3aa,4a,5b,6aa)]- (9CI)](http://img.cochemist.com/ccimg/64100/64091-14-1_b.png)
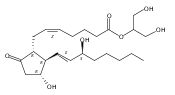
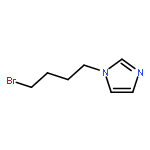
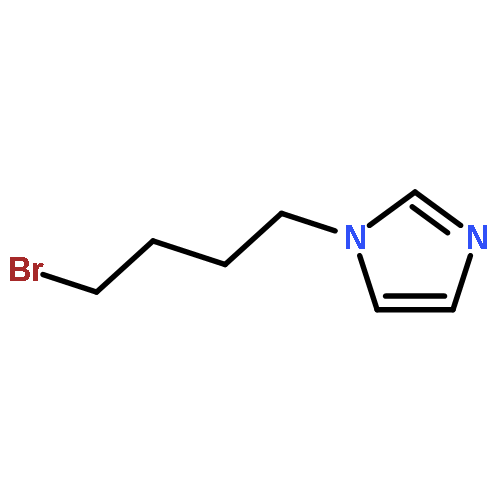
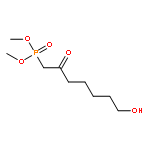
![1-Propanamine, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/115400/115306-75-7.png)
![1-Propanamine, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/115400/115306-75-7_b.png)


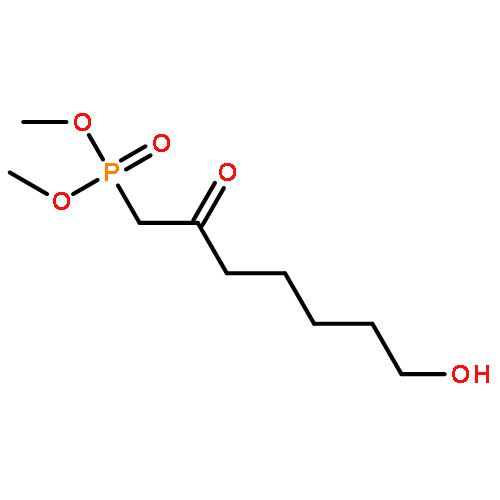

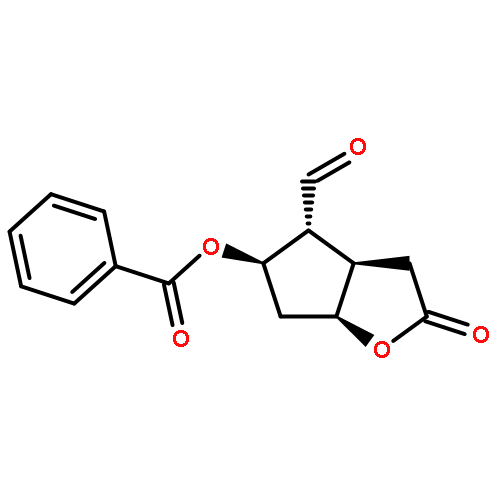


![Bicyclo[3.1.1]hept-3-en-2-one, 6,6-dimethyl-, (1R)-](http://img.cochemist.com/ccimg/35500/35408-03-8.png)
![Bicyclo[3.1.1]hept-3-en-2-one, 6,6-dimethyl-, (1R)-](http://img.cochemist.com/ccimg/35500/35408-03-8_b.png)


![(3aR,4R,5R,6aS)-5-Hydroxy-4-((S,E)-3-hydroxyoct-1-en-1-yl)hexahydro-2H-cyclopenta[b]furan-2-one](http://img.cochemist.com/ccimg/26100/26054-67-1.png)
![(3aR,4R,5R,6aS)-5-Hydroxy-4-((S,E)-3-hydroxyoct-1-en-1-yl)hexahydro-2H-cyclopenta[b]furan-2-one](http://img.cochemist.com/ccimg/26100/26054-67-1_b.png)
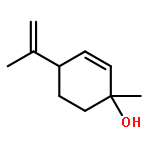
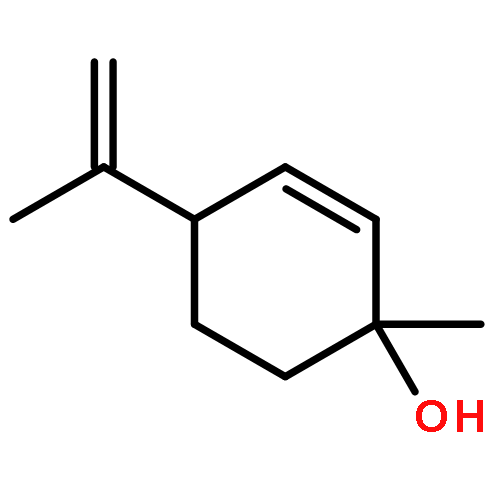
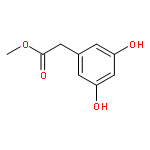
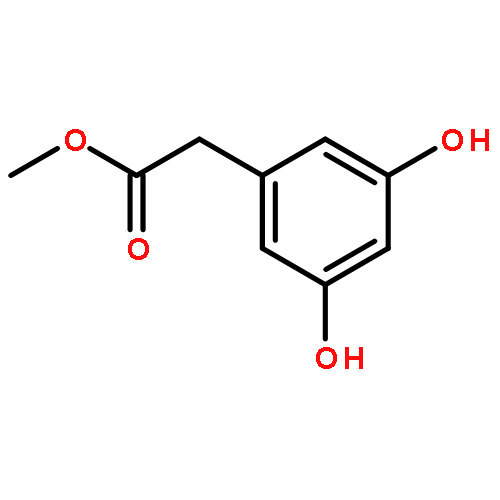
![BICYCLO[3.1.1]HEPT-2-ENE-2,4-DIOL, 6,6-DIMETHYL-, DIACETATE, (1R,5S)-](http://img.cochemist.com/ccimg/487600/487578-94-9.png)
![BICYCLO[3.1.1]HEPT-2-ENE-2,4-DIOL, 6,6-DIMETHYL-, DIACETATE, (1R,5S)-](http://img.cochemist.com/ccimg/487600/487578-94-9_b.png)
![2-Propanamine, 1-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (2R)-](http://img.cochemist.com/ccimg/175800/175717-73-4.png)
![2-Propanamine, 1-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (2R)-](http://img.cochemist.com/ccimg/175800/175717-73-4_b.png)
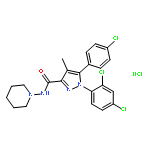
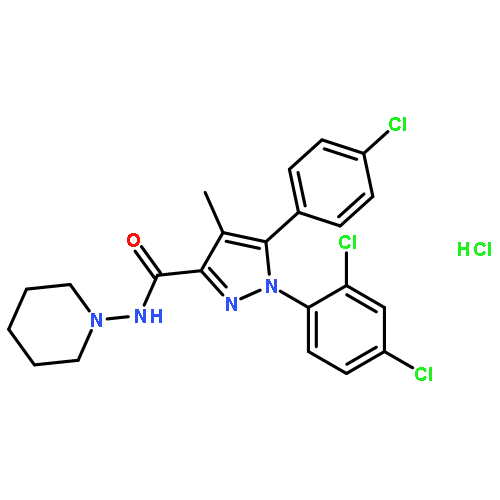
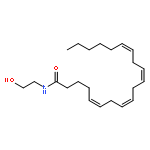
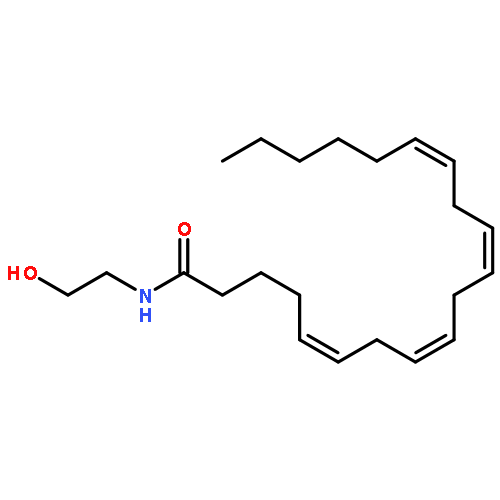
![Bicyclo[3.1.1]hept-3-ene-2,2-diol, 6,6-dimethyl-, diacetate, (1R,5R)-](http://img.cochemist.com/ccimg/82100/82078-77-1.png)
![Bicyclo[3.1.1]hept-3-ene-2,2-diol, 6,6-dimethyl-, diacetate, (1R,5R)-](http://img.cochemist.com/ccimg/82100/82078-77-1_b.png)