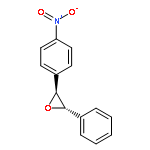
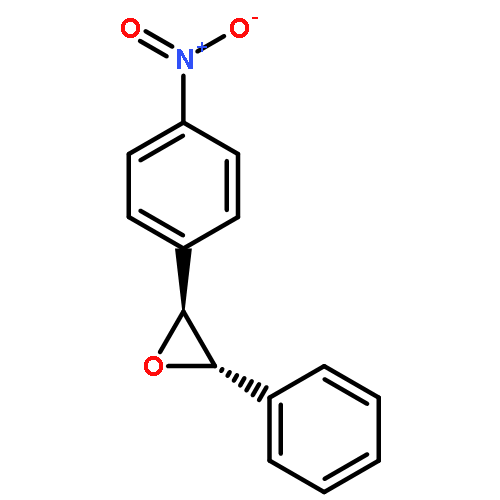
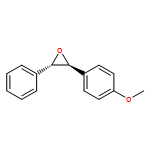
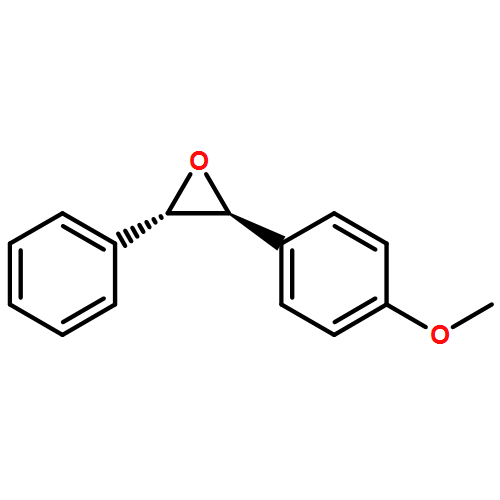
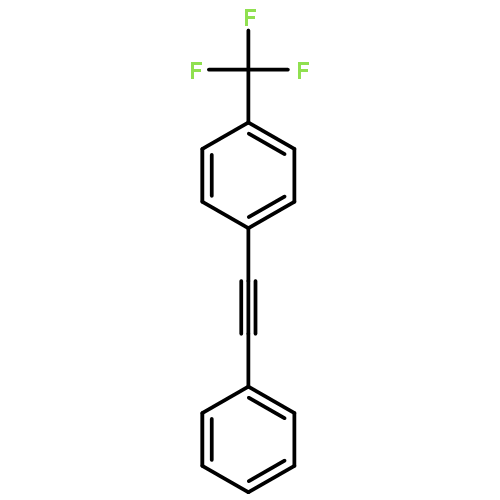
Co-reporter:Hemantkumar M. Savanur, Rajesh G. Kalkhambkar, Kenneth K. Laali
Applied Catalysis A: General 2017 Volume 543(Volume 543) pp:
Publication Date(Web):5 August 2017
DOI:10.1016/j.apcata.2017.06.015
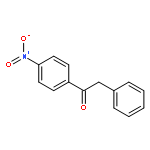
•[PAIM][NTf2]/[BMIM][X]/Pd-catalyst for Suzuki and Heck cross-coupling without additive.•[PAIM][NTf2]/[BMIM][X] for Horner-Emmons and Wittig mono- and bis-olefinations.•The Wittig-Suzuki, Wittig-Heck, Horner-Emmons-Suzuki, and Horner-Emmons-Heck hyphenated one-pot protocols.•Synthesis of a diverse pool of biaryls, diaryl-ethenes, and aryl-enoates.•Feasibility to recycle and reuse [BMIM][X].Facile, high yielding, one-pot methods for the synthesis of a library of diversely substituted bi-aryls, diarylethenes, and aryl-enoates, via Suzuki and Heck reactions, and by sequential Wittig-Suzuki, Wittig-Heck, Horner-Emmons-Suzuki, and Horner-Emmons-Heck reactions are reported. The reactions employ piperidine-appended imidazolium ionic liquid [PAIM][NTf2] as a task-specific basic-IL, butyl-methyl-imidazolium ionic liquid [BMIM][X] (X = PF6, BF4) as solvent, and catalytic amounts of Pd(OAc)2, with no other additives. Wittig and Horner-Emmons reactions are effected by reacting substituted benzaldehydes with 4-bromobenzyl-PPh3 (or bromomethyl-PPh3) phosphonium salts, or diethylphosphonate with bromobenzaldehydes respectively, to form the corresponding ethenes. Subsequent cross-coupling reactions are accomplished by addition of aryl-boronic acid or phenyl-ethenes along with Pd(OAc)2 to bring about the aforementioned hyphenated transformations. The feasibility to perform double-olefination via Wittig and Horner-Emmons reactions with dialdehydes to form highly conjugated bis-styryl and bis-enoate compounds is also shown. The [BMIM][X] solvent is recycled and reused.Download high-res image (195KB)Download full-size image
Co-reporter:Gabriela L. Borosky, Kenneth K. Laali
Journal of Fluorine Chemistry 2017 Volume 197(Volume 197) pp:
Publication Date(Web):1 May 2017
DOI:10.1016/j.jfluchem.2017.04.002
•SF5-bearing carbocations.•Relative electronegativity of SF5 compared to other strongly electron-withdrawing groups.•Evidence for fluoronium ion character in SF5-bearing benzenium ions and benzylic carbocations.The effect of the SF5 group as a substituent on various classes of carbocation was probed computationally employing DFT to examine geometrical changes, relative energies, and charge delocalization modes. Relative electron withdrawing power of the SF5 group was compared with CF3, NO2, CN, and SCF3 through isodesmic reactions. NPA charge densities were employed to gauge the influence of SF5 group on the charge delocalization modes in the carbocations.Download high-res image (97KB)Download full-size image
Co-reporter:Kenneth K. Laali;Arezu Jamalian;Gabriela L. Borosky
Journal of Physical Organic Chemistry 2016 Volume 29( Issue 7) pp:346-351
Publication Date(Web):
DOI:10.1002/poc.3541
Imidazolium ionic liquids (IMILs) with a piperidine moiety appended via variable length methylene spacers (with n = 1–4) were studied computationally to assess their potential to act as internal base for N-heterocyclic carbene (NHC) generation. Proton transfer energies computed by B3LYP/6-311+G(2d,p) were least endothermic for the basic-IL with n = 3, whose optimized structure showed the shortest C2-H----N(piperidine) distance. Inclusion of counter anion (Cl or NTf2) caused dramatic conformational changes to enable close contact between the acidic C2-H and the anions. To examine the prospect for internal C2-H----N coordination, multinuclear NMR data (1H, 15N, and 13C) were computed by gauge independent atomic orbitals–density functional theory (GIAO-DFT) in the gas phase and in several solvents by the PCM method for comparison with the experimental NMR data for the basic ILs (with n = 2–4) synthesized in the laboratory. These studies indicate that interactions with solvent and counter ion are dominant forces that could disrupt internal C2-H----N coordination/proton transfer, making carbene generation from these basic-ILs unlikely without an added external base. Therefore, the piperidine-appended IMILs appear suitable for application as dual solvent/base in organic/organometallic transformations that require the use of mild base, without the necessity to alkylate at C-2 to prevent N-heterocyclic carbene formation. Copyright © 2016 John Wiley & Sons, Ltd.
Co-reporter:Hemantkumar M. Savanur, Rajesh G. Kalkhambkar, Gopalakrishnan Aridoss, Kenneth K. Laali
Tetrahedron Letters 2016 Volume 57(27–28) pp:3029-3035
Publication Date(Web):6 July 2016
DOI:10.1016/j.tetlet.2016.05.103
•Use of readily available shelf-stable ILs and mild conditions.•Comparable or superior results compared to less table/less common ILs.•Meet positive improvement criteria with respect to reactant ratios, catalyst amounts, reaction times, and yields.•Recycling and reuse of the IL solvent.A facile, high yielding, and simple method for the synthesis of a library of dihydropyrimidinones (and -thiones) via the one-pot Biginelli reaction of aldehydes is reported. The method employs the Brønsted acidic IL [bmim(SO3H][OTf] or Zn(NTf2)2 as catalyst along with the readily available imidazolium-ILs as solvent, and exhibits good potential for the recycling and reuse of the IL solvent.
Co-reporter:Hemantkumar M. Savanur, Rajesh G. Kalkhambkar, Kenneth K. Laali
Tetrahedron Letters 2016 Volume 57(Issue 6) pp:663-667
Publication Date(Web):10 February 2016
DOI:10.1016/j.tetlet.2015.12.108
A facile, high yielding, and simple method for the synthesis of a library of symmetrical biaryls by homocoupling of arenediazonium salts is reported, employing catalytic amounts of Pd(OAc)2 and the readily available imidazolium ionic liquids (ILs), without oxidants, ligands, additives, or volatile solvents. Simple product isolation and recycling/reuse of the IL represent additional advantages of this method.
Co-reporter:Kenneth K. Laali, Benjamin M. Rathman, Scott D. Bunge, Xin Qi, Gabriela L. Borosky
Journal of Fluorine Chemistry 2016 Volume 191() pp:29-41
Publication Date(Web):November 2016
DOI:10.1016/j.jfluchem.2016.09.009
•Synthesis of α-fluorinated curcuminoids by direct mono- and difluorination.•Synthesis of ring fluorinated curcuminoids.•Isolation and characterization of curcuminoid-BF2 adducts.•X-ray structure analysis.•Bioassay and computational docking study.A series of α-carbonyl fluorinated curcuminoids were synthesized by direct mono- and difluorination with Selectfluor (F-TEDA-BF4) at r.t. without using a base or additive. Ring fluorinated/trifluoromethylated curcuminoid-BF2 adducts were synthesized by reaction of the corresponding benzaldehydes with acetylacetone-BF2. Decomplexation of CUR-BF2 adducts under microwave irradiation gave the corresponding curcuminoids. Multinuclear NMR and X-ray analysis confirm that curcuminoids bearing fluorines or trifluoromethyl groups in the aryl rings as well as those that are monofluorinated at the active methylene position all exist as enol tautomers. The α,α-difluorination brings about significant conformational change as these curcuminoids become fixed in the 1,3-diketone configuration. The X-ray structures of CUR-BF2 complexes are consistent with the formation of symmetrical adducts with equal BO bond distances. The anti-proliferative activity of these compounds were tested by in-vitro bioassay against several different cancer cell lines. The corresponding CUR-BF2 adducts exhibited exceptionally high activities at micromolar and in some cases nanomolar concentrations that greatly surpass the activity of parent curcumin. Computational docking calculations were performed to gauge binding energies of these compounds in HER2 protein, and in 20S proteasome, for comparison with the bioassay-derived activity data.
Co-reporter:A. Srinivas Reddy, Kenneth K. Laali
Tetrahedron Letters 2015 Volume 56(Issue 33) pp:4807-4810
Publication Date(Web):12 August 2015
DOI:10.1016/j.tetlet.2015.06.067
Application of a piperidine-appended dimethyl-imidazolium-NTf2 ionic liquid as dual solvent and base in the Sonogashira cross-coupling reaction of aryl-iodides with terminal acetylenes under mild conditions has been demonstrated. The method employs PdCl2(PPh3)2 without copper and external base. It is applicable to the synthesis of SF5-substituted diaryl- and aryl-alkyl-acetylenes, and can also be utilized for efficient homo-coupling of terminal acetylenes under aerobic conditions. The potential for recycling and reuse of this designer-IL offers an added advantage.
Co-reporter:A. Srinivas Reddy, Kenneth K. Laali
Tetrahedron Letters 2015 Volume 56(Issue 41) pp:5495-5499
Publication Date(Web):7 October 2015
DOI:10.1016/j.tetlet.2015.07.084
Structurally diverse ketones, 1,3-diketones, and β-ketoesters, were selectively monofluorinated with Selectfluor (F-TEDA-BF4) (1 equiv) in [BMIM][PF6] as solvent and [PMIM(SO3H)][OTf] as promoter under mild conditions. In selected cases, the monofluorinated products were transformed to the gem-difluoro derivatives by employing an additional equivalent of Selectfluor, and gem-difluoro-derivatives were synthesized directly from the substrates by employing 2 equiv of Selectfluor. The method was extended to monofluorination of representative α-nitroketones and β-ketonitriles using [BMIM][NTf2] without the need for promoters. The described method offers the added advantage of recycling and reuse of the IL solvent.Figure options
Co-reporter:Takao Okazaki;Kenneth K. Laali;Scott D. Bunge;Sonya K. Adas
European Journal of Organic Chemistry 2014 Volume 2014( Issue 8) pp:1630-1644
Publication Date(Web):
DOI:10.1002/ejoc.201301538
Abstract
The reagent 4-(pentafluorosulfanyl)benzenediazonium tetrafluoroborate (1) was synthesized and isolated as a stable salt for the first time; its application in a wide assortment of transformations was subsequently investigated. A series of novel SF5-bearing alkenes, alkynes, and biaryl derivatives were synthesized by employing Heck–Matsuda, Sonogashira, and Suzuki coupling protocols. Dediazoniation with TMSX (X = Hal; N3; and CN) and NH4SCN in [BMIM][BF4] as solvent furnished the corresponding p-SF5–C6H4X derivatives. The azide derivative p-SF5–C6H4N3 entered into click chemistry with phenylacetylenes to furnish the corresponding triazoles. The 4,4′-bis(pentafluorosulfanyl)biphenyl was synthesized by homo-coupling using Pd(OAc)2. The corresponding azo compounds were obtained through azo-coupling with reactive aromatic nucleophiles (1,3-dimethoxybenzene, 1,3,5-trimethoxybenzene, 1,2,4-trimethoxybenzene, aniline and phenol). Fluorodediazoniation in [BMIM][PF6] and [BMIM][BF4] selectively furnished the fluoro derivative p-SF5–C6H4F. Dediazoniation in [BMIM][NTf2] gave p-SF5–C6H4OS(O)(CF3)=NSO2CF3 as the major and p-SF5–C6H4-NTf2 as the minor products. Homolytic dediazoniation in MeCN/NaI gave the unsymmetrical biaryls p-SF5–C6H4-Ar (ArH = mesitylene and p-xylene) along with p-SF5–C6H4I. Analysis of the dediazoniation product mixtures indicated that dediazoniation of p-SF5–C6H4N2+ BF4– in low nucleophilicity, highly ionizing, solvents (TfOH, TFE, HFIP, TFAH) is mainly heterolytic, while in MeOH it is mainly homolytic. The isodesmic reaction p-SF5–C6H4+ + R-C6H5 SF5–C6H5 + p-R-C6H4+ (with R = NO2, CF3, H) was gauged by DFT at various levels and by PCM.
Co-reporter:Takao Okazaki, Kenneth K. Laali
Journal of Fluorine Chemistry 2014 Volume 165() pp:96-100
Publication Date(Web):September 2014
DOI:10.1016/j.jfluchem.2014.06.024
•Alternative methods for mono- and dinitration of ArSF5 are reported.•Competitive nitration of PhSF5 vs PhNO2 and PhSF5 vs PhCF3.•Relative energies of the benzenium ion intermediates by DFT calculations.PhSF51 reacts with NO2+BF4−/TfOH in CH2Cl2 (DCM) at room temperature to give 1-nitro-3-(pentafluorosulfanyl)benzene 2 in near quantitative yield. The dinitro derivative 4 is synthesized from 2 by reaction with NO2+BF4−/TfOH at 70 °C. The p-MeC6H4SF5 is mononitrated at room temperature with NO2+BF4−/DCM and dinitrated with NO2+BF4−/TfOH. Substrate selectivity (kPhSF5/kRPhkPhSF5/kRPh) in competitive nitration for PhSF5/PhCF3 and PhSF5/PhNO2 with NO2+BF4− in DCM at room temperature was determined at 21.3 and ∼1 respectively. Relative stability of the corresponding benzenium ions were gauged by DFT from the isodesmic proton transfer reaction SF5–C6H6+ + R-C6H5 → SF5–C6H5 + R-C6H6+ (R = CF3 and NO2). These studies indicate that reactivity of ArSF5 in SEAr is similar to ArNO2.
Co-reporter:Kenneth K. Laali, Ganesh C. Nandi, Scott D. Bunge
Tetrahedron Letters 2014 Volume 55(Issue 15) pp:2401-2405
Publication Date(Web):9 April 2014
DOI:10.1016/j.tetlet.2014.02.110
Presence of TMSX (X = Cl, Br, I) unleashes the oxidative character of Selectfluor and provides a mild dihalogenation method for 1-arylallenes. Preference for 2,3-addition was observed with TMSCl in MeCN irrespective of the nature of the substituent on the aryl moiety, whereas 1,2-addition was preferred in [BMIM][BF4]. With TMSBr and TMSI only products corresponding to 2,3-addition were observed. Reactions carried out with TMSBr in IL solvents gave the corresponding monobromoalkenes as a major product along with the isomeric dibromo-alkenes. Reaction with NH4SCN provided convenient access to dithiocyanate derivatives. The same products were formed via TMS-NCS/Selectfluor. Formation of common products via TMSNCS and NH4SCN points to the formation and interplay of SCN+/NCS+ as incipient electrophiles.
Co-reporter:Kenneth K. Laali, Daniel Insuasty, Rodrigo Abonia, Braulio Insuasty, Scott D. Bunge
Tetrahedron Letters 2014 Volume 55(Issue 31) pp:4395-4399
Publication Date(Web):30 July 2014
DOI:10.1016/j.tetlet.2014.05.094
A library of hydroxyquinolin-3-ylmethylimidazolium adducts were prepared in high yields from the reaction of [BMIM][Cl] with various substituted quinoline-3-carbaldehydes and 2-oxoquinoline-3-carbaldehydes under mild conditions by using sodium acetate in MeCN under ultrasound irradiation. The use of sodium acetate and imidazolium chloride was crucial for the success of these CC bond forming reactions. Attempted coupling with thiazolium bromide led instead to quinoline-3-carboxylic acid.
Co-reporter:Kenneth K. Laali, Arezu Jamalian, Chunqing Zhao
Tetrahedron Letters 2014 Volume 55(Issue 49) pp:6643-6646
Publication Date(Web):3 December 2014
DOI:10.1016/j.tetlet.2014.10.071
Selectfluor reacts with N-chloromethylated DABCO monocation BF4 or NTf2 salts in MeCN (rt to 80 °C) to give symmetrical [N−H−N]+ trication salts. The same dimeric adducts are formed via the reaction of Selectfluor with Et3N, piperidine, or a basic-IL (imidazolium with an alkyl-piperidine tether). The resulting stable salts were studied by multinuclear NMR, 15N/1H HMBC, electrospray-MS, and by chemical reactivity. This hitherto unreported reactivity behavior contrasts the well documented ‘transfer fluorination’ by Selectfluor to quinuclidine and the quinuclidinic nitrogen of cinchona alkaloids.
Co-reporter:Takao Okazaki, Kenneth K. Laali, A. Srinivas Reddy
Journal of Fluorine Chemistry 2014 Volume 165() pp:91-95
Publication Date(Web):September 2014
DOI:10.1016/j.jfluchem.2014.06.021
•The SF5-analog of Umemoto salt was prepared in just two steps.•The methodology illustrates the potential utility of 4-(pentafluorosulfanyl)benzenediazonium tetrafluoroborate as building block for SF5-aromatics.•Trifluoromethylating ability of the SF5-derivative of Umemoto salt toward reactive aromatics was surveyed.The SF5-analog of the Umemoto salt was synthesized in just two steps by combining our recently reported SF5-biaryl synthesis, via Suzuki coupling with 4-(pentafluorosulfanyl)benzenediazonium tetrafluoroborate, with Magnier's one-pot synthesis of dibenzothiophenium salts employing CF3SO2Na and triflic anhydride. The trifluoromethylating power of this novel onium salt toward reactive arenes was tested in a survey study on small scale.
Co-reporter:Rudolf J. Wehmschulte, Kenneth K. Laali, Gabriela L. Borosky, and Douglas R. Powell
Organometallics 2014 Volume 33(Issue 9) pp:2146-2149
Publication Date(Web):May 1, 2014
DOI:10.1021/om5003792
We report the first successful synthesis of the long-sought arene-solvated bridgehead silylium ion [Si4Me3(CH2)6][CHB11Cl11] by hydride abstraction from the tetrasilaadamantyl derivative HSi4Me3(CH2)6 with the trityl salt [Ph3C][CHB11Cl11] in toluene, benzene, or bromobenzene solution. The silylium···arene complex was characterized by X-ray diffraction analysis and by NMR spectroscopy and was studied computationally. NMR studies show that the silylium species undergoes a dynamic solvate exchange in BrPh/MePh mixed solvent.
Co-reporter:Rok Prebil, Kenneth K. Laali, and Stojan Stavber
Organic Letters 2013 Volume 15(Issue 9) pp:2108-2111
Publication Date(Web):April 2, 2013
DOI:10.1021/ol4001476
Novel multifunctional ionic liquids (ILs) are generated by addition of HBr or HCl to alkylammonium nitrates ([RNH3+] [NO3–]) and to 3-methyl-1-(butyl-4-sulfonyl)imidazolium nitrate ([BMIM(SO3H)][NO3]). The resulting [RNH3+] [NO3–]/HX and mono (3-methyl-1-(butyl-4-sulfonyl)imidazolium) monohalogenide mononitrate ([BMIM(SO3H)][NO3)x(X)y] (X = Br, Cl)) systems act as solvent and promoter for aerobic oxidative halogenation of arenes under mild conditions in high yields that can be repeated over several cycles.
Co-reporter:Kenneth K. Laali;Ganesh C. Ni;Gabriela L. Borosky;G. G. K. S. Narayana Kumar
European Journal of Organic Chemistry 2013 Volume 2013( Issue 24) pp:5455-5463
Publication Date(Web):
DOI:10.1002/ejoc.201300553
Abstract
Propargylic cations, generated by the ionization of propargyl alcohols with catalytic amounts of Bi(OTf)3, react with aryl-substituted allenes to generate incipient allylic cations that are quenched in the presence of TMSCl to form a number of sterically crowded chloro-enynes as a mixture of Z and E isomers with a strong preference for the Z alkenes. Several other metallic triflates, M(OTf)3 (M = Sc, Yb, La), as well as bismuth nitrate Bi(NO3)3·5H2O also promote this reaction with similar conversions and stereoselectivity. Although trapping with TMSBr and TMSI gave intractable mixtures, trapping with TMSN3 in a couple of cases led to the isolation of the corresponding isomeric azido-enynes, albeit in lower isolated yields and lower stereoselectivity. Competitive formation of the Meyer–Schuster rearrangement products was also observed. Sterically crowded chloro-allenes did not form adducts with propargyl alcohols, instead they underwent a skeletal rearrangement under the influence of Bi(OTf)3 to form 2-chloro-butadienes. The results of DFT calculations indicated that the relative anti/syn energies of the propargyl cation and the energy difference between the Z/E isomeric products are too small to explain the stereochemical preference observed for the enynes. A study of the transition state in the crucial C–C bond-forming step by DFT indicated that rotation of the benzylic portion away from the propargyl cation may be a key factor in favoring the anti isomer of the allylic cation.
Co-reporter:Kenneth K. Laali, Gabriela L. Borosky
Journal of Fluorine Chemistry 2013 Volume 151() pp:26-31
Publication Date(Web):July 2013
DOI:10.1016/j.jfluchem.2013.04.003
•Carbocations derived from protonation, methylation and bromine addition to FC(R1)CR2 (SMe) were studied computationally.•Unsymmetrical thiiranium cations were identified as lowest energy minima followed closely by α-thiocarbenium ions.•Computed NPA charges and GIAO-NMR data underscore the importance of α-thiocarbenium ions and imply significant carbosulfonium ion character.Carbocations derived from protonation, methylation, and bromine addition to HFCCH(SMe) (1) were studied by DFT [B3LYP/6-31G*, B3LYP/6-311G(3df,p), and B3LYP/aug-cc-pVTZ] and by MP2/6-31G* to examine relative carbocation stabilizing effects of α-SMe versus α-fluorine. The α-SMe carbocations 1aE+ and the unsymmetrical thiiranium ions 1cE+ were found to be considerably more stable than the corresponding α-fluorocarbenium ions 1bE+. Study of protonation and methylation of FC(R1)CR2(SMe) [R1 = H, R2 = SMe (2); R1 = F, R2 = H (3); R1 = F, R2 = SMe (4); R1 = Me, R2 = H (5); R1 = Me, R2 = SMe (6)] by B3LYP/6-31G* identified the corresponding unsymmetrical thiiranium cations as lowest energy minima, followed closely by the α-thiocarbenium ions. With 2, 4, and 6, skeletally rearranged α-thiocarbenium ions are formed by SMe migration. The α-SMe and the thiiranium cations are also favored relative to α-fluorocarbenium ions in protonation and methylation of the cyclic analogs 8 and 9. Computed NPA charges and the GIAO-derived 13C and 19F NMR chemical shifts underscore the significance of α-thiocarbocations and thiiranium ions in electrophilic addition to FC(R1)CR2(SMe).
Co-reporter:Ganesh C. Nandi, Benjamin M. Rathman, Kenneth K. Laali
Tetrahedron Letters 2013 Volume 54(Issue 46) pp:6258-6263
Publication Date(Web):13 November 2013
DOI:10.1016/j.tetlet.2013.09.028
Co-reporter:G.G.K.S. Narayana Kumar, Kenneth K. Laali
Tetrahedron Letters 2013 Volume 54(Issue 8) pp:965-969
Publication Date(Web):20 February 2013
DOI:10.1016/j.tetlet.2012.12.023
A series of doubly-propargylated N-methylcarbazole and carbazole derivatives were synthesized in good to excellent isolated yields in (bmim)PF6 using triflic acid (10 mol %). The use of bismuth nitrate (20 mol %) instead of triflic acid allowed stepwise propargylation employing two different propargylic alcohols, to prepare mixed bis-propargylated carbazoles. Simple product isolation, mild reaction conditions, and repeated recycling and reuse of (bmim)PF6 are added advantages of this IL-mediated synthetic method.
Co-reporter:Ganesh C. Nandi, Kenneth K. Laali
Tetrahedron Letters 2013 Volume 54(Issue 17) pp:2177-2179
Publication Date(Web):24 April 2013
DOI:10.1016/j.tetlet.2013.02.051
A mild and selective method is presented for the conversion of aromatic and heteroaromatic aldehydes to nitriles via the Schmidt reaction with TMSN3 by using [BMIM(SO3H)][OTf] as catalyst and [BMIM][PF6] as solvent. The method offers high yields and simple product isolation, and avoids the use of liquid superacids or corrosive Lewis acids commonly employed for this transformation. It also offers some potential for recycling/reuse of the IL solvent.
Co-reporter:G.G.K.S. Narayana Kumar, Gopalakrishnan Aridoss, Kenneth K. Laali
Tetrahedron Letters 2012 Volume 53(Issue 24) pp:3066-3069
Publication Date(Web):13 June 2012
DOI:10.1016/j.tetlet.2012.04.026
Propargylation of a variety of substituted indoles as well as mono- and di-propargylation of carbazole are achieved in 80–94% isolated yields by employing catalytic amounts (10 mol %) of the readily available Bi(NO3)3·5H2O in [bmim][PF6] ionic liquid (IL), with repeated recycling and reuse of the IL.
Co-reporter:Rajesh G. Kalkhambkar, Kenneth K. Laali
Tetrahedron Letters 2012 Volume 53(Issue 32) pp:4212-4215
Publication Date(Web):8 August 2012
DOI:10.1016/j.tetlet.2012.05.155
A facile, high yielding, method for the synthesis of a library of 2-aryl- and 2-heteroaryl-benzoxazoles and benzothiazoles from readily available Schiff bases is reported employing catalytic amounts of Pd(OAc)2 in imidazolium ionic liquids (bmim)BF4 and (bmim)PF6 without ligands and/or additives. Simple product isolation and recycling/re-use of the IL are additional advantages of this method.
Co-reporter:Gabriela L. Borosky;Kenneth K. Laali
Journal of Physical Organic Chemistry 2012 Volume 25( Issue 8) pp:720-728
Publication Date(Web):
DOI:10.1002/poc.2924
A computational density functional theory study on the structural and electronic properties of several polycyclic aromatic hydrocarbon (PAH) ortho-quinones was performed and the possible mechanism of DNA-adduct formation was analyzed to evaluate its thermodynamic viability. Molecular docking techniques were applied to examine the noncovalent interactions developed when a model PAH ortho-quinone intercalates between the DNA double helix. Quantum-chemical ONIOM (our Own N-layer Integrated molecular Orbital molecular Mechanics) calculations within the structure of a DNA fragment were carried out to evaluate the significant steps of noncovalent complex and covalent adduct formation. The solvent effect was also considered by employing a continuum solvation model. The present calculations suggest that initial noncovalent interactions of the PAH o-quinone within the DNA double helix could determine the feasibility of benzo[a]pyrene-7,8-dione-DNA covalent adduct formation, and that dispersion-corrected functionals are more suitable for locating the noncovalent complex. Copyright © 2012 John Wiley & Sons, Ltd.
Co-reporter:Jonathan Jacoway, G.G.K.S. Narayana Kumar, Kenneth K. Laali
Tetrahedron Letters 2012 Volume 53(Issue 50) pp:6782-6785
Publication Date(Web):12 December 2012
DOI:10.1016/j.tetlet.2012.09.137
A suspension of bismuth nitrate pentahydrate (BN) in [bmim][PF6] or [bmim][BF4] imidazolium ionic liquid (IL) is an effective reagent for ring nitration of activated aromatics under mild conditions without the need for external promoters. Nitration can also be effected in 1,2-DCE, MeCN, or MeNO2 without additives. Nitration of activated arenes (anisole, toluene, ethylbenzene, cumene, p-xylene, mesitylene, durene, and 1,3-dimethoxybenzene) is considerably faster (time to completion) in BN/[bmim][PF6] relative to BN/1,2-DCE and there are also differences in isomer distributions (for anisole, toluene, and ethylbenzene). With introduction of strongly deactivating substituents (–CHO; –MeCO; –NO2) the BN/IL system is no longer active but reactions still proceed with BN/1,2-DCE in reasonable yields. The ready availability and low cost of BN, simple operation, and absence of promoters, coupled to recycling and reuse of the IL, provide an attractive alternative to classical nitration methods for activated arenes. Switching from Bi(NO3)3·5H2O/[bmim][PF6] to Bi(NO3)3·5H2O/1,2-DCE increases the scope of the substrates that can be nitrated.
Co-reporter:Gopalakrishnan Aridoss, Chunqing Zhao, Gabriela L. Borosky, and Kenneth K. Laali
The Journal of Organic Chemistry 2012 Volume 77(Issue 8) pp:4152-4155
Publication Date(Web):April 5, 2012
DOI:10.1021/jo300242s
A series of 1-aryl/alkyl-1H-1,2,3,4-tetrazoles, 5-substituted 1H-tetrazoles, and 1,5- and 2,5-disubstituted 1H-tetrazoles were studied by a combination of experimental NMR (natural abundance 15N, 15N/1H HMBC, and 13C) and computational GIAO-NMR techniques to explore substituent effects on 15N (and 13C) NMR chemical shifts in the tetrazole (TA) moiety. Computed 15N chemical shifts via GIAO-B3LYP/6-311+G(2d,p) calculations gave satisfactory results in comparison with experimental data. Whereas N-alkylation leads to large 15N chemical shift changes, changes in the N1-aryl derivatives bearing diverse substituent(s) are generally small except for polar ortho-substituents (COOH, NO2). Large Δδ15N values were computed in N1-aryl derivatives for p-COH2+ and p-OMeH+ as extreme examples of electron-withdrawing substituents on a TA moiety.
Co-reporter:Gopalakrishnan Aridoss ;Kenneth K. Laali
European Journal of Organic Chemistry 2011 Volume 2011( Issue 15) pp:2827-2835
Publication Date(Web):
DOI:10.1002/ejoc.201100128
Abstract
1-Aryl/alkyl-1H-1,2,3,4-tetrazoles can conveniently be synthesized in one-pot reactions from the corresponding amines by reaction with TMSN3 and CH(OEt)3 using the readily available, recyclable, Brønsted acidic ionic liquids [EtNH3][NO3] IL-1 and [PMIM(SO3 H)][OTf] IL-2 under mild conditions in high yields. Based on comparative reactions, whereas both ILs are excellent promoters, reactions are completed with shorter reaction times and in higher yields with IL-2. Among 24 examples provided, identical products were obtained via the two ILs, except in the case of 2-aminobenzoic acid where tetrazole was formed with IL-2 and 2-ethylquinazolin-4(3H)-one was formed with IL-1. By leaving out TMS-N3 from the reaction, the in-situ formed CH(OEt)2+ and EtC(OEt)2+ (via their corresponding orthoesters) react under sonication with o-phenylenediamine bearing various substituents, o-aminothiophenol and o-aminophenol to form a wide array of benzazoles (benzimidazole, benzothiazole and benzoxazole) and quinazolin-4(3H)-one in high yields (18 examples). The two ILs reacted differently in reaction with 2-aminobenzamide, whereas quinazolin-4(3H)one was formed with IL-2/CH(OEt)3, the “unexpected” N-ethylquinazolin-4(3H)one was isolated with IL-1/CH(OEt)3. The latter was also formed from 2-aminobenzoic acid in IL-1/CH(OEt)3. Mechanistic implications are addressed. The reported protocols enable rapid assembly of a host of heterocyclic systems in high yields with the added advantage of recycling and re-use of the ILs.
Co-reporter:Gopalakrishnan Aridoss ;Kenneth K. Laali
European Journal of Organic Chemistry 2011 Volume 2011( Issue 31) pp:6343-6355
Publication Date(Web):
DOI:10.1002/ejoc.201100957
Abstract
A series of 5-substituted 1H-tetrazoles were synthesized through [3+2] cycloaddition reactions between nitriles RCN and NaN3 in the presence of Cu–Zn alloy nanopowder as catalyst. The 1,5-dibutyl, 1-butyl-5-hexyl, 2,5-dibutyl, and 2-butyl-5-hexyl derivatives were then used as building blocks to synthesize several novel tetrazolium ionic liquids (ILs) with EtSO4–, OTf–, and NTf2– counterions. Whereas alkylation of the 2,5-dialkyltetrazoles selectively gave the N-4 alkylated onium salts, with the 1,5-dialkyl derivatives approximately 1:1 mixtures of two tetrazolium salts were formed by alkylation at N-3 and N-4. The triflate and ethyl sulfate salts are room-temperature ILs that are hydrophilic, whereas the NTf2 salts are low-melting ILs and are hydrophobic. The resulting tetrazolium-based ionic liquids were studied by various multinuclear and 2D NMR techniques including natural abundance 15N and 1H/15N correlations.
Co-reporter:Gabriela L. Borosky;Takao Okazaki;Kenneth K. Laali
European Journal of Organic Chemistry 2011 Volume 2011( Issue 9) pp:1771-1775
Publication Date(Web):
DOI:10.1002/ejoc.201001655
Abstract
15N and 13C NMR chemical shifts were computed by GIAO-DFT and GIAO-MP2 for a series of p-substituted benzenediazonium mono- and dications in order to probe the electronic effects of the substituents on the diazonium moiety. Optimized geometries and N/N vibrational frequencies werealso considered for comparison. The GIAO-DFT derived15N chemical shifts correlate more closely with the experimental values as compared to GIAO-MP2. Energyminimizations at the B3LYP/6-311+G(2d,p), M062X-6-311+G(2d,p), MP2/6-311+G(2d,p), G2(MP2), and CBS-Q levels were carried out. Relative dication stability order HCO+ > HOMe+ > HN(Me)2+ > HOH+ > HCN+ > HNO2+ was derived from isodesmic proton transfer reactions. The Nβ-protonated dications were less stable than the corresponding p-R+ dications. Among the regioisomeric Nβ-protonated dications (with R = –F, –Cl, and –CN), those with the R group in the para position were preferred. For the regioisomeric, ring-protonated benzenium-diazonium dications, the meta-protonated dications were more favored (by DFT and MP2). Influence of the counterion and solvent on the computed 15N NMR chemical shifts in PhN2+ X– were also assessed.
Co-reporter:Rajesh G. Kalkhambkar, Sarah N. Waters, Kenneth K. Laali
Tetrahedron Letters 2011 Volume 52(Issue 8) pp:867-871
Publication Date(Web):23 February 2011
DOI:10.1016/j.tetlet.2010.12.028
The utility of Brønsted-acidic imidazolium ionic liquid [BMIM(SO3H)][OTf] as catalyst for the high yield synthesis of a wide variety of amides under mild conditions via the Ritter reaction of alcohols with nitriles has been demonstrated. As alternative methods for the carbocation generation step, NOPF6 immobilized in [BMIM][PF6] ionic liquid was used in the Ritter reaction of bromides with nitriles and for the synthesis of adamantyl amides from adamantane and nitriles.
Co-reporter:Rajesh G. Kalkhambkar, Kenneth K. Laali
Tetrahedron Letters 2011 Volume 52(Issue 15) pp:1733-1737
Publication Date(Web):13 April 2011
DOI:10.1016/j.tetlet.2011.02.021
The utility of arenediazonium salts immobilized in imidazolium-ILs [BMIMPF6 and BMIMBF4] for facile, high yielding, synthesis of olefins via the Pd(OAc)2-catalyzed Matsuda–Heck arylation reaction has been demonstrated. The reaction can also be performed as a two-step process in the IL starting from ArNH2, by sequential in-situ diazotization–arylation. Simple product isolation and the recycling/re-use of the IL are additional advantages of this one-pot method.
Co-reporter:Rajesh G. Kalkhambkar, Kenneth K. Laali
Tetrahedron Letters 2011 Volume 52(Issue 42) pp:5525-5529
Publication Date(Web):19 October 2011
DOI:10.1016/j.tetlet.2011.08.077
Polyfluoroarenes can be cross-coupled with simple aromatics (benzene, toluene, and anisole), in good isolated yields, by using Pd(OAc)2 dissolved in imidazolium ILs [(bmim)PF6 and (bmim)BF4] as solvent, without the need for an oxidant and an additive. The reaction is catalyzed by HOAc and it is subject to a primary isotope effect (KH/KD = 4.87). Competitive cross-coupling reactions of 1,2,4,5-tetrafluorobenzene with benzene/toluene, benzene/anisole, and anisole/toluene gave KB/KT = 5.1, KB/KA = 5.7, and KA/KT = 5.0, respectively, indicative of a remote substituent effect on Pd insertion into the phenyl C–H bond. Mild reaction conditions, simple product isolation and recycling/reuse of the IL are additional advantages of this method.
Co-reporter:Rajesh G. Kalkhambkar, Scott D. Bunge, Kenneth K. Laali
Tetrahedron Letters 2011 Volume 52(Issue 40) pp:5184-5187
Publication Date(Web):5 October 2011
DOI:10.1016/j.tetlet.2011.07.135
The synthetic utility of N-triflylimidazole as an in situ reagent for facile, high yielding, synthesis of various aliphatic, aromatic, and heteroaromatic nitriles from the corresponding aldoximes has been demonstrated. With benzaldoximes, in the presence of certain substitutents (2-F; 2-OMe; 3-CF3; 2-Me-5-F) a different course of reaction was observed, leading instead to novel 1:1 aldoxime-bis(N-triflyl)imidazole covalent adducts, in which the aldoxime oxygen atom is bonded to the imidazole C-2 ring carbon. For these aldoximes, conversion to nitrile could be effected by reaction with Tf2O in the absence of imidazole. The molecular structure of the adduct formed from 2-methyl-5-fluoro-benzaldoxime was confirmed by X-ray analysis. Plausible mechanisms for the formation of 1:1 covalent adducts have been considered. Various attempts to isolate such adducts via the reaction of an authentic sample of bis(N-triflyl)imidazolium trifate with aldoxime were unsuccessful. Remarkably, whereas isolated benzaldoxime adducts undergo deprotonation/methylation with NaH/MeI, an authentic sample of bis(N-triflyl)imidazolium triflate did not undergo H/Me exchange under these conditions. These transformations are discussed.
Co-reporter:Gopalakrishnan Aridoss, Kenneth K. Laali
Tetrahedron Letters 2011 Volume 52(Issue 51) pp:6859-6864
Publication Date(Web):21 December 2011
DOI:10.1016/j.tetlet.2011.10.021
Propargylic alcohols are activated toward 1,3-diketones by Lewis or Brønsted acidic ionic liquids (ILs) without an added catalyst, but significantly better conversions are achieved with metallic triflates [in particular Sc(OTf)3 and Ln(OTf)3] and bismuth nitrate in imidazolium ILs. The scope of this condensation reaction was investigated with a variety of propargylic alcohols and a host of acyclic and cyclic dicarbonyl compounds. Concomitant cycloisomerization leading to tetrasubstituted furans was observed with the propargylic alcohols 1b and 1c in reaction with 1,3-diketone 2b and the β-ketoester 2c. With propargylic alcohol 1c, propargylation, cycloisomerization, or dienone formation were observed, depending on the structure of the 1,3-dicarbonyl compound. The [BMIM][PF6]/Bi(NO3)3·5H2O system proved efficient for propargylation, vinylation, and alkylation of 4-hydroxycoumarins. The recycling and reuse of the IL are added advantages of this method.
Co-reporter:Gopalakrishnan Aridoss and Kenneth K. Laali
The Journal of Organic Chemistry 2011 Volume 76(Issue 19) pp:8088-8094
Publication Date(Web):August 22, 2011
DOI:10.1021/jo201374a
Acting as in situ sources of triflyl nitrate (TfONO2) and trifluoroacetyl nitrate (CF3COONO2), the EAN/Tf2O and EAN/TFAA systems, generated via metathesis in the readily available ethylammonium nitrate (EAN) ionic liquid as solvent, are powerful electrophilic nitrating reagents for a wide variety of aromatic and heteroaromatic compounds. Comparative nitration experiments indicate that EAN/Tf2O is superior to EAN/TFAA for nitration of strongly deactivated systems. Both systems exhibit low substrate selectivity (KT/KB = 5–10) in between values reported for covalent nitrates and preformed nitronium salts.
Co-reporter:Priscila M. Lalli, Yuri E. Corilo, Patrícia V. Abdelnur, Marcos N. Eberlin and Kenneth K. Laali
Organic & Biomolecular Chemistry 2010 vol. 8(Issue 11) pp:2580-2585
Publication Date(Web):29 Mar 2010
DOI:10.1039/C001985B
The ion/molecule chemistry of four representative propagyl/allenyl cations 1–4 of the general formula R1CH+-CC-R (a) ↔ R1CHCC+-R (b), that is, the reactive C3H3+ ions of m/z 39 from EI of propargyl chloride (H2C+-CC–H, 1a), isomeric C4H5+ ions of m/z 53 from EI of 3-butyne-2-ol (2a, H2C+-CC-CH3) and 2-butyne-1-ol (CH3-CH+-CC–H, 3a), and Ph-C3H2+ ions of m/z 115 from 3-phenyl-2-propyn-1-ol (H2C+-CC-Ph, 4a) was studied via pentaquadrupole mass spectrometry. With pyridine, proton transfer was observed as the predominant process for 1 and the sole reaction channel for the isomeric 2 and 3, whereas 4 reacted preferentially by adduct formation. These outcomes were rationalized using DFT calculations from isodesmic proton transfer reactions. Similar reaction tendencies were observed with acetonitrile and acrylonitrile, with adduct formation appearing again as a minor pathway for 1, 2 and 3, and as a major reaction channel for 4. With 1,3-dioxolane, hydride abstraction was a dominant reaction, with proton transfer and adduct formation competing as side reactions. With 2,2-dimethyl-1,3-dioxolane, an interplay of reactions including methyl anion abstraction, proton transfer, hydride abstraction and adduct formation were observed depending on the ion structure, with 4 reacting again mainly by adduct formation. Proton transfer was also observed as a dominant process in reactions with ethanol for 1, 2 and 3, with 4 being nearly unreactive whereas no adduct formation was observed for any of the carbocations studied. Limited reactivity was exhibited by these ions in cycloaddition reaction with isoprene.
Co-reporter:Gabriela L. Borosky;Kenneth K. Laali
Journal of Physical Organic Chemistry 2010 Volume 23( Issue 9) pp:810-818
Publication Date(Web):
DOI:10.1002/poc.1666
Abstract
A density functional theory (DFT) study aimed at understanding structure–reactivity relationships in the oxidized metabolites of cyclopenta-fused polycyclic aromatic hydrocarbons (CP-PAHs) is reported. Epoxidation at various positions was examined in order to identify the most stable epoxide in each class of CP-PAHs. Relative energies of the carbocations resulting from O-protonation and epoxide ring opening were analyzed and compared, taking into account the available biological activity data on these compounds. Geometrical, electronic, and conformational issues were considered. Charge delocalization modes in the resulting carbocations were deduced via the natural population analysis (NPA)-derived changes in charges. Computational results pointed to the importance of the unsaturated cyclopenta ring on the reactivity of these compounds. The reported bioactivity of this highly mutagenic/carcinogenic family of PAHs was observed to parallel their relative carbocation stabilities. A different behavior was observed in crowded non-planar structures possessing a distorted aromatic system. A covalent adduct formed between a CP-PAH epoxide and a purine base was computed inside a DNA fragment employing the ONIOM method. Copyright © 2010 John Wiley & Sons, Ltd.
Co-reporter:Kenneth K. Laali;Takao Okazaki;Toshikazu Kitagawa;Teruo Shinmyozu
European Journal of Organic Chemistry 2009 Volume 2009( Issue 26) pp:4451-4457
Publication Date(Web):
DOI:10.1002/ejoc.200900479
Abstract
Mono- and diprotonation of the tri-, tetra-, and pentabridged cyclophanes [33](1,3,5)cyclophane (4), [34](1,2,3,5)cyclophane (5), and [35](1,2,3,4,5)cyclophane (6) were realized, and NMR spectroscopic studies of the resulting carbocations are reported. Unlike [2.2]paracyclophane and its fluorinated analogs that are protonated at a position ipso to the ethano-bridge, the monocations derived from 4, 5, and 6 are protonated at the unsubstituted ring positions. Protonation regioselectivity in the dications is pseudo-meta for 4 and 5 at unsubstituted ring positions, but for more-crowded 6 the second protonation occurs at a position that is ipso to the trimethylene bridge. Transannular π–π interactions in the monocations are manifested in the observed proton deshielding in the unprotonated π-deck. DFT and GIAO-DFT were employed to study the mono- and dications for comparison with the solution studies in superacids. GIAO-derived ΔNICS(1)zz data for the [3n]cyclophane monocations imply decreased aromaticity in the cofacial unprotonated arene, consistent with transannular donor–acceptor interactions.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2009)
Co-reporter:Arezu Jamalian, Benjamin Rathman, Gabriela L. Borosky, Kenneth K. Laali
Applied Catalysis A: General (22 September 2014) Volume 486() pp:1-11
Publication Date(Web):22 September 2014
DOI:10.1016/j.apcata.2014.08.009
Co-reporter:Priscila M. Lalli, Yuri E. Corilo, Patrícia V. Abdelnur, Marcos N. Eberlin and Kenneth K. Laali
Organic & Biomolecular Chemistry 2010 - vol. 8(Issue 11) pp:NaN2585-2585
Publication Date(Web):2010/03/29
DOI:10.1039/C001985B
The ion/molecule chemistry of four representative propagyl/allenyl cations 1–4 of the general formula R1CH+-CC-R (a) ↔ R1CHCC+-R (b), that is, the reactive C3H3+ ions of m/z 39 from EI of propargyl chloride (H2C+-CC–H, 1a), isomeric C4H5+ ions of m/z 53 from EI of 3-butyne-2-ol (2a, H2C+-CC-CH3) and 2-butyne-1-ol (CH3-CH+-CC–H, 3a), and Ph-C3H2+ ions of m/z 115 from 3-phenyl-2-propyn-1-ol (H2C+-CC-Ph, 4a) was studied via pentaquadrupole mass spectrometry. With pyridine, proton transfer was observed as the predominant process for 1 and the sole reaction channel for the isomeric 2 and 3, whereas 4 reacted preferentially by adduct formation. These outcomes were rationalized using DFT calculations from isodesmic proton transfer reactions. Similar reaction tendencies were observed with acetonitrile and acrylonitrile, with adduct formation appearing again as a minor pathway for 1, 2 and 3, and as a major reaction channel for 4. With 1,3-dioxolane, hydride abstraction was a dominant reaction, with proton transfer and adduct formation competing as side reactions. With 2,2-dimethyl-1,3-dioxolane, an interplay of reactions including methyl anion abstraction, proton transfer, hydride abstraction and adduct formation were observed depending on the ion structure, with 4 reacting again mainly by adduct formation. Proton transfer was also observed as a dominant process in reactions with ethanol for 1, 2 and 3, with 4 being nearly unreactive whereas no adduct formation was observed for any of the carbocations studied. Limited reactivity was exhibited by these ions in cycloaddition reaction with isoprene.
Co-reporter:Gopalakrishnan Aridoss, Viorel D. Sarca, James F. Ponder Jr, Jessica Crowe and Kenneth K. Laali
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 7) pp:NaN2529-2529
Publication Date(Web):2011/02/21
DOI:10.1039/C0OB00872A
Metallic triflates M(OTf)3 (M = Bi, Sc, Yb), immobilized in imidazolium ionic liquids [BMIM][BF4], [BMIM][PF6] and [BMIM][OTf] are efficient systems for one-pot reactions of propargylic alcohols 1,3-diphenyl-2-propyn-1-ol Ia, 1-methyl-3-phenyl-2-propyn-1-ol Ib, and 2-pentyn-1-ol Ic, with a wide range of arenes bearing activating substituents, under mild conditions. The [BMIM][PF6]/B(C6F5)3 and [BMIM][PF6]/TfOH systems were superior in propargylation with Ib and Ic, while reaction of 3-phenyl-2-propyn-1-ol Id with activated aromatics resulted in the formation of diaryl-propanones instead. Propargylation of anisole with Ib under M(OTf)3 catalysis is highly para selective, but with TfOH or B(C6F5)3 as catalyst the ortho isomer was also formed. Steric influence of the propargylic moiety on substrate selectivity is reflected in the lack of ortho propargylation for phenol and ethylbenzene by using propargylic alcohol Ia, and notable formation of the ortho isomer employing alcohol Ib. In the later case para selectivity could be increased by running the reaction at r. t. for 10 h. The Bi(OTf)3-catalyzed reaction of 1,3-dimethoxybenzene with Ia led to minor formation of dipropargylated derivative, along with the monopropargyl product. Propargylation of the less reactive arenes (mesitylene, ethylbenzene, toluene), using Sc(OTf)3 as catalyst, led increasingly to the formation of dipropargylic ethers and propargyl ketones, with no ring propargylation product with toluene. Concomitant formation of dipropargylic ether was also observed in Yb(OTf)3-catalyzed propargylation of β-naphthol, whereas propargylation of 2-nitro and 4-nitro-aniline led to N-propargylation. The recycling/reuse of the IL was demonstrated in representative cases with no appreciable decrease in the conversions over 3 cycles. It was also shown that recycled IL could be used to propargylate a different aromatic compound. The efficacy of IL/M(OTf)3 and IL/TfOH systems for cross-breeding two propargylic alcohols or a propargylic alcohol with a non-propargylic alcohol and/or self-coupling, to form a wide variety of functionalized ethers is also demonstrated.



























![Benzene, 1-[(1E)-3-chloro-1-propenyl]-4-fluoro-](http://img.cochemist.com/ccimg/145800/145798-29-4.png)
![Benzene, 1-[(1E)-3-chloro-1-propenyl]-4-fluoro-](http://img.cochemist.com/ccimg/145800/145798-29-4_b.png)


![1-(chloromethyl)-4-aza-1-azoniabicyclo[2.2.2]octane tetrafluoroborate](http://img.cochemist.com/ccimg/140700/140681-69-2.png)
![1-(chloromethyl)-4-aza-1-azoniabicyclo[2.2.2]octane tetrafluoroborate](http://img.cochemist.com/ccimg/140700/140681-69-2_b.png)




















![Benzene, 1-[(1E)-3-chloro-1-propen-1-yl]-4-methyl-](/data/chemimg/3742100/96000-21-4.png)
![Benzene, 1-[(1E)-3-chloro-1-propen-1-yl]-4-methyl-](/data/chemimg/3742100/96000-21-4_b.png)








![2-[4-(HYDRAZINOMETHYL)PHENOXY]ACETAMIDE](http://img.cochemist.com/ccimg/73800/73744-44-2.png)
![2-[4-(HYDRAZINOMETHYL)PHENOXY]ACETAMIDE](http://img.cochemist.com/ccimg/73800/73744-44-2_b.png)














![Benzene, 1-[(1E)-3-bromo-1-propen-1-yl]-4-chloro-](/data/chemimg/1874500/60691-90-9.png)
![Benzene, 1-[(1E)-3-bromo-1-propen-1-yl]-4-chloro-](/data/chemimg/1874500/60691-90-9_b.png)
![1-FLUORO-4-[4-(4-FLUOROPHENYL)BUTA-1,3-DIYNYL]BENZENE](http://img.cochemist.com/ccimg/55700/55606-94-5.png)
![1-FLUORO-4-[4-(4-FLUOROPHENYL)BUTA-1,3-DIYNYL]BENZENE](http://img.cochemist.com/ccimg/55700/55606-94-5_b.png)


![Benzene, 1-[(1E)-3-bromo-1-propen-1-yl]-4-fluoro-](/data/chemimg/3235100/53041-12-6.png)
![Benzene, 1-[(1E)-3-bromo-1-propen-1-yl]-4-fluoro-](/data/chemimg/3235100/53041-12-6_b.png)






![4-(CHLOROMETHYL)-1-AZA-4-AZONIABICYCLO[2.2.2]OCTANE;CHLORIDE](http://img.cochemist.com/ccimg/36300/36273-11-7.png)
![4-(CHLOROMETHYL)-1-AZA-4-AZONIABICYCLO[2.2.2]OCTANE;CHLORIDE](http://img.cochemist.com/ccimg/36300/36273-11-7_b.png)


![Benzonitrile, 4-[(2R,3R)-3-phenyloxiranyl]-, rel-](http://img.cochemist.com/ccimg/31900/31836-52-9.png)
![Benzonitrile, 4-[(2R,3R)-3-phenyloxiranyl]-, rel-](http://img.cochemist.com/ccimg/31900/31836-52-9_b.png)



![Benzene, [(1Z)-2,3-dibromo-1-propenyl]-](http://img.cochemist.com/ccimg/21100/21003-54-3.png)
![Benzene, [(1Z)-2,3-dibromo-1-propenyl]-](http://img.cochemist.com/ccimg/21100/21003-54-3_b.png)