Co-reporter:Joanna M. Swarbrick ; Richard Graeff ; Hongmin Zhang ; Mark P. Thomas ; Quan Hao ;Barry V. L. Potter
Journal of Medicinal Chemistry 2014 Volume 57(Issue 20) pp:8517-8529
Publication Date(Web):September 16, 2014
DOI:10.1021/jm501037u
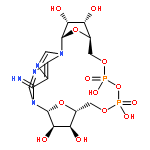
Cyclic adenosine 5′-diphosphate ribose (cADPR) analogs based on the cyclic inosine 5′-diphosphate ribose (cIDPR) template were synthesized by recently developed stereo- and regioselective N1-ribosylation. Replacing the base N9-ribose with a butyl chain generates inhibitors of cADPR hydrolysis by the human ADP-ribosyl cyclase CD38 catalytic domain (shCD38), illustrating the nonessential nature of the “southern” ribose for binding. Butyl substitution generally improves potency relative to the parent cIDPRs, and 8-amino-N9-butyl-cIDPR is comparable to the best noncovalent CD38 inhibitors to date (IC50 = 3.3 μM). Crystallographic analysis of the shCD38:8-amino-N9-butyl-cIDPR complex to a 2.05 Å resolution unexpectedly reveals an N1-hydrolyzed ligand in the active site, suggesting that it is the N6-imino form of cADPR that is hydrolyzed by CD38. While HPLC studies confirm ligand cleavage at very high protein concentrations, they indicate that hydrolysis does not occur under physiological concentrations. Taken together, these analogs confirm that the “northern” ribose is critical for CD38 activity and inhibition, provide new insight into the mechanism of cADPR hydrolysis by CD38, and may aid future inhibitor design.
Co-reporter:Huanchen Wang, Himali Y. Godage, Andrew M. Riley, Jeremy D. Weaver, Stephen B. Shears, Barry V.L. Potter
Chemistry & Biology 2014 Volume 21(Issue 5) pp:689-699
Publication Date(Web):22 May 2014
DOI:10.1016/j.chembiol.2014.03.009
•Chemical synthesis of 5-PP-InsP4 and a diphosphorylated analog•Chemical synthesis of inositol polyphosphate analogs with hydrophobic groups•An inositol pyrophosphate kinase has a surface-mounted, substrate capture site•Structural and biochemical characterization of a catch-and-pass catalytic cycleDiphosphoinositol pentakisphosphate kinase 2 (PPIP5K2) is one of the mammalian PPIP5K isoforms responsible for synthesis of diphosphoinositol polyphosphates (inositol pyrophosphates; PP-InsPs), regulatory molecules that function at the interface of cell signaling and organismic homeostasis. The development of drugs that inhibit PPIP5K2 could have both experimental and therapeutic applications. Here, we describe a synthetic strategy for producing naturally occurring 5-PP-InsP4, as well as several inositol polyphosphate analogs, and we study their interactions with PPIP5K2 using biochemical and structural approaches. These experiments uncover an additional ligand-binding site on the surface of PPIP5K2, adjacent to the catalytic pocket. This site facilitates substrate capture from the bulk phase, prior to transfer into the catalytic pocket. In addition to demonstrating a “catch-and-pass” reaction mechanism in a small molecule kinase, we demonstrate that binding of our analogs to the substrate capture site inhibits PPIP5K2. This work suggests that the substrate-binding site offers new opportunities for targeted drug design.Figure optionsDownload full-size imageDownload high-quality image (204 K)Download as PowerPoint slide
Co-reporter:Dr. Wolfgang Dohle;Dr. Mathew P. Leese;Dr. Fabrice L. Jourdan;Dr. Christopher J. Chapman;Dr. Ernest Hamel;Dr. Eric Ferris; Barry V. L. Potter
ChemMedChem 2014 Volume 9( Issue 8) pp:1783-1793
Publication Date(Web):
DOI:10.1002/cmdc.201402025
Abstract
Tetrahydroisoquinoline (THIQ)-based “chimeric” microtubule disruptors were optimised through modification of the N-benzyl motif, in concert with changes at C3 and C7, resulting in the identification of compounds with improved in vitro antiproliferative activities (e.g. 15: GI50 20 nM in DU-145). The broad anticancer activity of these novel structures was confirmed in the NCI 60-cell line assay, with 12 e,f displaying MGM values in the 40 nM region. In addition, their profiles as inhibitors of tubulin polymerisation and colchicine binding to tubulin were confirmed. Compound 15, for example, inhibited tubulin polymerisation with an IC50 of 1.8 μM, close to that of the clinical drug combretastatin A-4, and also proved effective at blocking colchicine binding. Additionally, compound 20 b was identified as the only phenol in the series to date showing both better in vitro antiproliferative properties than its corresponding sulfamate and excellent antitubulin data (IC50=1.6 μM). Compound 12 f was selected for in vivo evaluation at the NCI in the hollow fibre assay and showed very good activity and wide tissue distribution, illustrating the value of this template for further development.
Co-reporter:Dr. Mathew P. Leese;Dr. Fabrice L. Jourdan;Dr. Meriel R. Major;Dr. Wolfgang Dohle;Dr. Mark P. Thomas;Dr. Ernest Hamel;Dr. Eric Ferris;Dr. Mary F. Mahon;Dr. Simon P. Newman;Dr. Atul Purohit;Dr. Barry V. L. Potter
ChemMedChem 2014 Volume 9( Issue 4) pp:798-812
Publication Date(Web):
DOI:10.1002/cmdc.201400017
Abstract
A SAR translation strategy adopted for the discovery of tetrahydroisoquinolinone (THIQ)-based steroidomimetic microtubule disruptors has been extended to dihydroisoquinolinone (DHIQ)-based compounds. A steroid A,B-ring-mimicking DHIQ core was connected to methoxyaryl D-ring mimics through methylene, carbonyl, and sulfonyl linkers, and the resulting compounds were evaluated against two cancer cell lines. The carbonyl-linked DHIQs in particular exhibit significant in vitro antiproliferative activities (e.g., 6-hydroxy-7-methoxy-2-(3,4,5-trimethoxybenzoyl)-3,4-dihydroisoquinolin-1(2H)-one (16 g): GI50 51 nM in DU-145 cells). The broad anticancer activity of DHIQ 16 g was confirmed in the NCI 60-cell line assay giving a mean activity of 33 nM. Furthermore, 6-hydroxy-2-(3,5-dimethoxybenzoyl)-7-methoxy-3,4-dihydroisoquinolin-1(2H)-one (16 f) and 16 g and their sulfamate derivatives 17 f and 17 g (2-(3,5-dimethoxybenzoyl)-7-methoxy-6-sulfamoyloxy-3,4-dihydroisoquinolin-1(2H)-one and 7-methoxy-2-(3,4,5-trimethoxybenzoyl)-6-sulfamoyloxy-3,4-dihydroisoquinolin-1(2H)-one, respectively) show excellent activity against the polymerization of tubulin, close to that of the clinical combretastatin A-4, and bind competitively at the colchicine binding site of tubulin. Compounds 16 f and 17 f were also shown to demonstrate in vitro anti-angiogenic activity. Additionally, X-ray and computational analyses of 17 f reveal that electrostatic repulsion between the two adjacent carbonyl groups, through conformational biasing, dictates the adoption of a “steroid-like” conformation that may partially explain the excellent in vitro activities.
Co-reporter:Dr. Andrew M. Riley;Dr. Sabine Windhorst;Dr. Hong-Yin Lin; Barry V. L. Potter
ChemBioChem 2014 Volume 15( Issue 1) pp:57-67
Publication Date(Web):
DOI:10.1002/cbic.201300583
Abstract
When applied extracellularly, myo-inositol hexakisphosphate (InsP6) and myo-inositol pentakisphosphate (InsP5) can inhibit the growth and proliferation of tumour cells. There is debate about whether these effects result from interactions of InsP6 and InsP5 with intracellular or extracellular targets. We synthesised FAM-InsP5, a fluorescent conjugate of InsP5 that allows direct visualisation of its interaction with cells. FAM-InsP5 was internalised by H1229 tumour cells, a finding that supports earlier reports that externally applied inositol phosphates can—perhaps surprisingly—enter into cells. Close examination of the process of FAM-InsP5 uptake suggests a mechanism of non-receptor-mediated endocytosis, which is blocked at 4 °C and probably involves interaction of the ligand with the glycocalyx. However, our results are difficult to reconcile with antiproliferative mechanisms that require direct interactions of externally applied InsP5 or InsP6 with cytosolic proteins, because internalised FAM-InsP5 appears in lysosomes and apparently does not enter the cytoplasm. Studies using FAM-InsP5 are less difficult and time-consuming than experiments using InsP5 or InsP6, a factor that allowed us to analyse cellular uptake across a range of human cell types, identifying strong cell-specific differences.
Co-reporter:Dr. Mathew P. Leese;Dr. Fabrice L. Jourdan;Dr. Meriel R. Major;Dr. Wolfgang Dohle;Dr. Ernest Hamel;Dr. Eric Ferris;Dr. Ann Fiore;Dr. Philip G. Kasprzyk; Barry V. L. Potter
ChemMedChem 2014 Volume 9( Issue 1) pp:
Publication Date(Web):
DOI:10.1002/cmdc.201390056
Co-reporter:Dr. Wolfgang Dohle;Dr. Mathew P. Leese;Dr. Fabrice L. Jourdan;Dr. Meriel R. Major;Dr. Ruoli Bai;Dr. Ernest Hamel;Dr. Eric Ferris;Dr. Philip G. Kasprzyk;Dr. Ann Fiore;Dr. Simon P. Newman;Dr. Atul Purohit; Barry V. L. Potter
ChemMedChem 2014 Volume 9( Issue 2) pp:350-370
Publication Date(Web):
DOI:10.1002/cmdc.201300412
Abstract
The syntheses and antiproliferative activities of novel substituted tetrahydroisoquinoline derivatives and their sulfamates are discussed. Biasing of conformational populations through substitution on the tetrahydroisoquinoline core at C1 and C3 has a profound effect on the antiproliferative activity against various cancer cell lines. The C3 methyl-substituted sulfamate (±)-7-methoxy-2-(3-methoxybenzyl)-3-methyl-6-sulfamoyloxy-1,2,3,4-tetrahydroisoquinoline (6 b), for example, was found to be ∼10-fold more potent than the corresponding non-methylated compound 7-methoxy-2-(3-methoxybenzyl)-6-sulfamoyloxy-1,2,3,4-tetrahydroisoquinoline (4 b) against DU-145 prostate cancer cells (GI50 values: 220 nM and 2.1 μM, respectively). Such compounds were also found to be active against a drug-resistant MCF breast cancer cell line. The position and nature of substitution of the N-benzyl group in the C3-substituted series was found to have a significant effect on activity. Whereas C1 methylation has little effect on activity, introduction of C1 phenyl and C3-gem-dimethyl substituents greatly decreases antiproliferative activity. The ability of these compounds to inhibit microtubule polymerisation and to bind tubulin in a competitive manner versus colchicine confirms the mechanism of action. The therapeutic potential of a representative compound was confirmed in an in vivo multiple myeloma xenograft study.
Co-reporter:Dr. Mathew P. Leese;Dr. Fabrice L. Jourdan;Dr. Meriel R. Major;Dr. Wolfgang Dohle;Dr. Ernest Hamel;Dr. Eric Ferris;Dr. Ann Fiore;Dr. Philip G. Kasprzyk; Barry V. L. Potter
ChemMedChem 2014 Volume 9( Issue 1) pp:85-108
Publication Date(Web):
DOI:10.1002/cmdc.201300261
Abstract
A structure–activity relationship (SAR) translation strategy was used for the discovery of tetrahydroisoquinoline (THIQ)-based steroidomimetic and chimeric microtubule disruptors based upon a steroidal starting point. A steroid A,B-ring-mimicking THIQ core was connected to methoxyaryl D-ring ring mimics through methylene, carbonyl and sulfonyl linkers to afford a number of steroidomimetic hits (e.g., 7-methoxy-2-(3- methoxybenzyl)-6-sulfamoyloxy-1,2,3,4-tetrahydroisoquinoline (20 c) GI50=2.1 μM). Optimisation and control experiments demonstrate the complementary SAR of this series and the steroid derivatives that inspired its design. Linkage of the THIQ-based A,B-mimic with the trimethoxyaryl motif prevalent in colchicine site binding microtubule disruptors delivered a series of chimeric molecules whose activity (GI50=40 nM) surpasses that of the parent steroid derivatives. Validation of this strategy was obtained from the excellent oral activity of 7-methoxy-6-sulfamoyloxy-2-(3,4,5-trimethoxybenzyl)-1,2,3,4-tetrahydroisoquinoline (20 z) relative to a benchmark steroidal bis- sulfamate in an in vivo model of multiple myeloma.
Co-reporter:Christelle Moreau ; Tanja Kirchberger ; Joanna M. Swarbrick ; Stephen J. Bartlett ; Ralf Fliegert ; Timur Yorgan ; Andreas Bauche ; Angelika Harneit ; Andreas H. Guse ;Barry V. L. Potter
Journal of Medicinal Chemistry 2013 Volume 56(Issue 24) pp:10079-10102
Publication Date(Web):December 4, 2013
DOI:10.1021/jm401497a
Adenosine 5′-diphosphoribose (ADPR) activates TRPM2, a Ca2+, Na+, and K+ permeable cation channel. Activation is induced by ADPR binding to the cytosolic C-terminal NudT9-homology domain. To generate the first structure–activity relationship, systematically modified ADPR analogues were designed, synthesized, and evaluated as antagonists using patch-clamp experiments in HEK293 cells overexpressing human TRPM2. Compounds with a purine C8 substituent show antagonist activity, and an 8-phenyl substitution (8-Ph-ADPR, 5) is very effective. Modification of the terminal ribose results in a weak antagonist, whereas its removal abolishes activity. An antagonist based upon a hybrid structure, 8-phenyl-2′-deoxy-ADPR (86, IC50 = 3 μM), is more potent than 8-Ph-ADPR (5). Initial bioisosteric replacement of the pyrophosphate linkage abolishes activity, but replacement of the pyrophosphate and the terminal ribose by a sulfamate-based group leads to a weak antagonist, a lead to more drug-like analogues. 8-Ph-ADPR (5) inhibits Ca2+ signalling and chemotaxis in human neutrophils, illustrating the potential for pharmacological intervention at TRPM2.
Co-reporter:Himali Y. Godage, Andrew M. Riley, Timothy J. Woodman, Mark P. Thomas, Mary F. Mahon, and Barry V. L. Potter
The Journal of Organic Chemistry 2013 Volume 78(Issue 6) pp:2275-2288
Publication Date(Web):February 25, 2013
DOI:10.1021/jo3027774
Acid hydrolysis of myo-inositol 1,3,5-orthoesters, apart from orthoformates, exclusively affords the corresponding 2-O-acyl myo-inositol products via a 1,2-bridged five-membered ring dioxolanylium ion intermediate observed by NMR spectroscopy. These C-2-substituted inositol derivatives provide valuable precursors for rapid and highly efficient routes to 2-O-acyl inositol 1,3,4,5,6-pentakisphosphates and myo-inositol 1,3,4,5,6-pentakisphosphate with biologically interesting and anticancer properties. Deuterium incorporation into the α-methylene group of such alkyl ester products (2-O-C(O)CD2R), when the analogous alkyl orthoester is treated with deuterated acid, is established utilizing the novel orthoester myo-inositol 1,3,5-orthobutyrate as an example. Such deuterated ester products provide intermediates for deuterium-labeled synthetic analogues. Investigation into this selective formation of 2-O-ester products and the deuterium incorporation is presented with proposed mechanisms from NMR experiments.
Co-reporter:L. W. Lawrence Woo;Paul M. Wood;Christian Bubert;Mark P. Thomas;Atul Purohit;Barry V. L. Potter
ChemMedChem 2013 Volume 8( Issue 5) pp:779-799
Publication Date(Web):
DOI:10.1002/cmdc.201300015
Abstract
4-{[(4-Cyanophenyl)(4H-1,2,4-triazol-4-yl)amino]methyl}phenyl sulfamate and its ortho-halogenated (F, Cl, Br) derivatives are first-generation dual aromatase and sulfatase inhibitors (DASIs). Structure–activity relationship studies were performed on these compounds, and various modifications were made to their structures involving relocation of the halogen atom, introduction of more halogen atoms, replacement of the halogen with another group, replacement of the methylene linker with a difluoromethylene linker, replacement of the para-cyanophenyl ring with other ring structures, and replacement of the triazolyl group with an imidazolyl group. The most potent in vitro DASI discovered is an imidazole derivative with IC50 values against aromatase and steroid sulfatase in a JEG-3 cell preparation of 0.2 and 2.5 nM, respectively. The parent phenol of this compound inhibits aromatase with an IC50 value of 0.028 nM in the same assay.
Co-reporter:Andrew M. Riley, Huanchen Wang, Jeremy D. Weaver, Stephen B. Shears and Barry V. L. Potter
Chemical Communications 2012 vol. 48(Issue 92) pp:11292-11294
Publication Date(Web):18 Sep 2012
DOI:10.1039/C2CC36044F
We synthesised analogues of diphosphoinositol polyphosphates (PP-InsPs) in which the diphosphate is replaced by an α-phosphonoacetic acid (PA) ester. Structural analysis revealed that 5-PA-InsP5 mimics 5-PP-InsP5 binding to the kinase domain of PPIP5K2; both molecules were phosphorylated by the enzyme. PA-InsPs are promising candidates for further studies into the biology of PP-InsPs.
Co-reporter:Kana M. Sureshan ; Andrew M. Riley ; Mark P. Thomas ; Stephen C. Tovey ; Colin W. Taylor ;Barry V. L. Potter
Journal of Medicinal Chemistry 2012 Volume 55(Issue 4) pp:1706-1720
Publication Date(Web):January 16, 2012
DOI:10.1021/jm201571p
Although adenophostin A (AdA), the most potent agonist of d-myo-inositol 1,4,5-trisphosphate receptors (IP3R), is thought to mimic IP3, the relative roles of the different phosphate groups and the adenosine motif have not been established. We synthesized all three possible bisphosphate analogues of AdA and glucose 3,4-bisphosphate (7, AdA lacking the 2′-AMP). 2′-Dephospho-AdA (6) was prepared via a novel regioselective dephosphorylation strategy. Assessment of the abilities of these bisphosphates to stimulate intracellular Ca2+ release using recombinant rat type 1 IP3R (IP3R1) revealed that 6, a mimic of Ins(4,5)P2, is only 4-fold less potent than IP3, while 7 is some 400-fold weaker and even 3″-dephospho-AdA (5) is measurably active, despite missing one of the vicinal bisphosphate groups normally thought to be crucial for IP3-like activity. Compound 6 is the most potent bisphosphate yet discovered with activity at IP3R. Thus, adenosine has a direct role independent of the 2′-phosphate group in contributing toward the potency of adenophostins, the vicinal bisphosphate motif is not essential for activity at the IP3R, as always thought, and it is possible to design potent agonists with just two of the three phosphates. A model with a possible adenine–R504 interaction supports the activity of 5 and 6 and also allows a reappraisal of the unexpected activity previously reported for the AdA regioisomer 2″-phospho-3″-dephospho-AdA 40.
Co-reporter:Christelle Moreau ; Tanja Kirchberger ; Bo Zhang ; Mark P. Thomas ; Karin Weber ; Andreas H. Guse ;Barry V. L. Potter
Journal of Medicinal Chemistry 2012 Volume 55(Issue 4) pp:1478-1489
Publication Date(Web):January 16, 2012
DOI:10.1021/jm201127y
Two nicotinamide adenine dinucleotide (NAD+) analogues modified at the 6 position of the purine ring were synthesized, and their substrate properties toward Aplysia californica ADP-ribosyl cyclase were investigated. 6-N-Methyl NAD+ (6-N-methyl nicotinamide adenosine 5′-dinucleotide 10) hydrolyzes to give the linear 6-N-methyl ADPR (adenosine 5′-diphosphoribose, 11), whereas 6-thio NHD+ (nicotinamide 6-mercaptopurine 5′-dinucleotide, 17) generates a cyclic dinucleotide. Surprisingly, NMR correlation spectra confirm this compound to be the N1 cyclic product 6-thio N1-cIDPR (6-thio cyclic inosine 5′-diphosphoribose, 3), although the corresponding 6-oxo analogue is well-known to cyclize at N7. In Jurkat T cells, unlike the parent cyclic inosine 5′-diphosphoribose N1-cIDPR 2, 6-thio N1-cIDPR antagonizes both cADPR- and N1-cIDPR-induced Ca2+ release but possesses weak agonist activity at higher concentration. 3 is thus identified as the first C-6 modified cADPR (cyclic adenosine 5′-diphosphoribose) analogue antagonist; it represents the first example of a fluorescent N1-cyclized cADPR analogue and is a new pharmacological tool for intervention in the cADPR pathway of cellular signaling.
Co-reporter:Stephen J. Mills, Camilla Persson, Gyles Cozier, Mark P. Thomas, Lionel Trésaugues, Christophe Erneux, Andrew M. Riley, Pär Nordlund, and Barry V. L. Potter
ACS Chemical Biology 2012 Volume 7(Issue 5) pp:822
Publication Date(Web):February 13, 2012
DOI:10.1021/cb200494d
Phosphoinositides regulate many cellular processes, and cellular levels are controlled by kinases and phosphatases. SHIP2 (SH2 (Src homology 2)-domain-containing inositol-phosphatase-2) plays a critical role in phosphoinositide signaling, cleaving the 5-phosphate from phosphatidylinositol 3,4,5-trisphosphate. SHIP2 is thought to be involved in type-2 diabetes and obesity, conditions that could therefore be open to pharmacological modulation of the enzyme. However, rational design of SHIP2 inhibitors has been limited by the absence of a high-resolution structure. Here, we present a 2.1 Å resolution crystal structure of the phosphatase domain of SHIP2 bound to the synthetic ligand biphenyl 2,3′,4,5′,6-pentakisphosphate (BiPh(2,3′,4,5′,6)P5). BiPh(2,3′,4,5′,6)P5 is not a SHIP2 substrate but inhibits Ins(1,3,4,5)P4 hydrolysis with an IC50 of 24.8 ± 3.0 μM, (Km for Ins(1,3,4,5)P4 is 215 ± 28 μM). Molecular dynamics simulations suggest that when BiPh(2,3′,4,5′,6)P5 binds to SHIP2, a flexible loop folds over and encloses the ligand. Compounds targeting such a closed conformation might therefore deliver SHIP2-specific drugs.
Co-reporter:Xiangdong Su, Heather A. Halem, Mark P. Thomas, Cecile Moutrille, Michael D. Culler, Nigel Vicker, Barry V.L. Potter
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 21) pp:6394-6402
Publication Date(Web):1 November 2012
DOI:10.1016/j.bmc.2012.08.056
The modulation of 11β-HSD1 activity with selective inhibitors has beneficial effects on various metabolic disorders including insulin resistance, dyslipidemia and obesity. Here we report the discovery of a series of novel adamantyl carboxamide and acetamide derivatives as selective inhibitors of human 11β-HSD1 in HEK-293 cells transfected with the HSD11B1 gene. Optimization based on an initially identified 11β-HSD1 inhibitor (3) led to the discovery of potent inhibitors with IC50 values in the 100 nM range. These compounds are also highly selective 11β-HSD1 inhibitors with no activity against 11β-HSD2 and 17β-HSD1. Compound 15 (IC50 = 114 nM) with weak inhibitory activity against the key human cytochrome P450 enzymes and moderate stability in incubation with human liver microsomes is worthy of further development. Importantly, compound 41 (IC50 = 280 nM) provides a new lead that incorporates an adamantyl group surrogate and should enable further series diversification.Series of adamantyl carboxamide and acetamide derivatives were identified, providing potent and selective inhibitors of the therapeutic target human 11β-hydroxysteroid dehydrogenase type 1.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Mathew P. Leese, Fabrice Jourdan, Wolfgang Dohle, Meriel R. Kimberley, Mark P. Thomas, Ruoli Bai, Ernest Hamel, Eric Ferrandis, and Barry V. L. Potter
ACS Medicinal Chemistry Letters 2012 Volume 3(Issue 1) pp:5
Publication Date(Web):October 31, 2011
DOI:10.1021/ml200232c
Structure–activity relationship translation offers an expeditious means for discovery of new active series. This approach was applied to discover tetrahydroisoquinoline (THIQ)-based steroidomimetic microtubule disruptors. The two A-ring elements of a three-point steroidal pharmacophore were incorporated into a THIQ-based A,B-ring mimic to which an H-bond acceptor was attached as the third motif. Optimization of the representative 6c through conformational biasing delivered a 10-fold gain in activity and a new series of microtubule disruptors (e.g., 9c) with antiproliferative activity in the nanomolar range. The THIQ derivatives match, or surpass, the activities of the steroidal series and exhibit improved physicochemical properties.Keywords: microtubule disruptor; steroidomimetic; structure−activity relationship; Tetrahydroisoquinoline
Co-reporter:L.W. Lawrence Woo, Bertrand Leblond, Atul Purohit, Barry V.L. Potter
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 8) pp:2506-2519
Publication Date(Web):15 April 2012
DOI:10.1016/j.bmc.2012.03.007
Estrone sulfamate (EMATE) is a potent irreversible inhibitor of steroid sulfatase (STS). In order to further expand SAR, the compound was substituted at the 2- and/or 4-positions and its 17-carbonyl group was also removed. The following general order of potency against STS in two in vitro systems is observed for the derivatives: The 4-NO2 > 2-halogens, 2-cyano > EMATE (unsubstituted) > 17-deoxyEMATE > 2-NO2 > 4-bromo > 2-(2-propenyl), 2-n-propyl > 4-(2-propenyl), 4-n-propyl > 2,4-(2-propenyl) = 2,4-di-n-propyl. There is a clear advantage in potency to place an electron-withdrawing substituent on the A-ring with halogens preferred at the 2-position, but nitro at the 4-position. Substitution with 2-propenyl or n-propyl at the 2- and/or 4-position of EMATE, and also removal of the 17-carbonyl group are detrimental to potency. Three cyclic sulfamates designed are not STS inhibitors. This further confirms that a free or N-unsubstituted sulfamate group (H2NSO2O–) is a prerequisite for potent and irreversible inhibition of STS as shown by inhibitors like EMATE and Irosustat. The most potent derivative synthesized is 4-nitroEMATE (2), whose IC50s in placental microsomes and MCF-7 cells are respectively 0.8 nM and 0.01 nM.
Co-reporter:Fabienne Pradaux-Caggiano, Xiangdong Su, Nigel Vicker, Mark P. Thomas, Deborah Smithen, Heather A. Halem, Michael D. Culler and Barry V. L. Potter
MedChemComm 2012 vol. 3(Issue 9) pp:1117-1124
Publication Date(Web):16 Jul 2012
DOI:10.1039/C2MD20091K
Regulation of tissue-specific glucocorticoid action by inhibiting 11β-HSD1 activity is regarded as a potential viable treatment for metabolic and cardiovascular diseases. Our effort to find an alternative to the highly favoured adamantyl moiety found in many potent 11β-HSD1 inhibitors led to the synthesis and SAR study of a series of phenyl ethanone thiadiazole derivatives. Potent compounds were identified with IC50 values in the range 100–300 nM in biological evaluation on an HEK293 cell line stably transfected with the human HSD11B1 gene. These compounds are selective with no activity against human 11β-HSD2. An SAR study revealed the favoured combination of phenyl substitution and the linker system being meta-methoxy with a sulphide linker (18) or a para-trifluoromethyl with sulphoxide linker (27). Docking of 3 into a crystal structure of the enzyme shows how the substituted phenyl group of this new series might mimic the adamantane motif. Potent inhibitors 18 and 27 are new templates for further optimization.
Co-reporter:Joanna M. Swarbrick and Barry V. L. Potter
The Journal of Organic Chemistry 2012 Volume 77(Issue 9) pp:4191-4197
Publication Date(Web):January 25, 2012
DOI:10.1021/jo202319f
Stable cyclic adenosine 5′-diphosphate ribose (cADPR) analogues are chemical biology tools that can probe the Ca2+ release mechanism and structure–activity relationships of this emerging potent second messenger. However, analogues with an intact “northern” ribose have been inaccessible due to the difficulty of generating the sensitive N1-ribosyl link. We report the first total synthesis of the membrane permeant, hydrolytically stable, cADPR receptor agonist 8-Br-N1-cIDPR via regio- and stereoselective N1-ribosylation of protected 8-bromoinosine.
Co-reporter:Fabrice Jourdan ; Mathew P. Leese ; Wolfgang Dohle ; Eric Ferrandis ; Simon P. Newman ; Surinder Chander ; Atul Purohit ;Barry V. L. Potter
Journal of Medicinal Chemistry 2011 Volume 54(Issue 13) pp:4863-4879
Publication Date(Web):May 23, 2011
DOI:10.1021/jm200483x
The synthesis and antiproliferative activities of analogues of 2-substituted estradiol-3,17-O,O-bis-sulfamates (E2bisMATEs) are discussed. Modifications of the C-17 substituent confirm that an H-bond acceptor is essential for high activity; its optimal linkage to C-17 and the local environment in which it resides are defined. In the non-sulfamoylated series 17β-acyl substitution delivers 48b, the most potent compound identified to date. In the sulfamate series a number of permutations of linker and H-bond acceptor deliver excellent activity, with 55, 61, 65, 49a, and 49b proving especially promising. The in vivo potential of these compounds was explored in the NCI hollow fiber assay and also in a mouse Matrigel model of antiangiogenesis in which 49 and 55 show significant inhibitory activity.
Co-reporter:Christelle Moreau, Gloria A. Ashamu, Victoria C. Bailey, Antony Galione, Andreas H. Guse and Barry V. L. Potter
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 1) pp:278-290
Publication Date(Web):25 Oct 2010
DOI:10.1039/C0OB00396D
Novel 8-substituted base and sugar-modified analogues of the Ca2+ mobilizing second messenger cyclic adenosine 5′-diphosphate ribose (cADPR) were synthesized using a chemoenzymatic approach and evaluated for activity in sea urchin egg homogenate (SUH) and in Jurkat T-lymphocytes; conformational analysis investigated by 1H NMR spectroscopy revealed that a C2′ endo/syn conformation of the “southern” ribose is crucial for agonist or antagonist activity at the SUH-, but not at the T cell-cADPR receptor.
Co-reporter:L. W. Lawrence Woo, Christian Bubert, Atul Purohit, and Barry V. L. Potter
ACS Medicinal Chemistry Letters 2011 Volume 2(Issue 3) pp:243
Publication Date(Web):December 29, 2010
DOI:10.1021/ml100273k
Single agents against multiple drug targets are highly topical. Hormone-dependent breast cancer (HDBC) may be more effectively treated by dual inhibition of aromatase and steroid sulfatase (STS), and several dual aromatase-sulfatase inhibitors (DASIs) have been recently reported. The best compounds from two leading classes of DASI, 3 and 9, are low nanomolar inhibitors. In search of a novel class of DASI, core motifs of two leading classes were combined to give a series of hybrid structures, with several compounds showing markedly improved dual inhibitory activities in the picomolar range in JEG-3 cells. Thus, DASIs 14 (IC50: aromatase, 15 pM; STS, 830 pM) and 15 (IC50: aromatase, 18 pM; STS, 130 pM) are the first examples of an exceptional new class of highly potent dual inhibitor that should encourage further development toward multitargeted therapeutic intervention in HDBC.Keywords (keywords): aromatase; cancer; dual inhibitors; Hybrid; sulfatase
Co-reporter:Dr. Paul M. Wood;Dr. L. W. Lawrence Woo;Dr. Mark P. Thomas;Dr. Mary F. Mahon;Dr. Atul Purohit; Barry V. L. Potter
ChemMedChem 2011 Volume 6( Issue 8) pp:1423-1438
Publication Date(Web):
DOI:10.1002/cmdc.201100145
Abstract
Concurrent inhibition of aromatase and steroid sulfatase (STS) may provide a more effective treatment for hormone-dependent breast cancer than monotherapy against individual enzymes, and several dual aromatase–sulfatase inhibitors (DASIs) have been reported. Three aromatase inhibitors with sub-nanomolar potency, better than the benchmark agent letrozole, were designed. To further explore the DASI concept, a new series of letrozole-derived sulfamates and a vorozole-based sulfamate were designed and biologically evaluated in JEG-3 cells to reveal structure–activity relationships. Amongst achiral and racemic compounds, 2-bromo-4-(2-(4-cyanophenyl)-2-(1H-1,2,4-triazol-1-yl)ethyl)phenyl sulfamate is the most potent DASI (aromatase: IC50=0.87 nM; STS: IC50=593 nM). The enantiomers of the phenolic precursor to this compound were separated by chiral HPLC and their absolute configuration determined by X-ray crystallography. Following conversion to their corresponding sulfamates, the S-(+)-enantiomer was found to inhibit aromatase and sulfatase most potently (aromatase: IC50=0.52 nM; STS: IC50=280 nM). The docking of each enantiomer and other ligands into the aromatase and sulfatase active sites was also investigated.
Co-reporter:Dr. Xiangdong Su;Dr. Nigel Vicker;Dr. Mark P. Thomas;Dr. Fabienne Pradaux-Caggiano;Dr. Heather Halem;Dr. Michael D. Culler; Barry V. L. Potter
ChemMedChem 2011 Volume 6( Issue 8) pp:1439-1451
Publication Date(Web):
DOI:10.1002/cmdc.201100144
Abstract
11β-Hydroxysteroid dehydrogenase type 1 (11β-HSD1) plays a key role in converting intracellular cortisone to physiologically active cortisol, which is implicated in the development of several phenotypes of metabolic syndrome. Inhibition of 11β-HSD1 activity with selective inhibitors has beneficial effects on various conditions, including diabetes, dyslipidemia and obesity, and therefore constitutes a promising strategy to discover novel therapies for metabolic and cardiovascular diseases. A series of novel adamantyl heterocyclic ketones provides potent and selective inhibitors of human 11β-HSD1. Lead compounds display low nanomolar inhibition against human and mouse 11β-HSD1 and are selective with no activity against 11β-HSD2 and 17β-HSD1. Selected potent 11β-HSD1 inhibitors show moderate metabolic stability upon incubation with human liver microsomes and weak inhibition of human CYP450 enzymes.
Co-reporter:Dr. L. W. Lawrence Woo;Dr. Dharshini Ganeshapillai;Dr. Mark P. Thomas;Dr. Oliver B. Sutcliffe;Dr. Bindu Malini;Dr. Mary F. Mahon;Dr. Atul Purohit; Barry V. L. Potter
ChemMedChem 2011 Volume 6( Issue 11) pp:
Publication Date(Web):
DOI:10.1002/cmdc.201190047
Co-reporter:Dr. L. W. Lawrence Woo;Dr. Dharshini Ganeshapillai;Dr. Mark P. Thomas;Dr. Oliver B. Sutcliffe;Dr. Bindu Malini;Dr. Mary F. Mahon;Dr. Atul Purohit; Barry V. L. Potter
ChemMedChem 2011 Volume 6( Issue 11) pp:2019-2034
Publication Date(Web):
DOI:10.1002/cmdc.201100288
Abstract
Structure–activity relationship studies were conducted on Irosustat (STX64, BN83495), the first steroid sulfatase (STS) inhibitor to enter diverse clinical trials for patients with advanced hormone-dependent cancer. The size of its aliphatic ring was expanded; its sulfamate group was N,N-dimethylated, relocated to another position and flanked by an adjacent methoxy group; and series of quinolin-2(1H)-one and quinoline derivatives of Irosustat were explored. The STS inhibitory activities of the synthesised compounds were assessed in a preparation of JEG-3 cells. Stepwise enlargement of the aliphatic ring from 7 to 11 members increases potency, although a further increase in ring size is detrimental. The best STS inhibitors in vitro had IC50 values between 0.015 and 0.025 nM. Other modifications made to Irosustat were found to either abolish or significantly weaken its activity. An azomethine adduct of Irosustat with N,N-dimethylformamide (DMF) was isolated, and crystal structures of Irosustat and this adduct were determined. Docking studies were conducted to explore the potential interactions between compounds and the active site of STS, and suggest a sulfamoyl group transfer to formylglycine 75 during the inactivation mechanism.
Co-reporter:Dr. Xiangdong Su;Dr. Fabienne Pradaux-Caggiano;Dr. Nigel Vicker;Dr. Mark P. Thomas;Dr. Heather Halem;Dr. Michael D. Culler; Barry V. L. Potter
ChemMedChem 2011 Volume 6( Issue 9) pp:1616-1629
Publication Date(Web):
DOI:10.1002/cmdc.201100182
Abstract
Elevated levels of active glucocorticoids have been implicated in the development of several phenotypes of metabolic syndrome, such as type 2 diabetes and obesity. 11β-Hydroxysteroid dehydrogenase type 1 (11β-HSD1) catalyses the intracellular conversion of inactive cortisone to cortisol. Selective 11β-HSD1 inhibitors have shown beneficial effects in various conditions, including diabetes, dyslipidemia and obesity. A series of adamantyl ethanone pyridyl derivatives has been identified, providing potent and selective inhibitors of human 11β-HSD1. Lead compounds display low nanomolar inhibition against human and mouse 11β-HSD1 and are selective for this isoform, with no activity against 11β-HSD2 and 17β-HSD1. Structure–activity relationship studies reveal that an unsubstituted pyridine tethered to an adamantyl ethanone motif through an ether or sulfoxide linker provides a suitable pharmacophore for activity. The most potent inhibitors have IC50 values around 34–48 nM against human 11β-HSD1, display reasonable metabolic stability in human liver microsomes, and weak inhibition of key human CYP450 enzymes.
Co-reporter:Mathew P. Leese, Fabrice Jourdan, Meriel R. Kimberley, Gyles E. Cozier, Nethaji Thiyagarajan, Chloe Stengel, Sandra Regis-Lydi, Paul A. Foster, Simon P. Newman, K. Ravi Acharya, Eric Ferrandis, Atul Purohit, Michael J. Reed and Barry V. L. Potter
Chemical Communications 2010 vol. 46(Issue 17) pp:2907-2909
Publication Date(Web):20 Mar 2010
DOI:10.1039/C002558E
A chimeric approach is used to discover microtubule disruptors with excellent in vitro activity and oral bioavailability; a ligand–protein interaction with carbonic anhydrase that enhances bioavailability is characterised by protein X-ray crystallography. Dosing of a representative chimera in a tumour xenograft model confirms the excellent therapeutic potential of the class.
Co-reporter:L. W. Lawrence Woo ; Toby Jackson ; Aurélien Putey ; Gyles Cozier ; Philip Leonard ; K. Ravi Acharya ; Surinder K. Chander ; Atul Purohit ; Michael J. Reed ;Barry V. L. Potter
Journal of Medicinal Chemistry 2010 Volume 53(Issue 5) pp:2155-2170
Publication Date(Web):February 11, 2010
DOI:10.1021/jm901705h
Single agents against multiple drug targets are of increasing interest. Hormone-dependent breast cancer (HDBC) may be more effectively treated by dual inhibition of aromatase and steroid sulfatase (STS). The aromatase inhibitory pharmacophore was thus introduced into a known biphenyl STS inhibitor to give a series of novel dual aromatase−sulfatase inhibitors (DASIs). Several compounds are good aromatase or STS inhibitors and DASI 20 (IC50: aromatase, 2.0 nM; STS, 35 nM) and its chlorinated congener 23 (IC50: aromatase, 0.5 nM; STS, 5.5 nM) are examples that show exceptional dual potency in JEG-3 cells. Both biphenyls share a para-sulfamate-containing ring B and a ring A, which contains a triazol-1-ylmethyl meta to the biphenyl bridge and para to a nitrile. At 1 mg/kg po, 20 and 23 reduced plasma estradiol levels strongly and inhibited liver STS activity potently in vivo. 23 is nonestrogenic and potently inhibits carbonic anhydrase II (IC50 86 nM). A complex was crystallized and its structure was solved by X-ray crystallography. This class of DASI should encourage further development toward multitargeted therapeutic intervention in HDBC.
Co-reporter:Fabrice Jourdan ; Mathew P. Leese ; Wolfgang Dohle ; Ernest Hamel ; Eric Ferrandis ; Simon P. Newman ; Atul Purohit ; Michael J. Reed ;Barry V. L. Potter
Journal of Medicinal Chemistry 2010 Volume 53(Issue 7) pp:2942-2951
Publication Date(Web):March 12, 2010
DOI:10.1021/jm9018806
The synthesis and antiproliferative activity of analogues of estradiol 3,17-O,O-bis-sulfamates (E2bisMATEs) are discussed. Modifications of the C-17 substituent reveal that an H-bond acceptor is essential for high antiproliferative activity. The local environment in which this H-bond acceptor lies can be varied to an extent. The C-17-oxygen linker can be deleted or substituted with an electronically neutral methylene group, and replacement of the terminal NH2 with a methyl group is also acceptable. Mesylates 10 and 14 prove equipotent to the E2bisMATEs 2 and 3, while sulfones 20 and 35 display enhanced in vitro antiproliferative activity. In addition, the SAR of 2-substituted estradiol-3-O-sulfamate derivatives as inhibitors of tubulin polymerization has been established for the first time. These agents inhibit the binding of radiolabeled colchicine to tubulin.
Co-reporter:Gyles E. Cozier, Mathew P. Leese, Matthew D. Lloyd, Matthew D. Baker, Nethaji Thiyagarajan, K. Ravi Acharya and Barry V. L. Potter
Biochemistry 2010 Volume 49(Issue 16) pp:
Publication Date(Web):March 18, 2010
DOI:10.1021/bi902178w
Carbonic anhydrase (CA) catalyzes the reversible hydration of carbon dioxide to hydrogen carbonate, and its role in maintaining pH balance has made it an attractive drug target. Steroidal sulfamate esters, inhibitors of the cancer drug target steroid sulfatase (STS), are sequestered in vivo by CA II in red blood cells, which may be the origin of their excellent drug properties. Understanding the structural basis of this is important for drug design. Structures of CA II complexed with 2-methoxyestradiol 3-O-sulfamate (3), 2-ethylestradiol 3,17-O,O-bis(sulfamate) (4), and 2-methoxyestradiol 17-O-sulfamate (5) are reported to 2.10, 1.85, and 1.64 Å, respectively. Inhibitor 3 interacts with the active site Zn(II) ion through the 3-O-sulfamate, while inhibitors 4 and 5 bind through their 17-O-sulfamate. Comparison of the IC50 values for CA II inhibition gave respective values of 56, 662, 2113, 169, 770, and 86 nM for estrone 3-O-sulfamate (1), 2-methoxyestradiol 3,17-O,O-bis(sulfamate) (2), 3, 4, 5, and 5′-((4H-1,2,4-triazol-4-yl)methyl)-3-chloro-2′-cyanobiphenyl-4-yl sulfamate (6), a nonsteroidal dual aromatase−sulfatase inhibitor. Inhibitors 2, 5, and 6 showed binding to a second adjacent site that is capable of binding both steroidal and nonsteroidal ligands. Examination of both IC50 values and crystal structures suggests that 2-substituents on the steroid nucleus hinder binding via a 3-O-sulfamate, leading to coordination through a 17-O-sulfamate if present. These results underline the influence of small structural changes on affinity and mode of binding, the degree of flexibility in the design of sulfamate-based inhibitors, and suggest a strategy for inhibitors which interact with both the active site and the second adjacent binding site simultaneously that could be both potent and selective.
Co-reporter:PaulM. Wood Dr.;L.W. Lawrence Woo Dr.;Jean-Robert Labrosse Dr.;MarkP. Thomas Dr.;MaryF. Mahon Dr.;SurinderK. Cher Dr.;Atul Purohit Dr.;MichaelJ. Reed ;BarryV.L. Potter
ChemMedChem 2010 Volume 5( Issue 9) pp:1577-1593
Publication Date(Web):
DOI:10.1002/cmdc.201000203
Abstract
The design and synthesis of a series of bicyclic ring containing dual aromatase–sulfatase inhibitors (DASIs) based on the aromatase inhibitor (AI) 4-[(4-bromobenzyl)(4H-1,2,4-triazol-4-yl)amino]benzonitrile are reported. Biological evaluation with JEG-3 cells revealed structure–activity relationships. The X-ray crystal structure of sulfamate 23 was determined, and selected compounds were docked into the aromatase and steroid sulfatase (STS) crystal structures. In the sulfamate-containing series, compounds containing a naphthalene ring are both the most potent AI (39, IC50 AROM=0.25 nM) and the best STS inhibitor (31, IC50 STS=26 nM). The most promising DASI is 39 (IC50 AROM=0.25 nM, IC50 STS=205 nM), and this was evaluated orally in vivo at 10 mg kg−1, showing potent inhibition of aromatase (93 %) and STS (93 %) after 3 h. Potent aromatase and STS inhibition can thus be achieved with a DASI containing a bicyclic ring system; development of such a DASI could provide an attractive new option for the treatment of hormone-dependent breast cancer.
Co-reporter:Xiangdong Su Dr.;Fabienne Pradaux-Caggiano Dr.;MarkP. Thomas Dr.;Michelle W.Y. Szeto Dr.;HeatherA. Halem Dr.;MichaelD. Culler Dr.;Nigel Vicker Dr.;Barry V.L. Potter
ChemMedChem 2010 Volume 5( Issue 7) pp:1026-1044
Publication Date(Web):
DOI:10.1002/cmdc.201000081
Abstract
11β-Hydroxysteroid dehydrogenases (11β-HSDs) are key enzymes regulating the pre-receptor metabolism of glucocorticoid hormones. The modulation of 11β-HSD type 1 activity with selective inhibitors has beneficial effects on various conditions including insulin resistance, dyslipidemia and obesity. Inhibition of tissue-specific glucocorticoid action by regulating 11β-HSD1 constitutes a promising treatment for metabolic and cardiovascular diseases. A series of novel adamantyl ethanone compounds was identified as potent inhibitors of human 11β-HSD1. The most active compounds identified (52, 62, 72, 92, 103 and 104) display potent inhibition of 11β-HSD1 with IC50 values in the 50–70 nM range. Compound 72 also proved to be metabolically stable when incubated with human liver microsomes. Furthermore, compound 72 showed very weak inhibitory activity for human cytochrome P450 enzymes and is therefore a candidate for in vivo studies. Comparison of the publicly available X-ray crystal structures of human 11β-HSD1 led to docking studies of the potent compounds, revealing how these molecules may interact with the enzyme and cofactor.
Co-reporter:Kana M. Sureshan, Andrew M. Riley, Ana M. Rossi, Stephen C. Tovey, Skarlatos G. Dedos, Colin W. Taylor and Barry V. L. Potter
Chemical Communications 2009 (Issue 10) pp:1204-1206
Publication Date(Web):04 Feb 2009
DOI:10.1039/B819328B
Ca2+ release by D-myo-inositol 1,4,5-trisphosphate receptors (IP3Rs) is widely considered to require the vicinal 4,5-bisphosphate motif of IP3, with P-5 and P-4 engaging the α and β domains of the binding site; using synthesis and mutagenesis we show that the adenine of synthetic glyconucleotides, through an interaction with Arg504, can replace the interaction of either P-1 or P-5 with the α-domain producing, respectively, the most potent bisphosphate agonist yet synthesised and the first agonist of IP3R without a vicinal bisphosphate motif; this will stimulate new approaches to IP3R ligand design.
Co-reporter:Bo Zhang;Werner Dammermann;Chiara Cordiglieri;Merle Nebel;Tanja Kirchberger;Naoto Kawakami;Francesca Odoardi;Frederike Schmid;Martin Hohenegger;Klaus Dornmair;James Dowden;Andreas H. Guse;Barry V. L. Potter;Alexander Flügel
PNAS 2009 Volume 106 (Issue 26 ) pp:10678-10683
Publication Date(Web):2009-06-30
DOI:10.1073/pnas.0809997106
The nucleotide NAADP was recently discovered as a second messenger involved in the initiation and propagation of Ca2+ signaling in lymphoma T cells, but its impact on primary T cell function is still unknown. An optimized, synthetic, small
molecule inhibitor of NAADP action, termed BZ194, was designed and synthesized. BZ194 neither interfered with Ca2+ mobilization by d-myo-inositol 1,4,5-trisphosphate or cyclic ADP-ribose nor with capacitative Ca2+ entry. BZ194 specifically and effectively blocked NAADP-stimulated [3H]ryanodine binding to the purified type 1 ryanodine receptor. Further, in intact T cells, Ca2+ mobilization evoked by NAADP or by formation of the immunological synapse between primary effector T cells and astrocytes
was inhibited by BZ194. Downstream events of Ca2+ mobilization, such as nuclear translocation of “nuclear factor of activated T cells” (NFAT), T cell receptor-driven interleukin-2
production, and proliferation in antigen-experienced CD4+ effector T cells, were attenuated by the NAADP antagonist. Taken together, specific inhibition of the NAADP signaling pathway
constitutes a way to specifically and effectively modulate T-cell activation and has potential in the therapy of autoimmune
diseases.
Co-reporter:Mathew P. Leese ; Fabrice L. Jourdan ; Keira Gaukroger ; Mary F. Mahon ; Simon P. Newman ; Paul A. Foster ; Chloe Stengel ; Sandra Regis-Lydi ; Eric Ferrandis ; Anna Di Fiore ; Giuseppina De Simone ; Claudiu T. Supuran ▽; Atul Purohit ; Michael J. Reed ;Barry V. L. Potter
Journal of Medicinal Chemistry 2008 Volume 51(Issue 5) pp:1295-1308
Publication Date(Web):February 9, 2008
DOI:10.1021/jm701319c
The synthesis, SAR, and preclinical evaluation of 17-cyanated 2-substituted estra-1,3,5(10)-trienes as anticancer agents are discussed. 2-Methoxy-17β-cyanomethylestra-1,3,5(10)-trien-3-ol (14), but not the related 2-ethyl derivative 7, and the related 3-O-sulfamates 8 and 15 display potent antiproliferative effects (MCF-7 GI50 300, 60 and 70 nM, respectively) against human cancer cells in vitro. Investigation of the SAR reveals that a sterically unhindered hydrogen bond acceptor attached to C-17 is most likely key to the enhanced activity. Compound 8 displayed significant in vitro antiangiogenic activity, and its ability to act as a microtubule disruptor was confirmed. Inhibitory activity of the sulfamate derivatives against steroid sulfatase and carbonic anhydrase II (hCAII) was also observed, and the interaction between 15 and hCAII was investigated by protein crystallography. The potential of these multimechanism anticancer agents was confirmed in vivo, with promising activity observed for both 14 and 15 in an athymic nude mouse MDA-MB-231 human breast cancer xenograft model.
Co-reporter:Bo Zhang ; Gerd K. Wagner ; Karin Weber ; Clive Garnham ; Anthony J. Morgan ; Antony Galione ; Andreas H. Guse ;Barry V. L. Potter
Journal of Medicinal Chemistry 2008 Volume 51(Issue 6) pp:1623-1636
Publication Date(Web):February 28, 2008
DOI:10.1021/jm7010386
The structural features needed for antagonism at the cyclic ADP-ribose (cADPR) receptor are unclear. Chemoenzymatic syntheses of novel 8-substituted 2′-deoxy-cADPR analogues, including 8-bromo-2′-deoxy-cADPR 7, 8-amino-2′-deoxy-cADPR 8, 8-O-methyl-2′-deoxy-cADPR 9, 8-phenyl-2′-deoxy-cADPR 10 and its ribose counterpart 8-phenyl-cADPR 5 are reported, including improved syntheses of established antagonists 8-amino-cADPR 2 and 8-bromo-cADPR 3. Aplysia californica ADP-ribosyl cyclase tolerates even the bulky 8-phenyl-nicotinamide adenine 5′-dinucleotide as a substrate. Structure−activity relationships of 8-substituted cADPR analogues in both Jurkat T-lymphocytes and sea urchin egg homogenate (SUH) were investigated. 2′-OH Deletion decreased antagonistic activity (at least for the 8-amino series), showing it to be an important motif. Some 8-substituted 2′-deoxy analogues showed agonist activity at higher concentrations, among which 8-bromo-2′-deoxy-cADPR 7 was, unexpectedly, a weak but almost full agonist in SUH and was membrane-permeant in whole eggs. Classical antagonists 2 and 3 also showed previously unobserved agonist activity at higher concentrations in both systems. The 2′-OH group, without effect on the Ca2+-mobilizing ability of cADPR itself, is an important motif for the antagonistic activities of 8-substituted cADPR analogues.
Co-reporter:Paul M. Wood ; L. W. Lawrence Woo ; Jean-Robert Labrosse ; Melanie N. Trusselle ; Sergio Abbate ; Giovanna Longhi ; Ettore Castiglioni ; France Lebon ; Atul Purohit ; Michael J. Reed ;Barry V. L. Potter
Journal of Medicinal Chemistry 2008 Volume 51(Issue 14) pp:4226-4238
Publication Date(Web):July 1, 2008
DOI:10.1021/jm800168s
To explore aromatase inhibition and to broaden the structural diversity of dual aromatase−sulfatase inhibitors (DASIs), we introduced the steroid sulfatase (STS) inhibitory pharmacophore to letrozole. Letrozole derivatives were prepared bearing bis-sulfamates or mono-sulfamates with or without adjacent substituents. The most potent of the achiral and racemic aromatase inhibitor was 40 (IC50 = 3.0 nM). Its phenolic precursor 39 was separated by chiral HPLC, and the absolute configuration of each enantiomer was determined using vibrational and electronic circular dichroism in tandem with calculations of the predicted spectra. Of the two enantiomers, (R)-phenol (39a) was the most potent aromatase inhibitor (IC50 = 0.6 nM, comparable to letrozole), whereas the (S)-sulfamate, (40b) inhibited STS most potently (IC50 = 553 nM). These results suggest that a new structural class of DASI for potential treatment of hormone-dependent breast cancer has been identified, and this is the first report of STS inhibition by an enantiopure nonsteroidal compound.
Co-reporter:Fabrice Jourdan, Christian Bubert, Mathew P. Leese, Andrew Smith, Eric Ferrandis, Sandra Regis-Lydi, Simon P. Newman, Atul Purohit, Michael J. Reed and Barry V. L. Potter
Organic & Biomolecular Chemistry 2008 vol. 6(Issue 22) pp:4108-4119
Publication Date(Web):22 Sep 2008
DOI:10.1039/B810300C
The potent activity of 2-substituted estra-1,3,5(10)-triene-3-O-sulfamates against the proliferation of cancer cellsin vitro and tumours in vivo highlights the therapeutic potential of such compounds. Optimal activity is derived from a combination of a 2-XMe group (where X = CH2, O or S), a 3-O-sulfamate group in the steroidal A-ring and a H-bond acceptor around C-17 of the D-ring. Herein, we describe the synthesis and anti-proliferative activities of a series of novel 2-substituted estra-1,3,5(10)-triene-3-O-sulfamates bearing heterocyclic substituents (oxazole, tetrazole, triazole) tethered to C-17. In vitro evaluation of these molecules revealed that high anti-proliferative activity in breast and prostate cancer cells lines (GI50 of 340–850 nM) could be retained when the heterocyclic substituent possesses H-bond acceptor properties. A good correlation between the calculated electron density of the heterocyclic ring and anti-proliferative activity was observed. Docking of the most active compounds into their putative site of action, the colchicine binding site of tubulin, suggests that they bind through a different mode to the previously described bis-sulfamate derivatives 1 and 2, which possess similar in vitro activity.
Co-reporter:Gillian M. Allan, Nigel Vicker, Harshani R. Lawrence, Helena J. Tutill, Joanna M. Day, Marion Huchet, Eric Ferrandis, Michael J. Reed, Atul Purohit, Barry V.L. Potter
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 8) pp:4438-4456
Publication Date(Web):15 April 2008
DOI:10.1016/j.bmc.2008.02.059
The 17β-hydroxysteroid dehydrogenases (17β-HSDs) catalyze the interconversion between the oxidized and reduced forms of androgens and estrogens at the 17 position. The 17β-HSD type 1 enzyme (17β-HSD1) catalyzes the reduction of estrone (E1) to estradiol and is expressed in malignant breast cells. Inhibitors of this enzyme thus have potential as treatments for hormone dependent breast cancer. Syntheses and biological evaluation of novel non-steroidal inhibitors designed to mimic the E1 template are reported using information from potent steroidal inhibitors. Of the templates investigated biphenyl ethanone was promising and led to inhibitors with IC50 values in the low micromolar range.
Co-reporter:Stephen J. Mills Dr.;Fabrice Veput Dr.;Melanie N. Trusselle Dr.;Stephen T. Safrany Dr.;Christophe Erneux Dr.;Barry V. L. Potter Dr.
ChemBioChem 2008 Volume 9( Issue 11) pp:1757-1766
Publication Date(Web):
DOI:10.1002/cbic.200800104
Abstract
Novel benzene polyphosphates were synthesised as inositol polyphosphate mimics and evaluated against type-I inositol 1,4,5-trisphosphate 5-phosphatase, which only binds soluble inositol polyphosphates, and against the PH domain of protein kinase Bα (PKBα), which can bind both soluble inositol polyphosphates and inositol phospholipids. The most potent trisphosphate 5-phosphatase inhibitor is benzene 1,2,4-trisphosphate (2, IC50 of 14 μM), a potential mimic of D-myo-inositol 1,4,5-trisphosphate, whereas the most potent tetrakisphosphate Ins(1,4,5)P3 5-phosphatase inhibitor is benzene 1,2,4,5-tetrakisphosphate, with an IC50 of 4 μM. Biphenyl 2,3′,4,5′,6-pentakisphosphate (4) was the most potent inhibitor evaluated against type I Ins(1,4,5)P3 5-phosphatase (IC50 of 1 μM). All new benzene polyphosphates are resistant to dephosphorylation by type I Ins(1,4,5)P3 5-phosphatase. Unexpectedly, all benzene polyphosphates studied bind to the PH domain of PKBα with apparent higher affinity than to type I Ins(1,4,5)P3 5-phosphatase. The most potent ligand for the PKBα PH domain, measured by inhibition of biotinylated diC8-PtdIns(3,4)P2 binding, is biphenyl 2,3′,4,5′,6-pentakisphosphate (4, Ki=27 nm). The approximately 80-fold enhancement of binding relative to parent benzene trisphosphate is explained by the involvement of a cation–π interaction. These new molecular tools will be of potential use in structural and cell signalling studies.
Co-reporter:Toby Jackson Dr.;L.W. Lawrence Woo Dr.;MelanieN. Trusselle Dr.;Atul Purohit Dr.;MichaelJ. Reed ;BarryV.L. Potter
ChemMedChem 2008 Volume 3( Issue 4) pp:603-618
Publication Date(Web):
DOI:10.1002/cmdc.200700266
Abstract
The synthesis and in vitro biological evaluation (JEG-3 cells) of a series of novel and potent aromatase inhibitors, prepared by microwave-enhanced Suzuki cross-coupling methodology, are reported. These compounds possess a biphenyl template incorporated with the haem-ligating triazolylmethyl moiety, either on its own or in combination with other substituent(s) at various positions on the phenyl rings. The most potent aromatase inhibitor reported herein has an IC50 value of 0.12 nM, although seven of its congeners are also highly potent (IC50≤0.5 nM). They all bear the (5-triazolylmethyl-2-cyano)biphenyl structural motif. Docking of representative compounds into a homology model of human aromatase assists in the rationalisation of the SAR derived from the in vitro biological results and supports a crucial role for a cyano group on the “A” phenyl ring, which is accessible to hydrogen bond interactions with Ser 478. Further development of these compounds as potential therapeutic agents for the treatment of hormone-dependent breast cancer is warranted given the high level of potency observed for this class of aromatase inhibitor in vitro.
Co-reporter:Christian Bubert Dr.;L.W. Lawrence Woo Dr.;OliverB. Sutcliffe Dr.;MaryF. Mahon Dr.;SurinderK. Cher Dr.;Atul Purohit Dr.;MichaelJ. Reed ;BarryV.L. Potter
ChemMedChem 2008 Volume 3( Issue 11) pp:1708-1730
Publication Date(Web):
DOI:10.1002/cmdc.200800164
Abstract
4-(((4-Cyanophenyl)(4H-1,2,4-triazol-4-yl)amino)methyl)phenyl sulfamate (6 a) was the first dual aromatase–sulfatase inhibitor (DASI) reported. Several series of its derivatives with various linker systems between the steroid sulfatase (STS) and the aromatase inhibitory pharmacophores were synthesised and evaluated in JEG-3 cells. The X-ray crystal structures of the aromatase inhibitors, DASI precursors 42 d and 60, and DASI 43 h were determined. Nearly all derivatives show improved in vitro aromatase inhibition over 6 a but decreased STS inhibition. The best aromatase inhibitor is 42 e (IC50=0.26 nM) and the best DASI is 43 e (IC50 aromatase=0.45 nM, IC50 STS=1200 nM). SAR for aromatase inhibition shows that compounds containing an alkylene- and thioether-based linker system are more potent than those that are ether-, sulfone-, or sulfonamide-based, and that the length of the linker has a limited effect on aromatase inhibition beyond two methylene units. Compounds 43 d–f were studied in vivo (10 mg kg−1, single, p.o.). The most potent DASI is 43 e, which inhibited PMSG-induced plasma estradiol levels by 92 % and liver STS activity by 98 % 3 h after dosing. These results further strengthen the concept of designing and developing DASIs for potential treatment of hormone-related cancers.
Co-reporter:Stephen J. Mills, David Komander, Melanie N. Trusselle, Stephen T. Safrany, Daan M. F. van Aalten and Barry V. L. Potter
ACS Chemical Biology 2007 Volume 2(Issue 4) pp:242
Publication Date(Web):April 13, 2007
DOI:10.1021/cb700019r
Protein kinase B (PKB/Akt) plays a key role in cell signaling. The PH domain of PKB binds phosphatidylinositol 3,4,5-trisphosphate translocating PKB to the plasma membrane for activation by 3-phosphoinositide-dependent protein kinase 1. The crystal structure of the headgroup inositol 1,3,4,5-tetrakisphosphate Ins(1,3,4,5)P 4–PKB complex facilitates in silico ligand design. The novel achiral analogue benzene 1,2,3,4-tetrakisphosphate (Bz(1,2,3,4)P 4) possesses phosphate regiochemistry different from that of Ins(1,3,4,5)P 4 and surprisingly binds with similar affinity as the natural headgroup. Bz(1,2,3,4)P 4 co-crystallizes with the PKBα PH domain in a fashion also predictable in silico. The 2-phosphate of Bz(1,2,3,4)P 4 does not interact with any residue, and the d5-phosphate of Ins(1,3,4,5)P 4 is not mimicked by Bz(1,2,3,4)P 4. Bz(1,2,3,4)P 4 is an example of a simple inositol phosphate surrogate crystallized in a protein, and this approach could be applied to design modulators of inositol polyphosphate binding proteins.
Co-reporter:Toby Jackson, L. W. Lawrence Woo, Melanie N. Trusselle, Surinder K. Chander, Atul Purohit, Michael J. Reed and Barry V. L. Potter
Organic & Biomolecular Chemistry 2007 vol. 5(Issue 18) pp:2940-2952
Publication Date(Web):12 Sep 2007
DOI:10.1039/B707768H
The synthesis and biological evaluation of a series of novel Dual Aromatase–Sulfatase Inhibitors (DASIs) are described. It is postulated that dual inhibition of the aromatase and steroid sulfatase enzymes, both responsible for the biosynthesis of oestrogens, will be beneficial in the treatment of hormone-dependent breast cancer. The compounds are based upon the Anastrozolearomatase inhibitor template which, while maintaining the haem ligating triazole moiety crucial for enzyme inhibition, was modified to include a phenol sulfamate ester motif, the pharmacophore for potent irreversible steroid sulfatase inhibition. Adaption of a synthetic route to Anastrozole was accomplished via selective radical bromination and substitution reactions to furnish a series of aromatase inhibitory pharmacophores. Linking these fragments to the phenol sulfamate ester moiety employed SN2, Heck and Mitsunobu reactions with phenolic precursors, from where the completed DASIs were achieved via sulfamoylation. In vitro, the lead compound, 11, had a high degree of potency against aromatase (IC50 3.5 nM), comparable with that of Anastrozole (IC50 1.5 nM) whereas, only moderate activity against steroid sulfatase was found. However, in vivo, 11 surprisingly exhibited potent dual inhibition. Compound 11 was modelled into the active site of a homology model of human aromatase and the X-ray crystal structure of steroid sulfatase.
Co-reporter:Himali Y. Godage, Andrew M. Riley, Timothy J. Woodman and Barry V. L. Potter
Chemical Communications 2006 (Issue 28) pp:2989-2991
Publication Date(Web):07 Jun 2006
DOI:10.1039/B605392K
Acid hydrolysis of myo-inositol 1,3,5-orthobenzoate leads regioselectively to 2-O-benzoyl-myo-inositol via a 1,2-bridged 2′-phenyl-1′,3′-dioxolan-2′-ylium ion observed by 1H and 13C NMR spectroscopy, providing the precursor for a highly efficient route to the anticancer agent myo-inositol 1,3,4,5,6-pentakisphosphate.
Co-reporter:Kana M. Sureshan, Melanie Trusselle, Stephen C. Tovey, Colin W. Taylor and Barry V. L. Potter
Chemical Communications 2006 (Issue 19) pp:2015-2017
Publication Date(Web):31 Mar 2006
DOI:10.1039/B517911D
Guanophostin A, the guanosine counterpart of the inositol 1,4,5-trisphosphate receptor agonist adenophostin A, has been synthesized and is the first synthetic adenophostin A-like analogue to be equipotent to its parent in stimulating intracellular Ca2+ release; its nucleotide moiety is proposed to interact with the receptor binding core by guanine base cation-π stacking with Arg504 and hydrogen bonding with Glu505 and interaction of the ribosyl 2′-phosphate group with the helix-dipole of α6.
Co-reporter:Christelle Moreau, Timothy J. Woodman and Barry V. L. Potter
Chemical Communications 2006 (Issue 10) pp:1127-1129
Publication Date(Web):25 Jan 2006
DOI:10.1039/B517916E
Cyclic 8-bromo-inosine-5′-diphosphate ribose (8-Br-N1-cIDPR) was cleanly degraded at acidic pH by N9 ribosyl scission and subsequent pyrophosphate cleavage to give 8-bromo-N1-ribosyl hypoxanthine 5′-monophosphate (8-Br-N1-IMP), a novel class of mononucleotide, as the sole product.
Co-reporter:Andrew M. Riley Dr.;Melanie Trusselle Dr.;Paul Kuad Dr.;Michal Borkovec Dr.;Jaiesoon Cho Dr.;Jae H. Choi Dr.;Xun Qian Dr.;Stephen B. Shears Dr.;Bernard Spiess Dr.;Barry V. L. Potter Dr.
ChemBioChem 2006 Volume 7(Issue 7) pp:
Publication Date(Web):6 JUN 2006
DOI:10.1002/cbic.200600037
myo-Inositol 1,3,4,5,6-pentakisphosphate (Ins(1,3,4,5,6)P5), an inositol polyphosphate of emerging significance in cellular signalling, and its C-2 epimer scyllo-inositol pentakisphosphate (scyllo-InsP5) were synthesised from the same myo-inositol-based precursor. Potentiometric and NMR titrations show that both pentakisphosphates undergo a conformational ring-flip at higher pH, beginning at pH 8 for scyllo-InsP5 and pH 9 for Ins(1,3,4,5,6)P5. Over the physiological pH range, however, the conformation of the inositol rings and the microprotonation patterns of the phosphate groups in Ins(1,3,4,5,6)P5 and scyllo-InsP5 are similar. Thus, scyllo-InsP5 should be a useful tool for identifying biologically relevant actions of Ins(1,3,4,5,6)P5, mediated by specific binding sites, and distinguishing them from nonspecific electrostatic effects. We also demonstrate that, although scyllo-InsP5 and Ins(1,3,4,5,6)P5 are both hydrolysed by multiple inositol polyphosphate phosphatase (MINPP), scyllo-InsP5 is not dephosphorylated by PTEN or phosphorylated by Ins(1,3,4,5,6)P5 2-kinases. This finding both reinforces the value of scyllo-InsP5 as a biological control and shows that the axial 2-OH group of Ins(1,3,4,5,6)P5 plays a part in substrate recognition by PTEN and the Ins(1,3,4,5,6)P5 2-kinases.
Co-reporter:Andrew M. Riley, H. Yasmin Godage, Mary F. Mahon, Barry V.L. Potter
Tetrahedron: Asymmetry 2006 Volume 17(Issue 2) pp:171-174
Publication Date(Web):23 January 2006
DOI:10.1016/j.tetasy.2005.12.008
Chiral desymmetrisation of myo-inositol 1,3,5-orthobenzoate via the formation of diastereoisomeric bis[(1S)-(−)-camphanate] esters provides a convenient and fast route to precursors for biologically important inositol phosphates and lipids, and to synthetic analogues and probes modified at O-1 or O-3 of the inositol ring.d-2,6-Di-O-[(−)-camphanoyl]-myo-inositol 1,3,5-orthobenzoateC33H38O12De >98%[α]D20=-19.4 (c 0.9, DMF)Source of chirality: (1S)-(−)-camphanic acidd-2,6-Di-O-benzyl-myo-inositol 1,3,5-orthobenzoateC27H26O6Ee >98%[α]D20=+3 (c 1, CHCl3)Source of chirality: the precursord-1-O-Benzoyl-2,6-di-O-benzyl-myo-inositolC27H28O7Ee >98%[α]D20=-118.9 (c 1, MeOH)Source of chirality: the precursord-3-O-Benzoyl-2,6-di-O-benzyl-myo-inositolC27H28O7Ee >98%[α]D20=+36.7 (c 1, MeOH)Source of chirality: the precursor
Co-reporter:Nigel Vicker Dr.;Harshani R. Lawrence Dr.;Gillian M. Allan Dr.;Christian Bubert Dr.;Andrew Smith Dr.;Helena J. Tutill;Atul Purohit Dr.;Joanna M. Day Dr.;Mary F. Mahon Dr.;Michael J. Reed ;Barry V. L. Potter
ChemMedChem 2006 Volume 1(Issue 4) pp:464-481
Publication Date(Web):17 FEB 2006
DOI:10.1002/cmdc.200500087
17β-Hydroxysteroid dehydrogenase type 1 (17β-HSD1), an oxidoreductase which has a preferential reductive activity using NADPH as cofactor, converts estrone to estradiol and is expressed in many steroidogenic tissues including breast and in malignant breast cells. As estradiol stimulates the growth and development of hormone-dependent breast cancer, inhibition of the final step of its synthesis is an attractive target for the treatment of this disease. The parallel synthesis of novel focused libraries of 16-substituted estrone derivatives and modified E-ring pyrazole steroids as new potent 17β-HSD1 inhibitors is described. Substituted 3-O-sulfamoylated estrone derivatives were used as templates and were immobilised on 2-chlorotrityl chloride resin to give resin-bound scaffolds with a multi-detachable linker. Novel focused libraries of 16-substituted estrone derivatives and new modified E-ring steroids were assembled from these immobilised templates using solid-phase organic synthesis and solution-phase methodologies. Among the derivatives synthesised, the most potent 17β-HSD1 inhibitors were 25 and 26 with IC50 values in T-47D human breast cancer cells of 27 and 165 nm, respectively. Parallel synthesis resulting in a library of C5′-linked amides from the pyrazole E-ring led to the identification of 62 with an IC50 value of 700 nM. These potent inhibitors of 17β-HSD1 have a 2-ethyl substituent which will decrease their estrogenic potential. Several novel 17β-HSD1 inhibitors emerged from these libraries and these provide direction for further template exploration in this area. A new efficient diastereoselective synthesis of 25 has also been developed to facilitate supply for in vivo evaluation, and an X-ray crystal structure of this inhibitor is presented.
Co-reporter:Stephen J. Mills Dr.;Hélène Dozol Dr.;Fabrice Veput Dr.;Katrien Backers Dr.;Timothy Woodman Dr.;Christophe Erneux Dr.;Bernard Spiess Dr.;Barry V. L. Potter Dr.
ChemBioChem 2006 Volume 7(Issue 11) pp:
Publication Date(Web):11 SEP 2006
DOI:10.1002/cbic.200600125
3-Hydroxybenzene 1,2,4-trisphosphate 4 is a new myo-inositol 1,4,5-trisphosphate analogue based on the core structure of benzene 1,2,4-trisphosphate 2 with an additional hydroxyl group at position-3, and is the first noninositol based compound to be a substrate for inositol 1,4,5-trisphosphate 5-phosphatase. In physicochemical studies on 2, when three equivalents of protons were added, the 31P NMR spectrum displayed monophasic behaviour in which phosphate-1 and phosphate-2 behaved independently in most of the studied pH range. For compound 4, phosphate-2 and phosphate-4 interacted with the 3-OH group, which does not titrate at physiological pH, displaying complex biphasic behaviour which demonstrated co-operativity between these groups. Phosphate-1 and phosphate-2 strongly interacted with each other and phosphate-4 experienced repulsion because of the interaction of the 3-OH group. Benzene 1,2,4-trisphosphate 2 is resistant to inositol 1,4,5-trisphosphate type I 5-phosphatase catalysed dephosphorylation. However, surprisingly, 3-hydroxybenzene 1,2,4-trisphosphate 4 was dephosphorylated by this 5-phosphatase to give the symmetrical 2,3-dihydroxybenzene 1,4-bisphosphate 16. The extra hydroxyl group is shown to form a hydrogen bond with the vicinal phosphate groups at −15 °C, and 1H NMR titration of the ring and hydroxyl protons in 4 shows the OH proton to be strongly stabilized as soon as the phosphate groups are deprotonated. The effect of the phenolic 3-OH group in compound 4 confirms a critical role for the 6-OH group of the natural messenger in the dephosphorylation mechanism that persists even in radically modified analogues.
Co-reporter:Charles N. Borissow, Steven J. Black, Michael Paul, Stephen C. Tovey, Skarlatos G. Dedos, Colin W. Taylor and Barry V. L. Potter
Organic & Biomolecular Chemistry 2005 vol. 3(Issue 2) pp:245-252
Publication Date(Web):13 Dec 2004
DOI:10.1039/B415229H
The synthesis of adenophostin A (2) and two analogues [etheno adenophostin (4) and 8-bromo adenophostin (5)] modified at the adenine moiety, is reported. A combination of NMR analysis and molecular modelling was used to compare their structures in solution and determined that they all adopt very similar conformations. The analogues were tested for their ability to mobilise Ca2+ from DT40 cells expressing recombinant Type 1 rat Ins(1,4,5)P3R which reveals etheno adenophostin as a high affinity fluorescent probe of the Ins(1,4,5)P3R. 8-Bromo adenophostin was only slightly less potent. The biological results support our current hypothesis regarding the binding mode of adenophostin A at the Ins(1,4,5)P3R, i. e. that a cation–π interaction between the base moiety and Arg 504 of the receptor in combination with H-bonding may be responsible for the high potency of adenophostin A relative to Ins(1,4,5)P3.
Co-reporter:Xiangdong Su, Harshani Lawrence, Dharshini Ganeshapillai, Adrian Cruttenden, Atul Purohit, Michael J Reed, Nigel Vicker, Barry V.L Potter
Bioorganic & Medicinal Chemistry 2004 Volume 12(Issue 16) pp:4439-4457
Publication Date(Web):15 August 2004
DOI:10.1016/j.bmc.2004.06.008
Extensive structural modifications to the 18β-glycyrrhetinic acid template are described and their effects on the SAR of the 11β-hydroxysteroid dehydrogenase isozymes type 1 and 2 from the rat are investigated. Isoform selective inhibitors have been discovered and compound 7N-(2-hydroxyethyl)-3β-hydroxy-11-oxo-18β-olean-12-en-30-oic acid amide is highlighted as a very potent selective inhibitor of 11β-hydroxysteroid dehydrogenase 2 with an IC50 = 4 pM.The discovery of novel potent selective inhibitors of 11β-HSD2 is described.
Co-reporter:Christopher J.W Mort, Marie E Migaud, Antony Galione, Barry V.L Potter
Bioorganic & Medicinal Chemistry 2004 Volume 12(Issue 2) pp:475-487
Publication Date(Web):15 January 2004
DOI:10.1016/j.bmc.2003.10.012
Cyclic ADP-ribose mobilizes intracellular Ca2+ in a variety of cells. To elucidate the nature of the interaction between the C3′ substituent of cADP-ribose and the cADPR receptor, three analogues of NAD+ modified in the adenosine ribase (xyloNAD+ 3′F-xyloNAD+ and 3′F-NAD+ were chemically synthesised from d-xylose and adenine starting materials. 3′F-NAD+ was readily converted to cyclic 3′F-ADP ribose by the action of the cyclase enzyme derived from the mollusc Aplysia californica. XyloNAD+ and 3′F-xyloNAD+ were cyclised only reluctantly and in poor yield to afford unstable cyclic products. Biological evaluation of cyclic 3′F-ADP ribose for calcium release in sea urchin egg homogenate gave an EC50 of 1.5±0.5 μM. This high value suggests that the ability of the C3′ substituent to donate a hydrogen bond is crucial for agonism.Graphic
Co-reporter:Nigel Vicker, Xiangdong Su, Harshani Lawrence, Adrian Cruttenden, Atul Purohit, Michael J. Reed, Barry V.L. Potter
Bioorganic & Medicinal Chemistry Letters 2004 Volume 14(Issue 12) pp:3263-3267
Publication Date(Web):21 June 2004
DOI:10.1016/j.bmcl.2004.03.107
Using 18β-glycyrrhetinic acid as a template, the synthesis of a series of secondary amides at the 30-position is described and the effects of these modifications on the SAR of the 11β-hydroxysteroid dehydrogenase isozymes type 1 and 2 from the rat are investigated. An isoform selective inhibitor has been discovered and compound 5, N-(2-hydroxyethyl)-3β-hydroxy-11-oxo-18β-olean-12-en-30-oic acid amide, is highlighted as a very potent and selective inhibitor of 11β-hydroxysteroid dehydrogenase 2 with an IC50 = 4 pM.The discovery of a novel potent selective inhibitor of 11β-HSD2 from a series of amides using 18β-glycyrrhetinic acid as a template is described.
Co-reporter:Mathew P. Leese, Simon P. Newman, Atul Purohit, Michael J. Reed, Barry V.L. Potter
Bioorganic & Medicinal Chemistry Letters 2004 Volume 14(Issue 12) pp:3135-3138
Publication Date(Web):21 June 2004
DOI:10.1016/j.bmcl.2004.04.027
A flexible, direct, high yielding synthesis of 2-alkylsulfanyl estrogens from estrone has been developed. 2-Methylsulfanyl estradiol (2-MeSE2) 7 displays a similar anti-proliferative activity to the established 2-methoxyestradiol (2-MeOE2) 1, whilst its 3-O-sulfamate derivative (2-MeSE2MATE) 9 exhibits greatly enhanced anti-proliferative activity, combined with significant inhibition of steroid sulfatase, an enzyme target for the treatment of hormone-dependent tumours.A flexible, direct, high yielding synthesis of 2-alkylsulfanyl estrogens has been developed. 2-Methylsulfanyl estradiol (2-MeSE2) and its 3-O-sulfamate derivative display a similar anti-proliferative activity to 2-methoxyestradiol (2-MeOE2).
Co-reporter:Nigel Vicker, Yaikat Ho, James Robinson, Lawrence L.W. Woo, Atul Purohit, Michael J. Reed, Barry V.L. Potter
Bioorganic & Medicinal Chemistry Letters 2003 Volume 13(Issue 5) pp:863-865
Publication Date(Web):10 March 2003
DOI:10.1016/S0960-894X(03)00009-X
We describe the docking of selected steroidal and non-steroidal estrone sulphatase inhibitors, including the Phase I clinical trial candidate 667COUMATE (6), into the active site of human carbonic anhydrase II (hCA II). The docking scores are compared with the inhibition of hCA II and show good correlation with biological activity.The docking of selected steroidal and non-steroidal estrone sulphatase inhibitors, including the Phase I clinical trial candidate 667COUMATE (6), into the active site of human carbonic anhydrase II (hCA II).
Co-reporter:Stephen J. Mills;Andrew M. Riley;Changsheng Liu;Mary F. Mahon;Barry V. L. Potter
Chemistry - A European Journal 2003 Volume 9(Issue 24) pp:
Publication Date(Web):11 DEC 2003
DOI:10.1002/chem.200305207
New and rapid syntheses of the enantiomeric intracellular signalling molecules d-myo-inositol 1,4,5,6-tetrakisphosphate (1 a) and D-myo-inositol 3,4,5,6-tetrakisphosphate (1 b) are described. The synthetic strategy employs the novel butane-2,3-diacetal-protected (BDA-protected) myo-inositol (±)-3 ab, directly accessible from myo-inositol on a large scale, and an optical resolution with diastereoisomeric (R)-(−)-acetylmandelate esters. The X-ray crystal structure of (±)-4, an unusual side product of acid-catalysed reaction of myo-inositol with butanedione is also presented, and the absolute configurations of 1 a and 1 b are definitively assigned by conversion of key precursors into (+)-bornesitol and L-iditol hexaacetate, respectively. Biological activity of synthetic 1 b was confirmed in comparison with the natural polyphosphate.
Co-reporter:Heidi J. Rosenberg, Andrew M. Riley, Rachel D. Marwood, Vanessa Correa, Colin W. Taylor, Barry V.L. Potter
Carbohydrate Research 2001 Volume 332(Issue 1) pp:53-66
Publication Date(Web):8 May 2001
DOI:10.1016/S0008-6215(01)00067-2
The synthesis of a series of tetrahydrofuranyl α- and β-xylopyranoside trisphosphates, designed by excision of three motifs of adenophostin A is reported. The synthetic route features improved preparations of allyl α-d-xylopyranoside and its 2-O-benzyl ether, and gives access to four diastereoisomeric trisphosphates, which show a range of abilities to mobilise Ca2+ from the intracellular stores of hepatocytes. A comparison of the potencies of the four trisphosphates provides useful information relating to the effects of stereochemical variation on the recognition of carbohydrate-based trisphosphates by d-myo-inositol 1,4,5-trisphosphate receptors. 1-O-[(3′S,4′R)-3-hydroxytetrahydrofuran-4-yl] α-d-xylopyranoside 3,4,3′-trisphosphate (8) is the most active member of the series with a potency close to Ins(1,4,5)P3; a β-linked analogue, 1-O-[(3′R,4′S)-3-hydroxytetrahydrofuran-4-yl] β-d-xylopyranoside 3,4,3′-trisphosphate, is ca. 20-fold weaker than Ins(1,4,5)P3, and the other compounds are much less active. While no compound attained a potency close to that of adenophostin A, we believe that 8 represents the minimal structure for potent Ca2+-releasing activity in this type of carbohydrate-based analogue.Graphic
Co-reporter:Graeme Horne;Barry V. L. Potter
Chemistry - A European Journal 2001 Volume 7(Issue 1) pp:
Publication Date(Web):5 JAN 2001
DOI:10.1002/1521-3765(20010105)7:1<80::AID-CHEM80>3.0.CO;2-B
d-myo-Inositol 1,3,4,5-tetrakisphosphate [Ins(1,3,4,5)P4] is produced rapidly from the established second messenger d-myo-inositol 1,4,5-trisphosphate [Ins(1,4,5)P4] in stimulated cells. Despite extensive investigations, in particular concerning its potential role in mediating cellular Ca2+ influx, no exact cellular function has been described for this inositol phosphate; however, binding sites have been identified in a number of tissues and it has been shown to act synergistically with Ins(1,4,5)P3. To assist in the elucidation of the mechanism of action and structural requirements within the Ins(1,3,4,5)P4 moiety that are necessary for recognition and activation of the receptor, structural analogues of this tetrakisphosphate are required. Routes for the synthesis of racemic 6-deoxy-myo-inositol 1,3,4,5-tetrakisphosphate [6-deoxy-dl-Ins(1,3,4,5)P4] and the chiral antipodes D- and l-6-deoxy-myo-inositol 1,3,4,5-tetrakisphosphate are described here. The racemic tetrakisphosphate was synthesised from dl-1,2-O-isopropylidene-myo-inositol in eight steps. Deoxygenation at C-6 was achieved following the Barton–McCombie procedure. Both chiral tetrakisphosphates were synthesised through resolution of racemic cis-diol 6-deoxy-1,4,5-tri-O-p-methoxybenzyl-myo-inositol with the chiral auxiliary (S)-(+)-O-acetylmandelic acid. Absolute configuration was confirmed by synthesis of the known D-6-deoxy-myo-inositol. Both D-6-deoxy-Ins(1,3,4,5)P4 and its enantiomer will be useful tools to unravel the enigmatic role of Ins(1,3,4,5)P4 in the polyphosphoinositide pathway of signal transduction.
Co-reporter:Satoshi Shuto ;Graeme Horne;Rachel D. Marwood;Barry V. L. Potter
Chemistry - A European Journal 2001 Volume 7(Issue 22) pp:
Publication Date(Web):12 NOV 2001
DOI:10.1002/1521-3765(20011119)7:22<4937::AID-CHEM4937>3.0.CO;2-G
The adenophostins exhibit approximately 10–100 times higher receptor binding and Ca2+ mobilising potencies in comparison with the natural second messenger D-myo-inositol 1,4,5-trisphosphate [Ins(1,4,5)P3]. Despite many synthetic attempts to determine the minimal structural requirement for this unusual behaviour of the adenophostins, few related simplified analogues displaying higher activity than that of Ins(1,4,5)P3 have been reported. However, biological evaluation of such analogues has revealed that one of the key factors for the enhanced biological activity is the adenine moiety. To further understand the effect that the adenine base has upon the activity of the adenophostins, congeners in which this functionality is replaced by uracil, benzimidazole, 2-methoxynaphthalene, 4-methylanisole and 4-methylnaphthalene using the common intermediate 1,2-di-O-acetyl-5-O-benzyl-3-O-(3,4-di-O-acetyl-2,6-di-O-benzyl-α-d-glucopyranosyl)-ribofuranose have been synthesised using a base replacement strategy. The synthesis of the uracil and benzimidazole analogues was achieved using the Vorbrüggen condensation procedure. The 1′-C-glycosidic analogues were prepared using Friedel–Crafts type C-aryl glycosidation reactions. Phosphate groups were introduced using the phosphoramidite method with subsequent removal of all-benzyl protecting groups by catalytic hydrogenation or catalytic hydrogen transfer. Apart from one analogue with an α-glycosidic linkage all compounds were more potent than Ins(1,4,5)P3 and most tended more towards adenophostin in activity. These analogues will be valuable tools to unravel the role that the adenine moiety plays in the potent activity of the adenophostins and demonstrate that this strategy is effective at producing highly potent ligands.
Co-reporter:Graeme Horne;Barry V. L. Potter
Chemistry - A European Journal 2001 Volume 7(Issue 5) pp:
Publication Date(Web):23 FEB 2001
DOI:10.1002/1521-3765(20010302)7:5<942::AID-CHEM942>3.0.CO;2-F
Co-reporter:Rachel D. Marwood, Satoshi Shuto, David J. Jenkins and Barry V. L. Potter
Chemical Communications 2000 (Issue 3) pp:219-220
Publication Date(Web):31 Jan 2000
DOI:10.1039/A909347H
The first totally synthetic base-modified analogues of the
natural product and potent D-myo-inositol
1,4,5-trisphosphate receptor agonist adenophostin A were efficiently
synthesised from D-xylose and D-glucose using
methodology employing base and surrogate base addition to a common
disaccharide intermediate.
Co-reporter:Andrew M. Riley and Barry V. L. Potter
Chemical Communications 2000 (Issue 11) pp:983-984
Publication Date(Web):17 May 2000
DOI:10.1039/B002312O
The first poly(ethylene glycol)-linked dimers of
D-myo-inositol 1,4,5-trisphosphate have been
synthesised as probes for multi-subunit binding proteins of this ubiquitous
second messenger.
Co-reporter:Heidi J. Rosenberg, Andrew M. Riley, Vanessa Correa, Colin W. Taylor, Barry V.L. Potter
Carbohydrate Research 2000 Volume 329(Issue 1) pp:7-16
Publication Date(Web):20 October 2000
DOI:10.1016/S0008-6215(00)00175-0
Epimeric C-glycoside based polyphosphates, α- and β-d-glucopyranosylmethanol 3,4,1′-trisphosphates (8 and 9) were prepared from d-glucose. The key intermediate, allyl 2,6-di-O-benzyl-α-d-glucopyranoside, was prepared in five steps (67% yield) from allyl α-d-glucopyranoside without the need for chromatography. Compounds 8 and 9 were shown to be full agonists at the Ins(1,4,5)P3 receptors of permeabilised hepatocytes, but with markedly different potencies. Such C-glycoside analogues are worthy of further development as Ins(1,4,5)P3 receptor ligands.
Co-reporter:Rachel D. Marwood, Andrew M. Riley, Vanessa Correa, Colin W. Taylor, Barry V.L. Potter
Bioorganic & Medicinal Chemistry Letters 1999 Volume 9(Issue 3) pp:453-458
Publication Date(Web):8 February 1999
DOI:10.1016/S0960-894X(99)00006-2
The synthesis of 3,4,3′-trisphosphate (7), a novel Ca2+ mobilising agonist at the Ins(1,4,5)P3 receptor, designed by excision of two motifs of adenophostin A is reported, defining a potential minimal structure for potent glucopyranoside-based agonists of Ins(1,4,5)P3 receptors.The synthesis of a novel carbohydrate-based Ca2+ mobilising agonist at the Ins(1,4,5)P3 receptor is described.
Co-reporter: Dr. Barry V. L. Potter;Dr. Dethard Lampe
Angewandte Chemie 1995 Volume 107(Issue 18) pp:
Publication Date(Web):21 JAN 2006
DOI:10.1002/ange.19951071804
Vor etwas mehr als einem Jahrzehnt berichteten Michael Berridge und seine Mitarbeiter in Nature: micromolar concentrations of Ins(1,4,5)P3 (1D-myo-inositol 1,4,5-trisphosphate) release Ca2+ from a non-mitochondrial intra-cellular Ca2+ store in pancreatic acinar cells. Our results strongly suggest that this is the same Ca2+ store that is released by acetylcholine”. Mit der Entdeckung eines niedermolekularen sekundären Botenstoffs, der die räumlich getrennten Ereignisse der Aktivierung von Rezeptoren an der Zelloberfläche und der intrazellulären Ca2+ -Mobilisierung verbindet, wurde eine neue Ära auf dem Gebiet der Signalvermittlung eingeleitet und eine Renaissance der Inosit-und Inositphosphatchemie stimuliert. Die Synthese von Inositpolyphosphaten bringt mehrere Probleme mit sich: die regiospezifische Einführung von Schutzgruppen am Inositring, die Racematspaltung der resultierenden Zwischenprodukte, die Phosphorylierung des Polyols, die Entfernung aller Phosphat-Schutzgruppen unter Vermeidung einer gleichzeitigen Wanderung von Phosphatgruppen sowie die Reinigung des wasserlöslichen Ziel-Polyanions. Mit der Lösung dieser Probleme in den letzten Jahren ist es jetzt möglich, über die Synthese von natürlichen Inositphosphaten hinaus (von denen ständig mehr gefunden werden) zur Entwicklung von chemisch modifizierten Inositphosphat-Analoga überzugehen, mit der Aussicht, Enzyminhibitoren, zweckmäßig modifizierte Rezeptorliganden und -antagonisten sowie vielleicht sogar Therapeutica für den pharmakologischen Eingriff in Signalübertragungsbahnen zu entwickeln.
Co-reporter:Mark P. Thomas, Barry V.L. Potter
The Journal of Steroid Biochemistry and Molecular Biology (September 2013) Volume 137() pp:27-49
Publication Date(Web):1 September 2013
DOI:10.1016/j.jsbmb.2012.12.014
Many enzymes catalyse reactions that have an oestrogen as a substrate and/or a product. The reactions catalysed include aromatisation, oxidation, reduction, sulfonation, desulfonation, hydroxylation and methoxylation. The enzymes that catalyse these reactions must all recognise and bind oestrogen but, despite this, they have diverse structures. This review looks at each of these enzymes in turn, describing the structure and discussing the mechanism of the catalysed reaction. Since oestrogen has a role in many disease states inhibition of the enzymes of oestrogen metabolism may have an impact on the state or progression of the disease and inhibitors of these enzymes are briefly discussed.This article is part of a Special Issue entitled ‘CSR 2013’.Highlights► The structures of oestrogen-metabolising enzymes are discussed and compared. ► Substrate recognition and reaction mechanisms of these enzymes are discussed. ► The impact of structure on inhibitor design is briefly mentioned.
Co-reporter:Nigel Vicker, Xiangdong Su, Dharshini Ganeshapillai, Andrew Smith, Atul Purohit, Michael J. Reed, Barry V.L. Potter
The Journal of Steroid Biochemistry and Molecular Biology (May 2007) Volume 104(Issues 3–5) pp:123-129
Publication Date(Web):1 May 2007
DOI:10.1016/j.jsbmb.2007.03.023
11β-Hydroxysteroid dehydrogenase type 1 (11β-HSD1) regulates glucocorticoid action at the pre-receptor stage by converting cortisone to cortisol. 11β-HSD1 is selectively expressed in many tissues including the liver and adipose tissue where metabolic events are important. Metabolic syndrome relates to a number of metabolic abnormalities and currently has a prevalence of >20% in adult Americans. 11β-HSD1 inhibitors are being investigated by many major pharmaceutical companies for type 2 diabetes and other abnormalities associated with metabolic syndrome. In this area of intense interest a number of structural types of 11β-HSD1 inhibitor have been identified. It is important to have an array of structural types as the physicochemical properties of the compounds will determine tissue distribution, HPA effects, and ultimately clinical utility. Here we report the discovery and synthesis of three structurally different series of novel 11β-HSD1 inhibitors that inhibit human 11β-HSD1 in the low micromolar range. Docking studies with 1–3 into the crystal structure of human 11β-HSD1 reveal how the molecules may interact with the enzyme and cofactor and give further scope for structure based drug design in the optimisation of these series.
Co-reporter:Xiangdong Su, Nigel Vicker, Harshani Lawrence, Andrew Smith, Atul Purohit, Michael J. Reed, Barry V.L. Potter
The Journal of Steroid Biochemistry and Molecular Biology (May 2007) Volume 104(Issues 3–5) pp:312-320
Publication Date(Web):1 May 2007
DOI:10.1016/j.jsbmb.2007.03.019
11β-Hydroxysteroid dehydrogenase type 1 (11β-HSD1) plays an important role in regulating the cortisol availability to bind to corticosteroid receptors within specific tissue. Recent advances in understanding the molecular mechanisms of metabolic syndrome indicate that elevation of cortisol levels within specific tissues through the action of 11β-HSD1 could contribute to the pathogenesis of this disease. Therefore, selective inhibitors of 11β-HSD1 have been investigated as potential treatments for metabolic diseases, such as diabetes mellitus type 2 or obesity. Here we report the discovery and synthesis of some 18β-glycyrrhetinic acid (18β-GA) derivatives (2–5) and their inhibitory activities against rat hepatic11β-HSD1 and rat renal 11β-HSD2. Once the selectivity over the rat type 2 enzyme was established, these compounds’ ability to inhibit human 11β-HSD1 was also evaluated using both radioimmunoassay (RIA) and homogeneous time resolved fluorescence (HTRF) methods. The 11-modified 18β-GA derivatives 2 and 3 with apparent selectivity for rat 11β-HSD1 showed a high percentage inhibition for human microsomal 11β-HSD1 at 10 μM and exhibited IC50 values of 400 and 1100 nM, respectively. The side chain modified 18β-GA derivatives 4 and 5, although showing selectivity for rat 11β-HSD1 inhibited human microsomal 11β-HSD1 with IC50 values in the low micromolar range.
Co-reporter:Christelle Moreau, Gloria A. Ashamu, Victoria C. Bailey, Antony Galione, Andreas H. Guse and Barry V. L. Potter
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 1) pp:NaN290-290
Publication Date(Web):2010/10/25
DOI:10.1039/C0OB00396D
Novel 8-substituted base and sugar-modified analogues of the Ca2+ mobilizing second messenger cyclic adenosine 5′-diphosphate ribose (cADPR) were synthesized using a chemoenzymatic approach and evaluated for activity in sea urchin egg homogenate (SUH) and in Jurkat T-lymphocytes; conformational analysis investigated by 1H NMR spectroscopy revealed that a C2′ endo/syn conformation of the “southern” ribose is crucial for agonist or antagonist activity at the SUH-, but not at the T cell-cADPR receptor.
Co-reporter:Toby Jackson, L. W. Lawrence Woo, Melanie N. Trusselle, Surinder K. Chander, Atul Purohit, Michael J. Reed and Barry V. L. Potter
Organic & Biomolecular Chemistry 2007 - vol. 5(Issue 18) pp:NaN2952-2952
Publication Date(Web):2007/09/12
DOI:10.1039/B707768H
The synthesis and biological evaluation of a series of novel Dual Aromatase–Sulfatase Inhibitors (DASIs) are described. It is postulated that dual inhibition of the aromatase and steroid sulfatase enzymes, both responsible for the biosynthesis of oestrogens, will be beneficial in the treatment of hormone-dependent breast cancer. The compounds are based upon the Anastrozolearomatase inhibitor template which, while maintaining the haem ligating triazole moiety crucial for enzyme inhibition, was modified to include a phenol sulfamate ester motif, the pharmacophore for potent irreversible steroid sulfatase inhibition. Adaption of a synthetic route to Anastrozole was accomplished via selective radical bromination and substitution reactions to furnish a series of aromatase inhibitory pharmacophores. Linking these fragments to the phenol sulfamate ester moiety employed SN2, Heck and Mitsunobu reactions with phenolic precursors, from where the completed DASIs were achieved via sulfamoylation. In vitro, the lead compound, 11, had a high degree of potency against aromatase (IC50 3.5 nM), comparable with that of Anastrozole (IC50 1.5 nM) whereas, only moderate activity against steroid sulfatase was found. However, in vivo, 11 surprisingly exhibited potent dual inhibition. Compound 11 was modelled into the active site of a homology model of human aromatase and the X-ray crystal structure of steroid sulfatase.
Co-reporter:Kana M. Sureshan, Andrew M. Riley, Ana M. Rossi, Stephen C. Tovey, Skarlatos G. Dedos, Colin W. Taylor and Barry V. L. Potter
Chemical Communications 2009(Issue 10) pp:NaN1206-1206
Publication Date(Web):2009/02/04
DOI:10.1039/B819328B
Ca2+ release by D-myo-inositol 1,4,5-trisphosphate receptors (IP3Rs) is widely considered to require the vicinal 4,5-bisphosphate motif of IP3, with P-5 and P-4 engaging the α and β domains of the binding site; using synthesis and mutagenesis we show that the adenine of synthetic glyconucleotides, through an interaction with Arg504, can replace the interaction of either P-1 or P-5 with the α-domain producing, respectively, the most potent bisphosphate agonist yet synthesised and the first agonist of IP3R without a vicinal bisphosphate motif; this will stimulate new approaches to IP3R ligand design.
Co-reporter:Mathew P. Leese, Fabrice Jourdan, Meriel R. Kimberley, Gyles E. Cozier, Nethaji Thiyagarajan, Chloe Stengel, Sandra Regis-Lydi, Paul A. Foster, Simon P. Newman, K. Ravi Acharya, Eric Ferrandis, Atul Purohit, Michael J. Reed and Barry V. L. Potter
Chemical Communications 2010 - vol. 46(Issue 17) pp:NaN2909-2909
Publication Date(Web):2010/03/20
DOI:10.1039/C002558E
A chimeric approach is used to discover microtubule disruptors with excellent in vitro activity and oral bioavailability; a ligand–protein interaction with carbonic anhydrase that enhances bioavailability is characterised by protein X-ray crystallography. Dosing of a representative chimera in a tumour xenograft model confirms the excellent therapeutic potential of the class.
Co-reporter:Fabrice Jourdan, Christian Bubert, Mathew P. Leese, Andrew Smith, Eric Ferrandis, Sandra Regis-Lydi, Simon P. Newman, Atul Purohit, Michael J. Reed and Barry V. L. Potter
Organic & Biomolecular Chemistry 2008 - vol. 6(Issue 22) pp:NaN4119-4119
Publication Date(Web):2008/09/22
DOI:10.1039/B810300C
The potent activity of 2-substituted estra-1,3,5(10)-triene-3-O-sulfamates against the proliferation of cancer cellsin vitro and tumours in vivo highlights the therapeutic potential of such compounds. Optimal activity is derived from a combination of a 2-XMe group (where X = CH2, O or S), a 3-O-sulfamate group in the steroidal A-ring and a H-bond acceptor around C-17 of the D-ring. Herein, we describe the synthesis and anti-proliferative activities of a series of novel 2-substituted estra-1,3,5(10)-triene-3-O-sulfamates bearing heterocyclic substituents (oxazole, tetrazole, triazole) tethered to C-17. In vitro evaluation of these molecules revealed that high anti-proliferative activity in breast and prostate cancer cells lines (GI50 of 340–850 nM) could be retained when the heterocyclic substituent possesses H-bond acceptor properties. A good correlation between the calculated electron density of the heterocyclic ring and anti-proliferative activity was observed. Docking of the most active compounds into their putative site of action, the colchicine binding site of tubulin, suggests that they bind through a different mode to the previously described bis-sulfamate derivatives 1 and 2, which possess similar in vitro activity.
Co-reporter:Andrew M. Riley, Huanchen Wang, Jeremy D. Weaver, Stephen B. Shears and Barry V. L. Potter
Chemical Communications 2012 - vol. 48(Issue 92) pp:NaN11294-11294
Publication Date(Web):2012/09/18
DOI:10.1039/C2CC36044F
We synthesised analogues of diphosphoinositol polyphosphates (PP-InsPs) in which the diphosphate is replaced by an α-phosphonoacetic acid (PA) ester. Structural analysis revealed that 5-PA-InsP5 mimics 5-PP-InsP5 binding to the kinase domain of PPIP5K2; both molecules were phosphorylated by the enzyme. PA-InsPs are promising candidates for further studies into the biology of PP-InsPs.

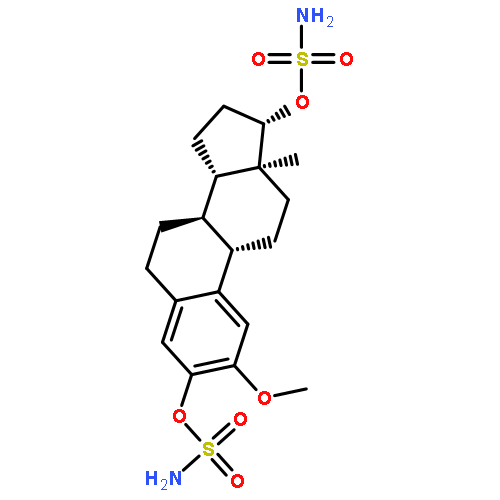
![Sulfamic acid,6,7,8,9,10,11-hexahydro-6-oxobenzo[b]cyclohepta[d]pyran-3-yl ester](http://img.cochemist.com/ccimg/288700/288628-05-7.png)
![Sulfamic acid,6,7,8,9,10,11-hexahydro-6-oxobenzo[b]cyclohepta[d]pyran-3-yl ester](http://img.cochemist.com/ccimg/288700/288628-05-7_b.png)
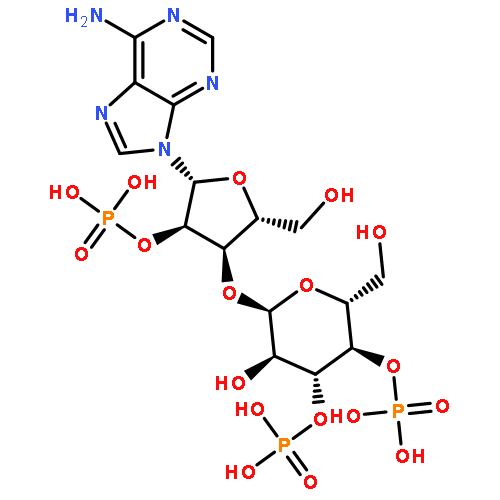
![6-Isoquinolinol, 1,2,3,4-tetrahydro-2-[(3-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/94100/94085-74-2.png)
![6-Isoquinolinol, 1,2,3,4-tetrahydro-2-[(3-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/94100/94085-74-2_b.png)






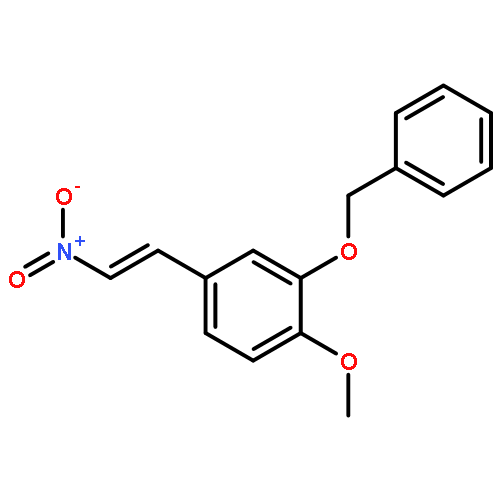
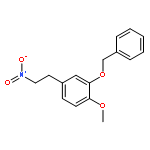
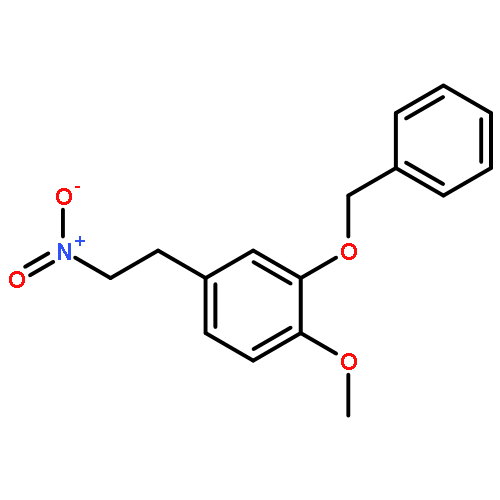


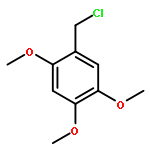
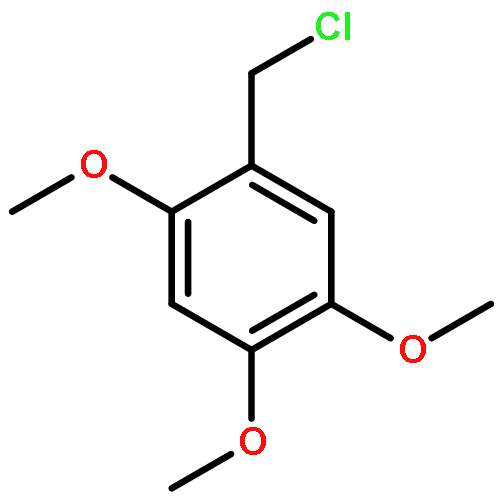
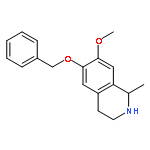
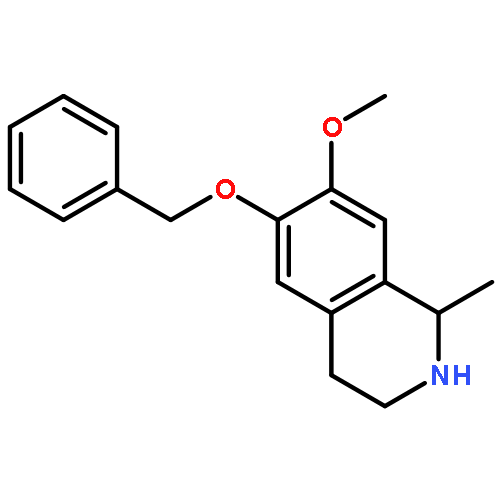


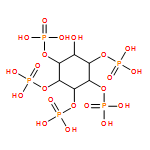
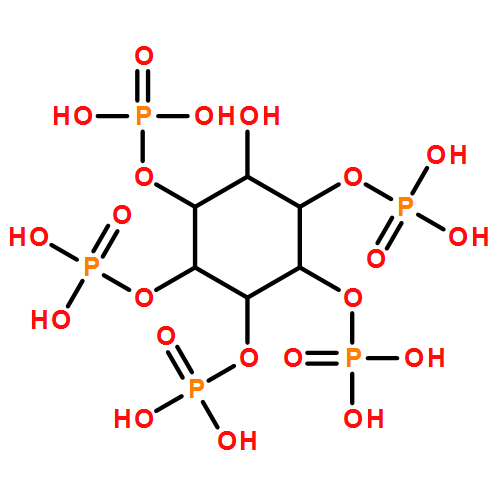
![[5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl [hydroxy-[(3,4,5-trihydroxyoxolan-2-yl)methoxy]phosphoryl] Hydrogen Phosphate](http://img.cochemist.com/ccimg/20800/20762-30-5.png)
![[5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl [hydroxy-[(3,4,5-trihydroxyoxolan-2-yl)methoxy]phosphoryl] Hydrogen Phosphate](http://img.cochemist.com/ccimg/20800/20762-30-5_b.png)


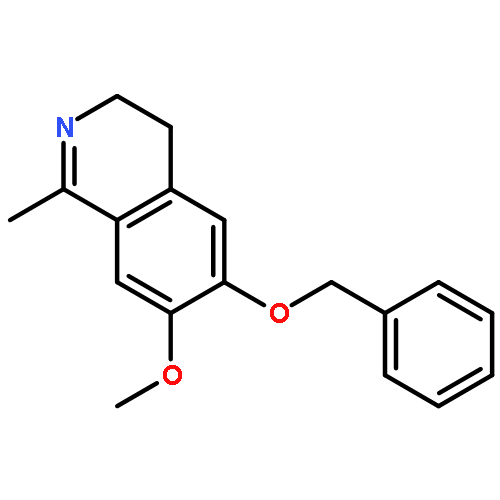
![[1,1'-Biphenyl]-3,3',4,4',5,5'-hexol](http://img.cochemist.com/ccimg/4400/4371-21-5.png)
![[1,1'-Biphenyl]-3,3',4,4',5,5'-hexol](http://img.cochemist.com/ccimg/4400/4371-21-5_b.png)
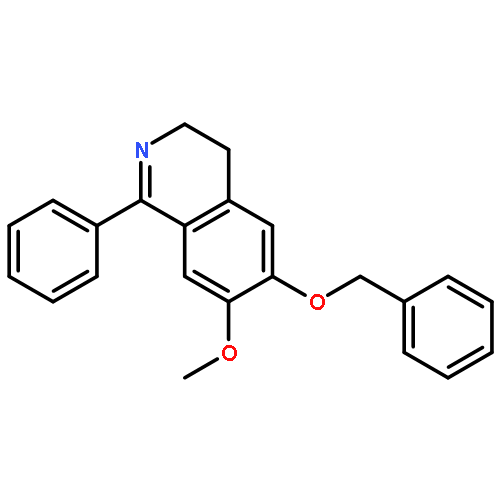
![Benzamide, N-[2-[4-methoxy-3-(phenylmethoxy)phenyl]ethyl]-](http://img.cochemist.com/ccimg/4100/4008-65-5.png)
![Benzamide, N-[2-[4-methoxy-3-(phenylmethoxy)phenyl]ethyl]-](http://img.cochemist.com/ccimg/4100/4008-65-5_b.png)

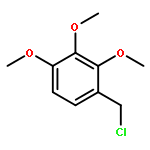
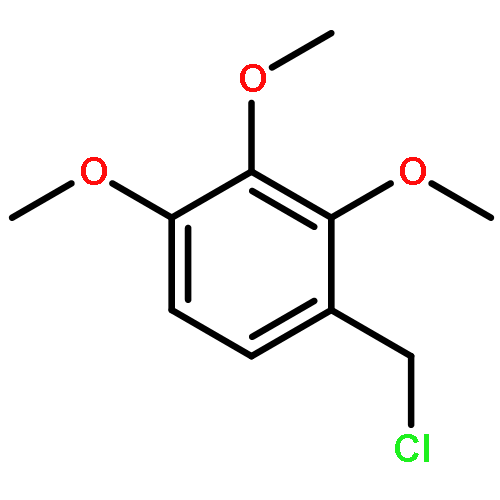
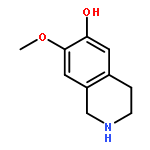
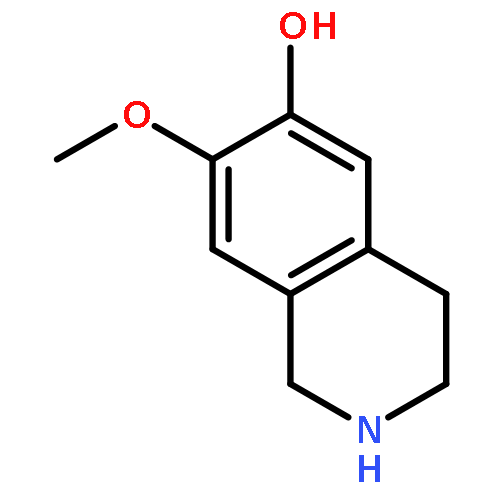
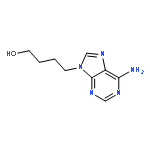




![Acetamide, N-[2-[4-methoxy-3-(phenylmethoxy)phenyl]ethyl]-](http://img.cochemist.com/ccimg/55200/55161-43-8.png)
![Acetamide, N-[2-[4-methoxy-3-(phenylmethoxy)phenyl]ethyl]-](http://img.cochemist.com/ccimg/55200/55161-43-8_b.png)


![Estra-1,3,5(10)-trien-17-one,3-[(aminosulfonyl)oxy]-](http://img.cochemist.com/ccimg/148700/148672-09-7.png)
![Estra-1,3,5(10)-trien-17-one,3-[(aminosulfonyl)oxy]-](http://img.cochemist.com/ccimg/148700/148672-09-7_b.png)