Co-reporter:Melissa Reckenthäler, Jörg-M. Neudörfl, Elif Zorlu, and Axel G. Griesbeck
The Journal of Organic Chemistry 2016 Volume 81(Issue 16) pp:7211-7216
Publication Date(Web):July 5, 2016
DOI:10.1021/acs.joc.6b01085
The photochemically induced coupling of aromatic ketones with cyclic ethers such as tetrahydrofuran, tetrahydropyran, and 1,4-dioxane was studied. Direct photolysis of the substrates with UV-A light centered at 350 nm does not lead to photoinduced hydrogen transfer whereas the addition of a mixture of the Lewis acid catalysts Ti(OiPr)4 and BF3 enables the formation of the hydroxyalkylation products.
Co-reporter:Axel G. Griesbeck;Andreas Maaßen;Maria Bräutigam;Markus Pietsch
European Journal of Organic Chemistry 2015 Volume 2015( Issue 20) pp:4349-4352
Publication Date(Web):
DOI:10.1002/ejoc.201500326
Abstract
The 1,2-hydroxy hydroperoxide 7 was obtained with high threo diastereoselectivity from the ester-conjugated allylic alcohol 6 through a porphyrin-sensitized singlet oxygen ene reaction. Compound 7 was used as substrate for peroxy(trans)acetalization with aldehydes, orthoesters, and acetals, catalyzed by boron and indium Lewis acids, as well as by PPTS as a Brønsted acid catalyst (for trioxane 10). The 1,2,4-trioxane 8 was formed with high diasteroselectivity with all-equatorially positioned substitutents at C-3, C-5, and C-6. Additionally, perorthoester 9 was obtained by indium(III)-catalyzed condensation of an orthoester with the hydroperoxide 7. The new compounds 8 and 9 exhibited moderate inhibition of human placental glutathione transferase (GST), comparable to that shown by the 5-unsubstituted model trioxane 11.
Co-reporter:Axel G. Griesbeck;Andreas Maaßen;Maria Bräutigam;Markus Pietsch
European Journal of Organic Chemistry 2015 Volume 2015( Issue 20) pp:
Publication Date(Web):
DOI:10.1002/ejoc.201590054
Co-reporter:Axel G. Griesbeck;Alan deKiff ;Margarethe Kleczka
Advanced Synthesis & Catalysis 2014 Volume 356( Issue 13) pp:2839-2845
Publication Date(Web):
DOI:10.1002/adsc.201400565
Co-reporter:Angelika Eske, Bernd Goldfuss, Axel G. Griesbeck, Alan de Kiff, Margarethe Kleczka, Matthias Leven, Jörg-M. Neudörfl, and Moritz Vollmer
The Journal of Organic Chemistry 2014 Volume 79(Issue 4) pp:1818-1829
Publication Date(Web):January 29, 2014
DOI:10.1021/jo5000434
The singlet oxygen reactivities and regioselectivities of the model compounds 1b–d were compared with those of the geminal (gem) selectivity model ethyl tiglate (1a). The kinetic cis effect is kE/kZ = 5.2 for the tiglate/angelate system 1a/1a′ without a change in the high gem regioselectivity. Further conjugation to vinyl groups enabled mode-selective processes, namely, [4 + 2] cycloadditions versus ene reactions. The site-specific effects of methylation on the mode selectivity and the regioselectivity of the ene reaction were studied for dienes 1e–g. A vinylogous gem effect was observed for the γ,δ-dimethylated and α,γ,δ-trimethylated substrates 1h and 1i, respectively. The corresponding phenylated substrates 1j–l showed similar mode selectivity, as monomethylated 1j exhibited exclusively [4 + 2] reactivity while the tandem products 12 and 14 were isolated from the di- and trimethylated substrates 1k and 1l, respectively. The vinylogous gem effect favors the formation of 1,3-dienes from the substrates, and thus, secondary singlet oxygen addition was observed to give hydroperoxy-1,2-dioxenes 19 and 20 in an ene–diene transmissive cycloaddition sequence. These products were reduced to give alcohols (16, 17, and 18) or furans (24 and 25), respectively, or treated with titanium(IV) alkoxides to give the epoxy alcohols 26 and 27. The vinylogous gem effect is rationalized by DFT calculations showing that biradicals are the low-energy intermediates and that no reaction path bifurcations compete.
Co-reporter:Axel G. Griesbeck and Alan de Kiff
Organic Letters 2013 Volume 15(Issue 9) pp:2073-2075
Publication Date(Web):April 24, 2013
DOI:10.1021/ol4009602
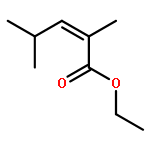
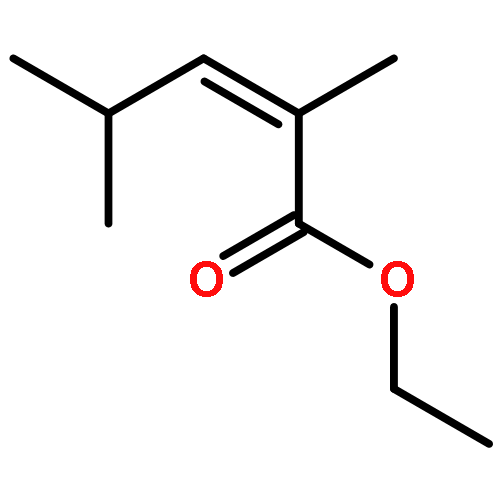
The singlet oxygen reactions of 4-methyl-2,4-hexadienoates E,E- and E,Z-4 proceed in a highly mode selective and regioselective domino process. The initial product is the allylic hydroperoxide 5 directed by a vinylogous gem effect. The subsequent 1O2 [4 + 2] cycloaddition delivers a 3:2 diastereoisomeric mixture of 1,2-dioxanes 8 in a one-pot process. The identical protocol delivers from the more reactive α-methylated substrates E,E-10 and E,Z-10 with excellent primary regioselectivity the 1,2-dioxane 13.
Co-reporter:Melissa Reckenthäler
Advanced Synthesis & Catalysis 2013 Volume 355( Issue 14-15) pp:2727-2744
Publication Date(Web):
DOI:10.1002/adsc.201300751
Co-reporter:Axel G. Griesbeck, Viktor Schlundt and Jörg M. Neudörfl
RSC Advances 2013 vol. 3(Issue 20) pp:7265-7270
Publication Date(Web):21 Mar 2013
DOI:10.1039/C3RA40555A
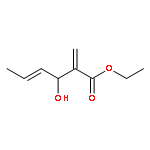
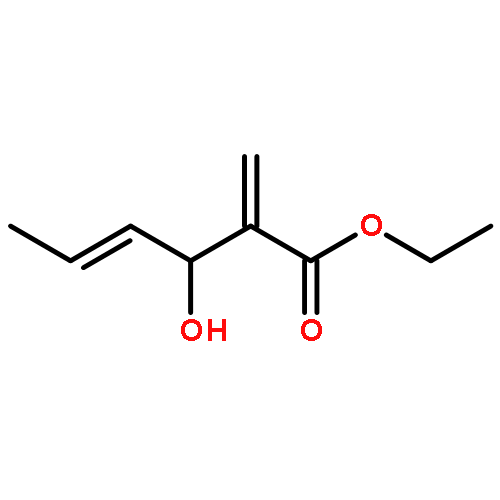
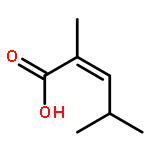
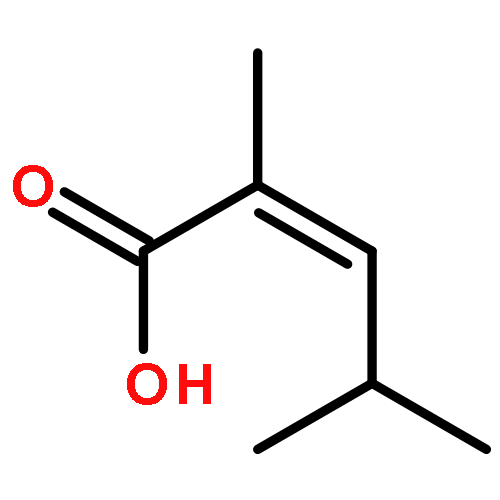
The substrate 4-hydroxy tiglic acid (1), which has low singlet oxygen reactivity, is converted to the 1,2-hydroperoxyalcohol 2 by photooxygenation in deuterated solvents and used for subsequent BF3-catalyzed peroxyacetalizations with acyclic and cyclic ketones. This route enables the synthesis of water-soluble trioxanes that are versatile building blocks for further functionalization, e.g. ester and amide formation.
Co-reporter:Axel G. Griesbeck, Bernd Goldfuss, Matthias Leven, Alan de Kiff
Tetrahedron Letters 2013 Volume 54(Issue 23) pp:2938-2941
Publication Date(Web):5 June 2013
DOI:10.1016/j.tetlet.2013.03.099
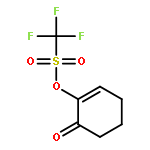
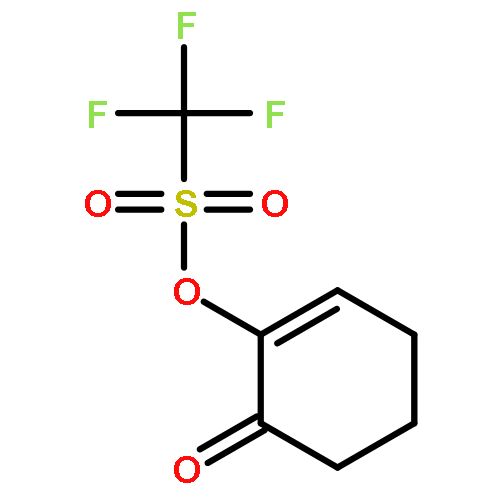
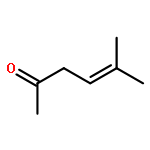
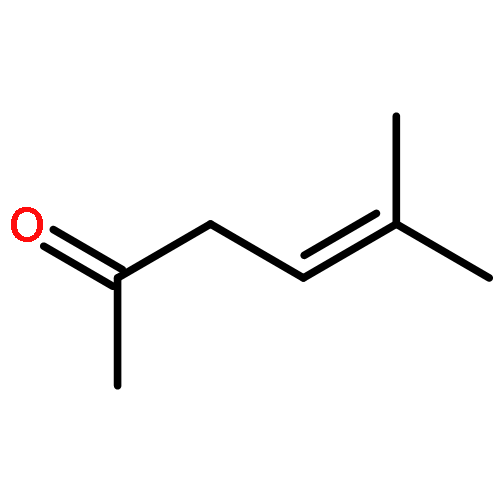
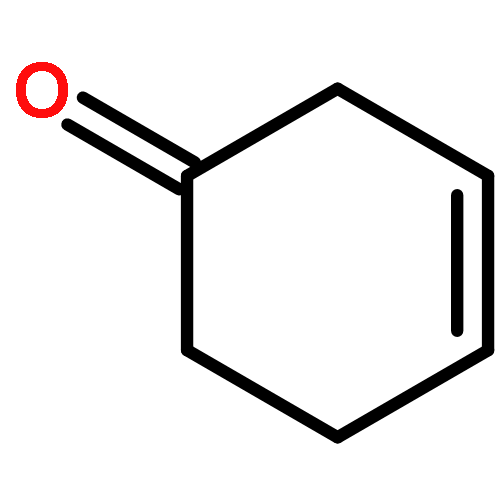
The photooxygenation of two β,γ-unsaturated ketones was studied by experimental and computational methods: 5-methyl-hex-4-en-2-one (1) and cyclohex-3-en-1-one (6) as model compounds for acyclic versus cyclic deconjugated enones. The open-chain substrate delivered a 1:1 mixture of regioisomers 2a,b following the established cis-selectivity model whereas the cyclic substrate reacts with 1O2 to give preferentially the conjugated product 7. This effect is in agreement with the mechanistic two-stage no-intermediate model and on a computational level corresponds to a regioselectivity control following the steepest decent pathway from the corresponding transition stages in a valley ridge potential energy surface region.The synthetic and computational analysis of singlet oxygen ene reactions with unsaturated ketones is described.
Co-reporter:Axel G. Griesbeck;Jörg Steinwascher
Research on Chemical Intermediates 2013 Volume 39( Issue 1) pp:33-42
Publication Date(Web):2013 January
DOI:10.1007/s11164-012-0629-3
Homogeneous as well as heterogeneous photocatalysts that are able to oxidize the azide anion with low competitive singlet oxygen quantum yields are used to generate azidyl radicals. These radicals add to electron-rich as well as electron-poor (Michael acceptors) alkenes, and carbon radicals are formed regioselectively. Trapping with triplet oxygen (type I photooxygenation) is diffusion controlled, and the initially formed peroxy radicals are reduced with regeneration of the photocatalyst. Fluorescence quenching studies reveal rapid photoinduced electron transfer in the first catalysis step. The lack of rearrangement products in the bicyclic terpene series (pinenes, limonene) accounts for rapid subsequent oxygen trapping and back electron transfer steps. The 1,2-azidohydroperoxidation enables synthesis of 1,2-azidoalcohols and 1,2-aminoalcohols by different reduction protocols. Substrate modification and combination of type II photooxygenation with electron transfer photocatalysis allows the synthesis of 1-amino-2,3-diols and 2-amino-1,3-diols.
Co-reporter:Axel G. Griesbeck, Nestor Nazarov, Jörg M. Neudörfl and Maria Heffen
Green Chemistry 2012 vol. 14(Issue 11) pp:3004-3006
Publication Date(Web):04 Sep 2012
DOI:10.1039/C2GC36089F
The intermolecular photodecarboxylation of arylacetic acids 3–5 in water by use of stoichiometric amounts of N-alkylated phthalimides 1a,b was investigated by NMR and online pH- and CO2-release sensing. Decreasing the electron donor capability of the arylacetic acids by 4-fluoro and trifluoromethyl substitution alters the efficiency of the photodecarboxylation and selectivity of the secondary reaction. A remarkable switch in reaction channel (A to B) was observed for perfluoropropionic acid.
Co-reporter:Dipl.-Chem. Darius Paul Kranz;Dr. Axel Georg Griesbeck;Dr. Ronald Alle;Dr. Raul Perez-Ruiz;Dr. Jörg Martin Neudörfl;Dr. Klaus Meerholz;Dr. Hans-Günther Schmalz
Angewandte Chemie International Edition 2012 Volume 51( Issue 24) pp:6000-6004
Publication Date(Web):
DOI:10.1002/anie.201201222
Co-reporter:Dipl.-Chem. Darius Paul Kranz;Dr. Axel Georg Griesbeck;Dr. Ronald Alle;Dr. Raul Perez-Ruiz;Dr. Jörg Martin Neudörfl;Dr. Klaus Meerholz;Dr. Hans-Günther Schmalz
Angewandte Chemie 2012 Volume 124( Issue 24) pp:6102-6106
Publication Date(Web):
DOI:10.1002/ange.201201222
Co-reporter:Dr. Axel G. Griesbeck;Dr. Johannes Uhlig;Dr. Thomas Sottmann;Dr. Lhoussaine Belkoura;Dr. Reinhard Strey
Chemistry - A European Journal 2012 Volume 18( Issue 50) pp:16161-16165
Publication Date(Web):
DOI:10.1002/chem.201202296
Abstract
Pluronic F-127 hydrogels are highly efficient microenvironments for photochemical reactions, as demonstrated for singlet oxygen reactions of monoalkenes. Nonpolar substrates are localized in the nanosized polymer compartment, which can be visualized by neutron scattering. The efficiency of 1O2 reactions is strongly increased for tiglate derivatives and the regioselectivity of the ene reaction of trisubstituted alkenes is completely switched in comparison with solution phase and inverted in comparison with intrazeolite photo-oxygenations.
Co-reporter:Alberto Soldevilla, Raúl Pérez-Ruiz, Yrene Díaz Miara and Axel Griesbeck
Chemical Communications 2010 vol. 46(Issue 21) pp:3747-3749
Publication Date(Web):23 Apr 2010
DOI:10.1039/C001622E
The fluorescence of caged phthalimide–serine couples 2 and 4 is up/down modulated by decarboxylative photorelease with fluorescence decrease (2) vs. moderate fluorescence increase (4) serving as reporter function.
Co-reporter:Axel G. Griesbeck, Sebastian Hanft and Yrene Díaz Miara
Photochemical & Photobiological Sciences 2010 vol. 9(Issue 10) pp:1385-1390
Publication Date(Web):03 Sep 2010
DOI:10.1039/C0PP00175A
The chiral chemosensor 1, based on a thiourea-activated phthalimide, is available by four reaction steps from 4-nitrophthalimide. 1 detects fluoride, chloride, acetate, and dihydrogen phosphate anions by changes in UV-vis absorption. Fluoride in excess induces deprotonation whereas the other anions show only complex formation in the ground state. 1H-NMR studies confirm the formation of these H-bonded complexes and the fluoride-induced receptor deprotonation in the recognition process. Moderate chiral recognition was observed for sodium D/L-lactate with Kass(D)/Kass(L) = 1.93.
Co-reporter:Axel G. Griesbeck, Melissa Reckenthäler and Johannes Uhlig
Photochemical & Photobiological Sciences 2010 vol. 9(Issue 6) pp:775-778
Publication Date(Web):15 Apr 2010
DOI:10.1039/C0PP00033G
The allylic hydroperoxide 2 (myrtenyl hydroperoxide), available from singlet oxygen photooxygenation of β-pinene (1), is converted into the azido bis-hydroperoxide 3 by an electron-transfer induced azidyl radical formation and trapping of the initial tertiary carbon radical by triplet oxygen. The azido bis-hydroperoxide 3 is reduced to the azido 1,2-diol 4 or the amino diol 5, respectively. Beside classical fluorescent PET sensitizers such as rhodamines, also nanosized semiconductor particles as well as lucigenin were applied as catalysts. The electron transfer rate of azide oxidation was determined for lucigenin by fluorescence quenching analysis.
Co-reporter:Raúl Pérez-Ruiz;Robert Fichtler;Yrene Diaz Miara
Journal of Fluorescence 2010 Volume 20( Issue 3) pp:657-664
Publication Date(Web):2010 May
DOI:10.1007/s10895-010-0598-0
The photophysical properties of a series of structurally related 4-aminophthalimides and the corresponding 5-aminophthalic hydrazides (luminols) are reported. Absorption, steady-state, and time-resolved fluorescence spectra of luminols exhibited substitution, solvent, and pH dependence. Singlet lifetimes have been determined by time-resolved laser flash spectroscopy. UV spectra in gas phase and DMSO solution were calculated by TD-DFT which revealed the existence of two low-energy excited singlet states with strong pH-sensitivity.
Co-reporter:Axel G. Griesbeck ; Jörg Neudörfl ; Achim Hörauf ; Sabine Specht ;Angela Raabe
Journal of Medicinal Chemistry 2009 Volume 52(Issue 10) pp:3420-3423
Publication Date(Web):April 29, 2009
DOI:10.1021/jm9002523
Three synthetic approaches to highly antimalarial peroxide dyads that are composed of the natural artemisinin part (either as dihydroartemisinin or artesunic acid components) and synthetic 1,2,4-trioxanes linked by ether or ester bridges are described. Photooxygenation is the key step to introduce the trioxane group initially or at the end of the reaction sequence, respectively. Dihydroartemisinin or artesunate coupling to hydroxyethyltrioxanes are the two processes that use intact peroxide units from the beginning, whereas the dihydroartemisinin-coupling to an allylic alcohol is a postphotooxygenation route, where the second trioxane ring is installed in the last step of the procedure.
Co-reporter:Raúl Pérez-Ruiz, Yrene Díaz, Bernd Goldfuss, Dirk Hertel, Klaus Meerholz and Axel G. Griesbeck
Organic & Biomolecular Chemistry 2009 vol. 7(Issue 17) pp:3499-3504
Publication Date(Web):07 Jul 2009
DOI:10.1039/B908433A
The anion chemosensor 1 based on a urea-activated phthalimide with a stereogenic centre was synthesized using an efficient procedure involving a Curtius rearrangement. Its photophysical properties were estimated in several solvents. Sensor 1 detected fluoride with absorption as well as fluorescence changes and was only observable for this case and not for other halides. The appearance of a new CT complex emission at a longer wavelength and no changes in the singlet lifetime of 1 in the presence of fluoride supported a fluorescence static quenching mechanism. 1H-NMR studies, together with theoretical calculations based on DFT methods at the B3lYP/6–31G* level of theory confirmed the formation of a [1–F]− complex through H-bonding interactions rather than receptor deprotonation in the recognition process. Reversibility of this process was observed upon addition of a protic solvent.
Co-reporter:Axel G. Griesbeck, Miyeon Cho
Tetrahedron Letters 2009 50(1) pp: 121-123
Publication Date(Web):
DOI:10.1016/j.tetlet.2008.10.094
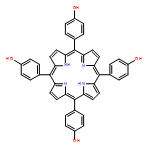
Co-reporter:AxelG. Griesbeck;Mathias Schäfer ;Johannes Uhlig
Advanced Synthesis & Catalysis 2008 Volume 350( Issue 13) pp:2104-2108
Publication Date(Web):
DOI:10.1002/adsc.200800282
Abstract
Polar meso-tetraarylporphyrins 2–4 were synthesized from tetrakis-4-hydroxyphenylporphyrin 1 as the central building block by consecutive base-induced reactions with glycidol. The decorating units form a polar hydrogen-bonded shell around the sensitizer core which is proposed as the binding site for polar substrates in photocatalyzed oxygenation reactions. As substrate, the polarity-sensor mesitylol (5) was applied and the reaction constrained in a polystyrene matrix. Increasing shell dimensions lead to increased diastereoselectivities for the allylic hydroperoxides 6 and thus clearly demonstrate the concept of shell-induced substrate stereoselectivity in singlet oxygen reactions.
Co-reporter:Raúl Pérez-Ruiz, Olga Hinze, Jörg-M. Neudörfl, Dirk Blunk, Helmut Görner and Axel G. Griesbeck
Photochemical & Photobiological Sciences 2008 vol. 7(Issue 7) pp:782-788
Publication Date(Web):29 May 2008
DOI:10.1039/B807889K
The solution photochemistry of the ortho allyloxy-substituted benzophenone 1 has been investigated in detail. Product analysis revealed formation of a diastereomeric mixture of dihydrobenzofuran derivatives 3 by cyclization via a short-lived intermediate 1,5-biradical and an unusual acetal 4 by a pseudo-Paternò-Büchi rearrangement. The latter reaction pathway was supported by means of laser flash photolysis, where a long-lived intermediate with a maximum absorption band at 380 nm was observed. Besides, theoretical calculations (TD-DFT) of this UV-transient resulted in a band with maximum intensity at 390 nm showing a good correlation between experimental results and theoretical calculations. For comparison, the meta-substituent substrate 2 was also investigated showing preferred triplet–triplet energy transfer.
Co-reporter:Axel G. Griesbeck Dr.;Dirk Blunk Dr.;Tamer T. El-Idreesy Dr.;Angela Raabe Dipl.-Chem.
Angewandte Chemie International Edition 2007 Volume 46(Issue 46) pp:
Publication Date(Web):17 OCT 2007
DOI:10.1002/anie.200701397
Resistance effects to the antimalarial agent artemisinin and its derivatives spurs on the search for new compounds with related cyclic peroxide structures. A flexible route to new bicyclic peroxides and perorthoesters is provided by a sequence of 1O2 ene reaction and subsequent Lewis acid catalyzed peroxyacetalization.
Co-reporter:Axel G. Griesbeck Dr.;Dirk Blunk Dr.;Tamer T. El-Idreesy Dr.;Angela Raabe Dipl.-Chem.
Angewandte Chemie 2007 Volume 119(Issue 46) pp:
Publication Date(Web):17 OCT 2007
DOI:10.1002/ange.200701397
Resistenzeffekte beeinträchtigen zunehmend die Anwendung von Artemisinpräparaten gegen Malaria, sodass die Entwicklung von Derivaten oder verwandten cyclischen Peroxidstrukturen dringender wird. Eine Sequenz aus 1O2-En-Reaktion und nachfolgender Lewis-Säure-katalysierter Peroxacetalisierung bietet eine vielseitige Route zu neuen bicyclischen Peroxiden und Perorthoestern (siehe Schema).
Co-reporter:Raúl Pérez-Ruiz, Miguel A. Miranda, Ronald Alle, Klaus Meerholz and Axel G. Griesbeck
Photochemical & Photobiological Sciences 2006 vol. 5(Issue 1) pp:51-55
Publication Date(Web):02 Dec 2005
DOI:10.1039/B513875B
The bicyclic oxetanes 1 and 2 resulting from photocycloaddition of aromatic aldehydes to 2,3-dihydrofuran, were efficiently cleaved by means of electron-transfer reduction, photoinduced by the electronically excited reductants 1-methoxynaphthalene (MN) and 2,7-dimethoxynaphthalene (DMN) in acetonitrile. The fluorescence quenching rates of DMN/MN by 1 and 2 were determined by static methods, the triplet quenching rates were determined by means of laser flash photolysis (LFP). The product analysis established a “photo-photo metathesis” where both cycloaddition and cycloreversion processes are induced by photochemical processes.
Co-reporter:Axel G. Griesbeck;Anna Bartoschek;Jörg Neudörfl;Claus Miara
Photochemistry and Photobiology 2006 Volume 82(Issue 5) pp:1233-1240
Publication Date(Web):30 APR 2007
DOI:10.1562/2006-03-03-RA-832
The ene reaction of chiral allylic alcohols is applied as a tool for the investigation of intrapolymer effects by means of the stereoselectivity of the singlet-oxygen addition. The diastereo selectivity strongly depends on the structure of the polymer, the substrate loading degree and also on the degree of conversion demonstrating additional supramolecular effects evolving during the reaction. The efficiency and the stability of polymer-bound sensitizers were evaluated by the ene reaction of singlet oxygen with citronellol. The ene reaction with chiral ammonium salts of tiglic acid was conducted under solution phase conditions or in polystyrene beads under chiral contact ion-pair conditions. The products thus obtained precipitate during the photoreaction as ammonium salts. Moderate asymmetric induction was observed for this procedure for the first time.
Co-reporter:Xichen Cai Dr.;Peter Cygon Dr.;Bernd Goldfuss Dr. Dr.;Heike Heckroth Dr.;Mamoru Fujitsuka Dr.;Tetsuro Majima Dr.
Chemistry - A European Journal 2006 Volume 12(Issue 17) pp:
Publication Date(Web):24 MAR 2006
DOI:10.1002/chem.200600011
Triplet 1,4-biradicals were generated by Norrish-Type-II hydrogen transfer from α-heteroatom-substituted β-branched butyrophenones 1–6 and detected by laser flash absorption measurements. For three oxy-substituted compounds 2–4 (Rα=OH, OCOMe, OCOOEt) comparable lifetimes were determined in acetonitrile (roughly 1.5 μs). In benzene, divergent trends were observed: for the hydroxy compound 2 a lower lifetime of 790 ns was determined, whereas for 3 and 4 the lifetimes increased to 4.9 μs. Photolyses of the α-amino-substituted compounds 1 and 6 resulted in transient species with significant lower lifetimes (for 1 160 ns in benzene and 450 ns in acetonitrile; for 6 <100 ns in both solvents). The mesyloxy substrate 5 undergoes rapid CO bond cleavage upon photolysis and no transient triplet species were detected. Computational (UB3 LYP/6–31G* and natural don orbital (NBO) analyses) results supported the assumption of a negative hyperconjugative interaction strongly stabilizing α-oxy-substituted over α-amino-substituted radicals.
Co-reporter:Axel G. Griesbeck and Tamer T. El-Idreesy
Photochemical & Photobiological Sciences 2005 vol. 4(Issue 2) pp:205-209
Publication Date(Web):16 Dec 2004
DOI:10.1039/B416159A
A solvent-free route for the photooxygenation of the 5-methoxyoxazoles (1) is described. The substrates were embedded in nanosized polystyrene particles generated by the emulsifier-free emulsion polymerization of styrene with divinylbenzene and porphyrine dyes as cross-polymerizable reagents. From the photooxygenation of 1, the 1,2,4-dioxazoles (2) were formed and isolated from the reaction cavities by ethanol extraction. From comparison of the substrate conversions, the efficiency of singlet oxygen generation from the porphyrine dyestuff and the stability of the sensitizing material were estimated.
Co-reporter:Axel G. Griesbeck, Tamer T. El-Idreesy, Lars-Oliver Höinck, Johann Lex, Reto Brun
Bioorganic & Medicinal Chemistry Letters 2005 Volume 15(Issue 3) pp:595-597
Publication Date(Web):1 February 2005
DOI:10.1016/j.bmcl.2004.11.043
A remarkable increase in antimalarial in vitro activity was achieved by integration of spiroadamantane motifs in 6-alkylidene 1,2,4-trioxanes 3a–h via diastereoselective photooxygenation of allylic alcohols and subsequent BF3-catalyzed peroxyacetalization with adamantanone to give the active compounds 3e–h.A remarkable increase in antimalarial in vitro activity was achieved by integration of spiroadamantane motifs in 6-alkylidene 1,2,4-trioxanes 3a–h via diastereoselective photooxygenation of allylic alcohols and subsequent BF3-catalyzed peroxyacetalization with adamantanone.
Co-reporter:Axel G. Griesbeck;Tamer T. El-Idreesy;Anna Bartoschek
Advanced Synthesis & Catalysis 2004 Volume 346(Issue 2-3) pp:
Publication Date(Web):29 MAR 2004
DOI:10.1002/adsc.200303181
Two reaction protocols are described which involve the use of polymer supports as reaction media for photooxygenation processes: 1) The use of polystyrene beads (PS) loaded with tetraphenyl- (TPP) or tetratolylporphyrin (TTP), swollen with the substrate in an appropriate organic solvent and subsequent irradiation under air. Products were isolated simply by dissolution in alcoholic solvents and filtration. 2) Covalently linked tetrastyrylporphyrin in polystyrene-divinylbenzene beads were synthesized by emulsifier-free emulsion polymerization and directly used for the photooxygenation protocol described above. The latter alternative allows also the use of less polar solvents for the extraction of the oxygenation products from the polymer beads. From the sensitizer loading degree, an optimal substrate/sensitizer molar ratio of 1,000–2,000 was determined and recyclization is possible for at least five times resulting in a minimum turnover number (with respect to the sensitizer TTP) of 5×104 (after five cycles). Both approaches were applied to the ene reaction of singlet oxygen with citronellol (1), the regioisomeric pinenes 3 and 5, and the allylic alcohols 9a–c, respectively, as well as to the [4+2]-cycloadditions of singlet oxygen to sorbinol (7) and the chiral diene 11.
Co-reporter:Axel G. Griesbeck, Samir Bondock and Johann Lex
Organic & Biomolecular Chemistry 2004 vol. 2(Issue 8) pp:1113-1115
Publication Date(Web):17 Mar 2004
DOI:10.1039/B401990C
The photocycloaddition of methyl pyruvate and methyl phenylglyoxylate, respectively, to 5-methoxy oxazoles bearing additional substituents at C-2 and C-4 leads to bicyclic oxetanes 2 and 3 with high to moderate (exo) diastereoselectivity that can be easily ring-opened to give bis-quaternary aspartic acid diester derivatives 4 and 5.
Co-reporter:Axel G. Griesbeck, Nesmine Maptue, Samir Bondock and Michael Oelgemöller
Photochemical & Photobiological Sciences 2003 vol. 2(Issue 4) pp:450-451
Publication Date(Web):21 Feb 2003
DOI:10.1039/B212357F
The XeCl excimer 308 nm radiation system is described as a powerful tool for synthetic organic photochemistry involving carbonyl and carbonyl-type chromophores. Scaleable photochemical reactions which have been tested and optimized for this system were photodecarboxylation, [2 + 2]-photocycloaddition, and Type III (electron transfer) photooxygenation reactions.
Co-reporter:Axel G. Griesbeck and Heike Heckroth
Photochemical & Photobiological Sciences 2003 vol. 2(Issue 11) pp:1130-1133
Publication Date(Web):15 Oct 2003
DOI:10.1039/B307658J
The photochemical fingerprint for the N-acetyl methyl ester of the aliphatic amino acid tert-leucine 1 was investigated. This reaction path was compared with the electron transfer active amino acids methionine (N-acetyl methyl ester derivative 2a as well as the methyl ester derivative 2b) and the cysteine derivatives 3a and 3b
(penicillamine derivative). Photofragmenation of the ester group dominated the photolysis of 1, whereas loss of methylmercaptane was observed for 2a and 3a. Vinylglycine derivatives 11 and 14 were formed to a minor extent. The gem-dimethylated compound 3b gave a solvent-characteristic product pattern with photoelimination products 18 and 19 in acetonitrile and loss of methylmercaptane to give 20 in methanol.
Co-reporter:Axel G. Griesbeck, Waldemar Adam, Anna Bartoschek and Tamer T. El-Idreesy
Photochemical & Photobiological Sciences 2003 vol. 2(Issue 8) pp:877-881
Publication Date(Web):09 Apr 2003
DOI:10.1039/B302255B
The kinetics of the chemical and physical quenching of the first excited singlet state of oxygen [1O2
(1Δg)] by unfunctionalized alkenes 1–4, allylic alcohols 5–7 and 9, allylic acetates 8 and 11, and the allylic ether 10 display small solvent-polarity effects on the reactivity. The regioselectivity of the singlet oxygen ene reaction is solvent independent for the unfunctionalized alkenes as well as the prenol-type substrates, the latter showing substantial solvent effects on the diastereoselectivity. Pronounced physical quenching is detected only for the allylic alcohols 5 and 6. These results are interpreted in terms of the interactions between singlet oxygen and the allylic hydroxy groups, conformationally promoted by allylic strain which lead either to chemical activation or to physical quenching. The results for substrate 9 in deuterated vs. non-deuterated methanol are in accord with hydrogen bonding between the allylic alcohol and 1O2, which directs the diastereoselectivity of the ene reaction with chiral allylic alcohols.
Co-reporter:Axel G. Griesbeck and Stefan Schieffer
Photochemical & Photobiological Sciences 2003 vol. 2(Issue 2) pp:113-117
Publication Date(Web):15 Jan 2003
DOI:10.1039/B211008C
The fluorescent 4,5-dimethoxyphthalimides 1–10 were applied as sensors for intra- and intermolecular photoinduced electron transfer processes. Strong intramolecular fluorescence quenching was detected for the thioether 2 and the tertiary amine 3. The fluorescence of the carboxylic acids 4–7 is pH-dependent accounting for PET-quenching of the singlet excited phthalimide at pH > pKs. At low pH, chromophore protonation might contribute to moderate fluorescence quenching. The arylated phthalimides 9 and 10 show remarkable low fluorescence independent of pH and substituent pattern. Intermolecular fluorescence quenching was detected for the combinations of 1 with dimethyl sulfide, and 1 with triethylamine but not with metal carboxylates.
Co-reporter:Axel G. Griesbeck and Anna Bartoschek
Chemical Communications 2002 (Issue 15) pp:1594-1595
Publication Date(Web):26 Jun 2002
DOI:10.1039/B204017D
A solvent-free photooxygenation process that uses organic substrates embedded in porphyrin-loaded polystyrene beads as solid support is described and applied for ene- and [4+2]-cycloaddition reactions involving singlet oxygen (1Δg).
Co-reporter:Axel G. Griesbeck, Wolfgang Kramer, Thomas Heinrich and Johann Lex
Photochemical & Photobiological Sciences 2002 vol. 1(Issue 4) pp:237-239
Publication Date(Web):06 Mar 2002
DOI:10.1039/B200448H
Medium-sized heterocyclic ring systems are available via a photo-elctron transfer (PET)-induced decarboxylation–cyclization sequence using a triad composed of electronically excited acceptor, linker and electron donor A*–L–D with target ring sizes varying from 8–16.
Co-reporter:Axel G. Griesbeck and Samir Bondock
Photochemical & Photobiological Sciences 2002 vol. 1(Issue 2) pp:81-83
Publication Date(Web):02 Jan 2002
DOI:10.1039/B107226A
The simple diastereoselectivity of the photocycloaddition of aliphatic aldehydes to (Z)- and (E)-cyclooctene was analyzed as a function of the substrate concentrations and applied to spin mapping.
Co-reporter:Axel G. Griesbeck;Thomas Heinrich;Michael Oelgemöller;Arne Molis;Axel Heidtmann
Helvetica Chimica Acta 2002 Volume 85(Issue 12) pp:4561-4578
Publication Date(Web):2 JAN 2003
DOI:10.1002/hlca.200290027
The synthesis of a variety of cyclic peptides from N-phthaloyl-protected di-, tri-, tetra-, and pentapeptides with different aminocarboxylic acid tethers by photodecarboxylation – initiated by intramolecular electron transfer – has been explored in aqueous media. The progress and the chemoselectivity of the follow-up processes after CO2 extrusion were traced by the respective pH/time-profiles, as well as by the overall change in pH after completion of the reaction. The competition between cyclization and simple oxidative decarboxylation depends on spacer length and geometry, H-bonding interaction between the electron accepting phthalimide CO groups and amide H-atoms, as well as the geometric reorganization coupled with the radical combination step and the formation of the lactam rings. With progressing reaction, hydrolysis of the phthalimide chromophore becomes an increasingly important side reaction due to the constant increase in pH. The use of phosphate-buffered aqueous media consequently improved the cyclization yields. The ground-state interactions between amide groups and the terminal COO− group with the imide CO groups were studied for the model system [N-(phthaloyl)glycyl]sarcosine (1) by NMR spectroscopy where the amide (E/Z)-equilibrium depends on the presence of carboxylate vs. free carboxylic acid, demonstrating the role of H-bonding and metal coordination.
Co-reporter:Axel G. Griesbeck Dr.;Uwe J. Meierhenrich Dr.
Angewandte Chemie International Edition 2002 Volume 41(Issue 17) pp:
Publication Date(Web):30 AUG 2002
DOI:10.1002/1521-3773(20020902)41:17<3147::AID-ANIE3147>3.0.CO;2-V
One of the most interesting phenomena on Earth is the chirality of biomolecules, the origin of which remains unknown. A challenge arising from this phenomenon is the selective, atom-economic synthesis of enantiomerically pure target molecules from nonchiral starting materials. Herein, new developments in the field of asymmetric photochemistry and photochirogenesis are described with special emphasis on absolute asymmetric synthesis. In this context, the elucidation of the ultimate cause of homochirality phenomena on earth and the possible correlation with physicochemical parameters are also presented.
Co-reporter:Axel G. Griesbeck Dr.;Uwe J. Meierhenrich Dr.
Angewandte Chemie 2002 Volume 114(Issue 17) pp:
Publication Date(Web):30 AUG 2002
DOI:10.1002/1521-3757(20020902)114:17<3279::AID-ANGE3279>3.0.CO;2-9
Eines der interessantesten Phänomene des Lebens auf der Erde ist die Chiralität von Biomolekülen, deren Ursprung nach wie vor nicht geklärt ist. Eine der großen Herausforderungen, die mit diesem Phänomen verknüpft ist, ist die selektive und atomökonomische Synthese enantiomerenreiner Zielverbindungen aus nicht-chiralen Ausgangsverbindungen. In diesem Beitrag werden neue Entwicklungen auf dem Gebiet der asymmetrischen Photochemie und der Photochirogenese sowie der absoluten asymmetrischen Synthese beschrieben. Außerdem werden die möglichen Ursachen der Homochiralität auf der Erde sowie der Zusammenhang mit physikochemischen Parametern dargestellt.
Co-reporter:Axel G. Griesbeck;Michael Oelgemöller;Johann Lex;Andreas Haeuseler;Michael Schmittel
European Journal of Organic Chemistry 2001 Volume 2001(Issue 10) pp:
Publication Date(Web):12 APR 2001
DOI:10.1002/1099-0690(200105)2001:10<1831::AID-EJOC1831>3.0.CO;2-7
The intramolecular and intermolecular photoinduced electron transfer reactions of a series of mercaptoacetic acid and mercaptopropionic acid derivatives were investigated. In the intermolecular series, the phthalimidoalkylsulfanylalkylcarboxylates 1a−j and 2 were transformed into the tricyclic ring systems 3a−j and 4, respectively, with high regioselectivities. The mercaptoacetic acid and 2-mercaptopropionic acid derived substrates 1a−g and 2 readily cyclized in good to excellent yields (60−98%) but with low diastereoselectivities (except for 1d), whereas the corresponding 3-mercaptopropionic acid derived substrates 1h−j gave the corresponding tricyclic products 3h−j after prolonged irradiation, but with poor yields (11−20%). The intermolecular version − i.e., photodecarboxylative addition to N-methylphthalimide (5) as electron acceptor − was successful with mercaptoacetic acid, and 2-mercaptopropionic acid substrates 6a−c and the addition products 7a−c were obtained in high yields (57−90%). No addition, however, was observed with 3-(methylsulfanyl)propionic acid (6d). The regioselectivity of decarboxylation proceeded in a controlled manner for the mercaptosuccinic acid derivatives in both the intramolecular (with 8a−c) and the intermolecular (with 9) versions. Comparison between sulfur-activated and nonactivated species (13, 15) or irradiation of 1a under nonactivating conditions showed that the carboxylate anion in the position α to the electron-donating sulfur atom acts as a superior leaving group. This efficiency is drastically reduced for carboxylate anions in the β position. With the former substrates, the photochemical cyclization proceeds with high product yields. Quantum yield measurements for decomposition (Φd as a measure for cyclization) supported these observations. CV measurements indicated preorientation prior to electron transfer in the intramolecular pathway.
Co-reporter:Axel G. Griesbeck Dr.;Samir Bondock M. Sc.;Murthy S. Gudipati Priv.-Doz. Dr.
Angewandte Chemie 2001 Volume 113(Issue 24) pp:
Publication Date(Web):18 DEC 2001
DOI:10.1002/1521-3757(20011217)113:24<4828::AID-ANGE4828>3.0.CO;2-Y
Die Multiplizität des angeregten Zustandes kontrolliert die Produktverteilung bei der Bildung der endo/exo-Oxetane 3 durch Photocycloaddition von Aldehyden 1 (R=Ph, Et, Me, iBu) an 2,3-Dihydrofuran 2 (Paternò-Büchi-Reaktion). Ob dabei der Singulett- oder der Triplettkanal dominiert, hängt stark von der Temperatur ab, die sich damit als entscheidende Einflussgröße auf die Selektivität der Reaktion entpuppt.
Co-reporter:Axel G. Griesbeck Dr.;Wolfgang Kramer Dr.;Johann Lex Dr.
Angewandte Chemie 2001 Volume 113(Issue 3) pp:
Publication Date(Web):30 JAN 2001
DOI:10.1002/1521-3757(20010202)113:3<586::AID-ANGE586>3.0.CO;2-#
Co-reporter:Axel G. Griesbeck Dr.;Samir Bondock;Murthy S. Gudipati Priv.-Doz. Dr.
Angewandte Chemie International Edition 2001 Volume 40(Issue 24) pp:
Publication Date(Web):18 DEC 2001
DOI:10.1002/1521-3773(20011217)40:24<4684::AID-ANIE4684>3.0.CO;2-9
The multiplicity of the excited state controls the product distribution in the formation of endo/exo-oxetane 3 in the photocycloaddition of aldehydes 1 (R = Ph, Et, Me, iBu) with dihydrofuran 2 (Paternò–Büchi reaction). Whether the singlet or triplet channel dominates is strongly dependent on the temperature, which is therefore critical for the selectivity of the reaction.
Co-reporter:Axel G. Griesbeck, Johann Lex, Kurt M. Saygin and Jörg Steinwascher
Chemical Communications 2000 (Issue 22) pp:2205-2206
Publication Date(Web):01 Nov 2000
DOI:10.1039/B005834N
The photoinduced electron transfer of azide anions in the
presence of an excited organic dyestuff, oxygen, and α- or
β-pinene, respectively, gave 2-azidohydroperoxides in excellent regio-
and good diastereoselectivity.
Co-reporter:Axel G. Griesbeck, Maren Fiege and Johann Lex
Chemical Communications 2000 (Issue 7) pp:589-590
Publication Date(Web):20 Mar 2000
DOI:10.1039/B000578I
The photocycloaddition of aliphatic and aromatic aldehydes
with 2,4,5-trimethyloxazole proceeds highly regio- and diastereoselectively
to give bicyclic oxetanes; hydrolytic cleavage of these adducts gives
selectively erythro
α-amino, β-hydroxy methyl
ketones.
Co-reporter:Axel G. Griesbeck;Maren Fiege;Murthy S. Gudipati;Robert Wagner
European Journal of Organic Chemistry 1998 Volume 1998(Issue 12) pp:
Publication Date(Web):23 DEC 1998
DOI:10.1002/(SICI)1099-0690(199812)1998:12<2833::AID-EJOC2833>3.0.CO;2-6
The photooxygenation of 2,4-dimethyl-1,3-pentadiene (1) was investigated in seven polar and nonpolar solvents by oxygen-uptake measurements. The overall deactivation rate ko ( = kr + kq) was additionally measured in chloroform solutions by singlet-oxygen (1Δg) phosphorescence quenching which showed excellent agreement with the data from the detailed steady-state kinetics. The difference in solvent-polarity effects on the [4 + 2] cycloaddition (major path, leading to the endoperoxide 2) and ene reaction (minor path, leading to the allylic hydroperoxide 3) are explained by competition between a concerted and a perepoxide mechanism. In all solvents the physical quenching of singlet oxygen by 1 is at least as efficient as the chemical quenching. The reaction of the endoperoxide 2 and 3,3,6,6-tetramethyl-1,2-dioxene 9 with carbonyl compounds in the presence of TMSOTf resulting in the dihydrofuran 8 and the 1,2,4-trioxane 10, was also studied.
Co-reporter:Axel G. Griesbeck;Thomas Deufel;Georg Hohlneicher;Rupert Rebentisch;Jörg Steinwascher
European Journal of Organic Chemistry 1998 Volume 1998(Issue 9) pp:
Publication Date(Web):7 DEC 1998
DOI:10.1002/(SICI)1099-0690(199809)1998:9<1759::AID-EJOC1759>3.0.CO;2-I
The twofold bridged sesquinorbornenes 2 and 6 were prepared using sequential [4 + 2] cycloadditions of benzoquinone with 1,5-dihydropentalenes 1 and 5. These syntheses were improved using dilution conditions or a more reactive substituted benzoquinone. Results from semiempirical and ab initio DFT calculations indicated remarkably high pyramidalization angles (ϕ = 46-47°) for the central C–C double-bond atoms. The chemical reactivity with triplet and singlet oxygen, dimethyldioxirane and N-methyl-1,2,4-triazoline-3,5-dione supports these structural assignments.
Co-reporter:Alberto Soldevilla, Raúl Pérez-Ruiz, Yrene Díaz Miara and Axel Griesbeck
Chemical Communications 2010 - vol. 46(Issue 21) pp:NaN3749-3749
Publication Date(Web):2010/04/23
DOI:10.1039/C001622E
The fluorescence of caged phthalimide–serine couples 2 and 4 is up/down modulated by decarboxylative photorelease with fluorescence decrease (2) vs. moderate fluorescence increase (4) serving as reporter function.
Co-reporter:Raúl Pérez-Ruiz, Yrene Díaz, Bernd Goldfuss, Dirk Hertel, Klaus Meerholz and Axel G. Griesbeck
Organic & Biomolecular Chemistry 2009 - vol. 7(Issue 17) pp:NaN3504-3504
Publication Date(Web):2009/07/07
DOI:10.1039/B908433A
The anion chemosensor 1 based on a urea-activated phthalimide with a stereogenic centre was synthesized using an efficient procedure involving a Curtius rearrangement. Its photophysical properties were estimated in several solvents. Sensor 1 detected fluoride with absorption as well as fluorescence changes and was only observable for this case and not for other halides. The appearance of a new CT complex emission at a longer wavelength and no changes in the singlet lifetime of 1 in the presence of fluoride supported a fluorescence static quenching mechanism. 1H-NMR studies, together with theoretical calculations based on DFT methods at the B3lYP/6–31G* level of theory confirmed the formation of a [1–F]− complex through H-bonding interactions rather than receptor deprotonation in the recognition process. Reversibility of this process was observed upon addition of a protic solvent.



![Spiro[cyclopropane-1,1'-[1H]isoindol]-3'(2'H)-one, 2'-(hydroxymethyl)-](http://img.cochemist.com/ccimg/192000/191983-21-8.png)
![Spiro[cyclopropane-1,1'-[1H]isoindol]-3'(2'H)-one, 2'-(hydroxymethyl)-](http://img.cochemist.com/ccimg/192000/191983-21-8_b.png)






![6,7-Dioxabicyclo[3.2.2]nonane, 1,5-bis(4-methylphenyl)-](http://img.cochemist.com/ccimg/157400/157367-59-4.png)
![6,7-Dioxabicyclo[3.2.2]nonane, 1,5-bis(4-methylphenyl)-](http://img.cochemist.com/ccimg/157400/157367-59-4_b.png)





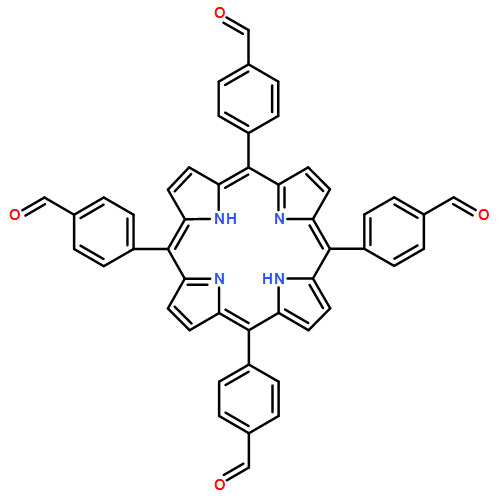
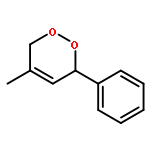
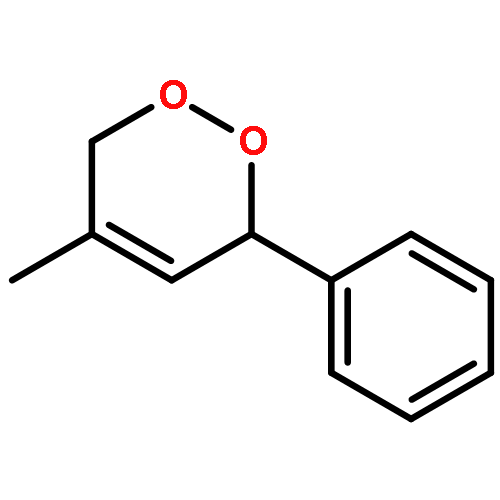




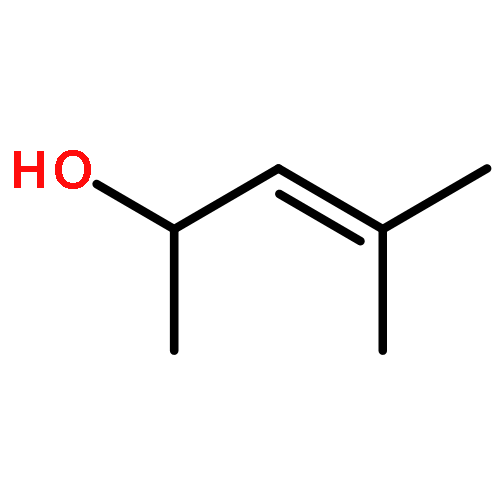
![Cyclopenta[b]pyrrole-2-carboxylic acid, octahydro-, (2R,3aR,6aR)- (9CI)](/data/chemimg/1077400/128900-19-6.png)
![Cyclopenta[b]pyrrole-2-carboxylic acid, octahydro-, (2R,3aR,6aR)- (9CI)](/data/chemimg/1077400/128900-19-6_b.png)











![21H,23H-Porphine, 5,10,15,20-tetrakis[4-(2-bromoethyl)phenyl]-](http://img.cochemist.com/ccimg/113500/113477-20-6.png)
![21H,23H-Porphine, 5,10,15,20-tetrakis[4-(2-bromoethyl)phenyl]-](http://img.cochemist.com/ccimg/113500/113477-20-6_b.png)
![GLYCINE, N-[11-(1,3-DIHYDRO-1,3-DIOXO-2H-ISOINDOL-2-YL)-1-OXOUNDECYL]-](http://img.cochemist.com/ccimg/112800/112717-35-8.png)
![GLYCINE, N-[11-(1,3-DIHYDRO-1,3-DIOXO-2H-ISOINDOL-2-YL)-1-OXOUNDECYL]-](http://img.cochemist.com/ccimg/112800/112717-35-8_b.png)












![Benzene, 1,1'-[1,6-bis(methylene)-1,6-hexanediyl]bis[4-methyl-](http://img.cochemist.com/ccimg/99300/99260-47-6.png)
![Benzene, 1,1'-[1,6-bis(methylene)-1,6-hexanediyl]bis[4-methyl-](http://img.cochemist.com/ccimg/99300/99260-47-6_b.png)
![3H,5H-Thiazolo[4,3-a]isoindol-5-one, 1,9b-dihydro-9b-hydroxy-](http://img.cochemist.com/ccimg/98200/98184-43-1.png)
![3H,5H-Thiazolo[4,3-a]isoindol-5-one, 1,9b-dihydro-9b-hydroxy-](http://img.cochemist.com/ccimg/98200/98184-43-1_b.png)


![1H-Isoindole-1,3(2H)-dione, 2-[(3-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/94900/94894-14-1.png)
![1H-Isoindole-1,3(2H)-dione, 2-[(3-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/94900/94894-14-1_b.png)
![Isoxazole, 3-phenyl-5-[(trimethylsilyl)oxy]-](http://img.cochemist.com/ccimg/94800/94744-98-6.png)
![Isoxazole, 3-phenyl-5-[(trimethylsilyl)oxy]-](http://img.cochemist.com/ccimg/94800/94744-98-6_b.png)


![Bicyclo[3.2.0]heptane,1,5-diphenyl-](http://img.cochemist.com/ccimg/94400/94383-67-2.png)
![Bicyclo[3.2.0]heptane,1,5-diphenyl-](http://img.cochemist.com/ccimg/94400/94383-67-2_b.png)

![PROPANOIC ACID, 3-[[(1,3-DIHYDRO-1,3-DIOXO-2H-ISOINDOL-2-YL)METHYL]THIO]-](http://img.cochemist.com/ccimg/91600/91570-07-9.png)
![PROPANOIC ACID, 3-[[(1,3-DIHYDRO-1,3-DIOXO-2H-ISOINDOL-2-YL)METHYL]THIO]-](http://img.cochemist.com/ccimg/91600/91570-07-9_b.png)


![Pyrido[2,1-a]isoindol-6(2H)-one, 1,3,4,10b-tetrahydro-10b-hydroxy-](http://img.cochemist.com/ccimg/88900/88892-69-7.png)
![Pyrido[2,1-a]isoindol-6(2H)-one, 1,3,4,10b-tetrahydro-10b-hydroxy-](http://img.cochemist.com/ccimg/88900/88892-69-7_b.png)





























![BENZENE, 1,1'-[1,6-BIS(METHYLENE)-1,6-HEXANEDIYL]BIS-](http://img.cochemist.com/ccimg/68400/68305-44-2.png)
![BENZENE, 1,1'-[1,6-BIS(METHYLENE)-1,6-HEXANEDIYL]BIS-](http://img.cochemist.com/ccimg/68400/68305-44-2_b.png)
![Benzene, [(1E)-3-methyl-1,3-butadienyl]-](http://img.cochemist.com/ccimg/68100/68036-69-1.png)
![Benzene, [(1E)-3-methyl-1,3-butadienyl]-](http://img.cochemist.com/ccimg/68100/68036-69-1_b.png)









![1H-Isoindole-1,3(2H)-dione, 2-[2-(4-hydroxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/65000/64985-42-8.png)
![1H-Isoindole-1,3(2H)-dione, 2-[2-(4-hydroxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/65000/64985-42-8_b.png)



















![BENZAMIDE, N-[(3S)-TETRAHYDRO-2,5-DIOXO-3-FURANYL]-](http://img.cochemist.com/ccimg/54900/54819-23-7.png)
![BENZAMIDE, N-[(3S)-TETRAHYDRO-2,5-DIOXO-3-FURANYL]-](http://img.cochemist.com/ccimg/54900/54819-23-7_b.png)






![ETHANE, [(TRIMETHYLSILYL)-ACI-NITRO]-](http://img.cochemist.com/ccimg/51200/51146-36-2.png)
![ETHANE, [(TRIMETHYLSILYL)-ACI-NITRO]-](http://img.cochemist.com/ccimg/51200/51146-36-2_b.png)















![4-[3-FLUORO-4-({[3-(2-METHYL-2-PROPANYL)-1-(6-QUINOLINYL)-1H-PYRA<WBR />ZOL-5-YL]CARBAMOYL}AMINO)PHENOXY]-N-METHYL-2-PYRIDINECARBOXAMIDE](http://img.cochemist.com/ccimg/35400/35386-78-8.png)
![4-[3-FLUORO-4-({[3-(2-METHYL-2-PROPANYL)-1-(6-QUINOLINYL)-1H-PYRA<WBR />ZOL-5-YL]CARBAMOYL}AMINO)PHENOXY]-N-METHYL-2-PYRIDINECARBOXAMIDE](http://img.cochemist.com/ccimg/35400/35386-78-8_b.png)
















![2H-Isoindole-2-aceticacid, 1,3-dihydro-a-[2-(methylthio)ethyl]-1,3-dioxo-, (aS)-](http://img.cochemist.com/ccimg/29600/29588-91-8.png)
![2H-Isoindole-2-aceticacid, 1,3-dihydro-a-[2-(methylthio)ethyl]-1,3-dioxo-, (aS)-](http://img.cochemist.com/ccimg/29600/29588-91-8_b.png)
































![1H-Isoindole-1,3(2H)-dione, 2-[(methylthio)methyl]-](http://img.cochemist.com/ccimg/20100/20044-28-4.png)
![1H-Isoindole-1,3(2H)-dione, 2-[(methylthio)methyl]-](http://img.cochemist.com/ccimg/20100/20044-28-4_b.png)










![尾-Alanine,N-[(1,3-dihydro-1,3-dioxo-2H-isoindol-2-yl)acetyl]- (9CI)](http://img.cochemist.com/ccimg/17900/17896-84-3.png)
![尾-Alanine,N-[(1,3-dihydro-1,3-dioxo-2H-isoindol-2-yl)acetyl]- (9CI)](http://img.cochemist.com/ccimg/17900/17896-84-3_b.png)


![1H-Isoindole-1,3(2H)-dione, 2-[(3S)-tetrahydro-2,5-dioxo-3-furanyl]-](http://img.cochemist.com/ccimg/16700/16652-95-2.png)
![1H-Isoindole-1,3(2H)-dione, 2-[(3S)-tetrahydro-2,5-dioxo-3-furanyl]-](http://img.cochemist.com/ccimg/16700/16652-95-2_b.png)




![1H-Isoindole-1,3(2H)-dione,2-[2-(1H-indol-3-yl)ethyl]-](http://img.cochemist.com/ccimg/15800/15741-71-6.png)
![1H-Isoindole-1,3(2H)-dione,2-[2-(1H-indol-3-yl)ethyl]-](http://img.cochemist.com/ccimg/15800/15741-71-6_b.png)











![Benzo[f]-1,3-benzodioxolo[6,5,4-cd]indol-5(6H)-one,8-methoxy-](http://img.cochemist.com/ccimg/13400/13395-02-3.png)
![Benzo[f]-1,3-benzodioxolo[6,5,4-cd]indol-5(6H)-one,8-methoxy-](http://img.cochemist.com/ccimg/13400/13395-02-3_b.png)













![5H-Pyrrolo[3,4-b]pyridine-5,7(6H)-dione,6-methyl-](http://img.cochemist.com/ccimg/6800/6789-51-1.png)
![5H-Pyrrolo[3,4-b]pyridine-5,7(6H)-dione,6-methyl-](http://img.cochemist.com/ccimg/6800/6789-51-1_b.png)
![4-[(3-methylphenyl)carbonyl]-8-{[3-(trifluoromethyl)phenyl]sulfonyl}-1-thia-4,8-diazaspiro[4.5]decane](http://img.cochemist.com/ccimg/6000/5959-80-8.png)
![4-[(3-methylphenyl)carbonyl]-8-{[3-(trifluoromethyl)phenyl]sulfonyl}-1-thia-4,8-diazaspiro[4.5]decane](http://img.cochemist.com/ccimg/6000/5959-80-8_b.png)













![1,3-BIS[3,5-BIS(TRIFLUOROMETHYL)PHENYL]UREA](http://img.cochemist.com/ccimg/3900/3824-74-6.png)
![1,3-BIS[3,5-BIS(TRIFLUOROMETHYL)PHENYL]UREA](http://img.cochemist.com/ccimg/3900/3824-74-6_b.png)






















![N-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)acetyl]glutamic acid](http://img.cochemist.com/ccimg/1200/1165-77-1.png)
![N-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)acetyl]glutamic acid](http://img.cochemist.com/ccimg/1200/1165-77-1_b.png)


![SPIRO[2.4]HEPTA-4,6-DIENE](http://img.cochemist.com/ccimg/800/765-46-8.png)
![SPIRO[2.4]HEPTA-4,6-DIENE](http://img.cochemist.com/ccimg/800/765-46-8_b.png)






![1H-Isoindole-1,3(2H)-dione, 2-[(4-fluorophenyl)methyl]-](http://img.cochemist.com/ccimg/400/318-49-0.png)
![1H-Isoindole-1,3(2H)-dione, 2-[(4-fluorophenyl)methyl]-](http://img.cochemist.com/ccimg/400/318-49-0_b.png)




























![GLYCINE, N-[6-(1,3-DIHYDRO-1,3-DIOXO-2H-ISOINDOL-2-YL)-1-OXOHEXYL]-](http://img.cochemist.com/ccimg/471900/471896-44-3.png)
![GLYCINE, N-[6-(1,3-DIHYDRO-1,3-DIOXO-2H-ISOINDOL-2-YL)-1-OXOHEXYL]-](http://img.cochemist.com/ccimg/471900/471896-44-3_b.png)
![GLYCINE, 1-[(1,3-DIHYDRO-1,3-DIOXO-2H-ISOINDOL-2-YL)ACETYL]-L-PROLYLGLYCYL-](http://img.cochemist.com/ccimg/471900/471896-32-9.png)
![GLYCINE, 1-[(1,3-DIHYDRO-1,3-DIOXO-2H-ISOINDOL-2-YL)ACETYL]-L-PROLYLGLYCYL-](http://img.cochemist.com/ccimg/471900/471896-32-9_b.png)
![Glycine, N-[6-(1,3-dihydro-1,3-dioxo-2H-isoindol-2-yl)-1-oxohexyl]glycyl-](http://img.cochemist.com/ccimg/471900/471896-12-5.png)
![Glycine, N-[6-(1,3-dihydro-1,3-dioxo-2H-isoindol-2-yl)-1-oxohexyl]glycyl-](http://img.cochemist.com/ccimg/471900/471896-12-5_b.png)

![6H-[1,4]Thiazino[3,4-a]isoindol-6-one, 3,4-dihydro-](http://img.cochemist.com/ccimg/205600/205593-22-2.png)
![6H-[1,4]Thiazino[3,4-a]isoindol-6-one, 3,4-dihydro-](http://img.cochemist.com/ccimg/205600/205593-22-2_b.png)




![1H-Isoindole-1,3(2H)-dione, 2-[(2-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/172400/172372-20-2.png)
![1H-Isoindole-1,3(2H)-dione, 2-[(2-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/172400/172372-20-2_b.png)


![1H-Isoindol-1-one,3-[(3,4-dimethoxyphenyl)methyl]-2,3-dihydro-3-hydroxy-2-methyl-](http://img.cochemist.com/ccimg/109400/109328-99-6.png)
![1H-Isoindol-1-one,3-[(3,4-dimethoxyphenyl)methyl]-2,3-dihydro-3-hydroxy-2-methyl-](http://img.cochemist.com/ccimg/109400/109328-99-6_b.png)
![Undecanoic acid, 11-[[(1,3-dihydro-1,3-dioxo-2H-isoindol-2-yl)acetyl]amino]-](http://img.cochemist.com/ccimg/102500/102454-05-7.png)
![Undecanoic acid, 11-[[(1,3-dihydro-1,3-dioxo-2H-isoindol-2-yl)acetyl]amino]-](http://img.cochemist.com/ccimg/102500/102454-05-7_b.png)

![1,10b-dihydro-10b-hydroxy-2-methyl-Pyrazino[2,1-a]isoindole-3,6(2H,4H)-dione](http://img.cochemist.com/ccimg/100200/100166-87-8.png)
![1,10b-dihydro-10b-hydroxy-2-methyl-Pyrazino[2,1-a]isoindole-3,6(2H,4H)-dione](http://img.cochemist.com/ccimg/100200/100166-87-8_b.png)
![Glycine,N-[(1,3-dihydro-1,3-dioxo-2H-isoindol-2-yl)acetyl]glycyl- (9CI)](http://img.cochemist.com/ccimg/94000/93923-88-7.png)
![Glycine,N-[(1,3-dihydro-1,3-dioxo-2H-isoindol-2-yl)acetyl]glycyl- (9CI)](http://img.cochemist.com/ccimg/94000/93923-88-7_b.png)


![Acetamide, N-[2-(4-methylphenyl)-2-oxoethyl]-](http://img.cochemist.com/ccimg/74200/74104-57-7.png)
![Acetamide, N-[2-(4-methylphenyl)-2-oxoethyl]-](http://img.cochemist.com/ccimg/74200/74104-57-7_b.png)




![HYDROPEROXIDE, (6,6-DIMETHYLBICYCLO[3.1.1]HEPT-2-EN-2-YL)METHYL](http://img.cochemist.com/ccimg/58500/58434-29-0.png)
![HYDROPEROXIDE, (6,6-DIMETHYLBICYCLO[3.1.1]HEPT-2-EN-2-YL)METHYL](http://img.cochemist.com/ccimg/58500/58434-29-0_b.png)










![1H-Isoindole-1,3(2H)-dione, 2-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/21300/21244-24-6.png)
![1H-Isoindole-1,3(2H)-dione, 2-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/21300/21244-24-6_b.png)
![Glycine, N-[3-(1,3-dihydro-1,3-dioxo-2H-isoindol-2-yl)-1-oxopropyl]-](http://img.cochemist.com/ccimg/17200/17136-19-5.png)
![Glycine, N-[3-(1,3-dihydro-1,3-dioxo-2H-isoindol-2-yl)-1-oxopropyl]-](http://img.cochemist.com/ccimg/17200/17136-19-5_b.png)


![GLYCINE, N-[(1,3-DIHYDRO-1,3-DIOXO-2H-ISOINDOL-2-YL)ACETYL]-N-METHYL-](http://img.cochemist.com/ccimg/13700/13648-29-8.png)
![GLYCINE, N-[(1,3-DIHYDRO-1,3-DIOXO-2H-ISOINDOL-2-YL)ACETYL]-N-METHYL-](http://img.cochemist.com/ccimg/13700/13648-29-8_b.png)











![5H-Pyrrolo[2,1-a]isoindol-5-one, 1,2,3,9b-tetrahydro-9b-hydroxy-](http://img.cochemist.com/ccimg/163000/162971-82-6.png)
![5H-Pyrrolo[2,1-a]isoindol-5-one, 1,2,3,9b-tetrahydro-9b-hydroxy-](http://img.cochemist.com/ccimg/163000/162971-82-6_b.png)