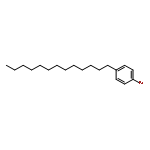
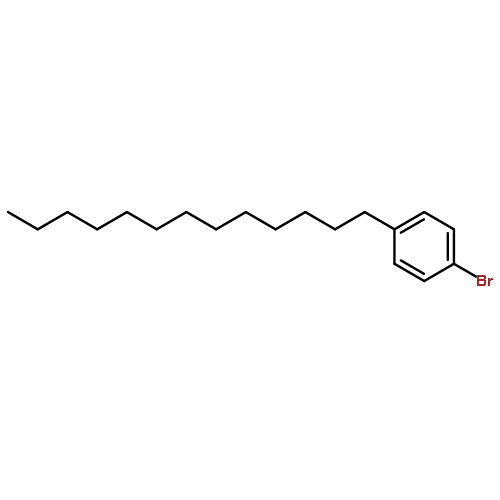
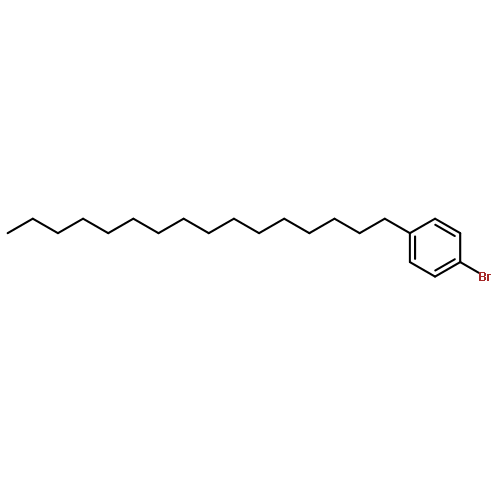
Co-reporter:Masahito Murai, Chisato Mizuta, Ryuji Taniguchi, and Kazuhiko Takai
Organic Letters November 17, 2017 Volume 19(Issue 22) pp:6104-6104
Publication Date(Web):October 30, 2017
DOI:10.1021/acs.orglett.7b02956
The combination of diiodomethylboronate ester, CrCl2 with TMEDA promoted borylcyclopropanation of unactivated alkenes under mild conditions. Compared with the typical Simmons–Smith cyclopropanation, the current protocol offers the following advantages: (1) the reaction proceeds stereoselectively with disubstituted alkenes even without hydroxy or alkoxy groups; (2) both electron-rich and electron-deficient alkenes can be applicable; and (3) the reaction does not require potentially flammable alkylzinc. These unique reactivity features result from the steric and electronic nature of the gem-dichromiomethane intermediates.
Co-reporter:Dr. Masahito Murai;Erika Uemura;Shunsuke Hori; Dr. Kazuhiko Takai
Angewandte Chemie 2017 Volume 129(Issue 21) pp:5956-5960
Publication Date(Web):2017/05/15
DOI:10.1002/ange.201701159
AbstractA regioselective cyclization of 1,n-diynes under rhenium catalysis was developed on the basis of a rare type of 1,1-difunctionalization of terminal alkynes with carbon nucleophiles, followed by sequential addition reactions of the resulting alkenylrhenium species. The reaction provides an efficient approach to the synthesis of complex cyclopentane-fused bi- and tricycles and spirocycles, which are useful building blocks for the construction of essential frameworks of biologically active compounds as well as functional materials, from simple starting materials by the formation of up to six new carbon–carbon bonds in a single step. The reaction proceeds under neutral conditions and does not require external ligands or additives. The key to this reactivity is the unique activation mode of the rhenium carbonyl complex, which prefers to interact with heteroatoms in polar carbon–heteroatom bonds as well as nonpolar carbon–carbon unsaturated π bonds.
Co-reporter:Masahito Murai;Atsushi Nishiyama;Naoki Nishinaka;Haruka Morita
Chemical Communications 2017 vol. 53(Issue 66) pp:9281-9284
Publication Date(Web):2017/08/15
DOI:10.1039/C7CC04296E
While cyclopropanes have been explored as synthetically valuable building blocks, their transformation without conjugated substituents or directly substituted heteroatoms remains challenging. The current study describes the iridium-catalysed ring-opening hydrosilylation of cyclopropanes. A nitrogen-based directing group was found to control the reactivity of iridium active species as well as the regiochemistry of carbon–carbon bond cleavage and hydrosilylation.
Co-reporter:Dr. Masahito Murai;Erika Uemura;Shunsuke Hori; Dr. Kazuhiko Takai
Angewandte Chemie International Edition 2017 Volume 56(Issue 21) pp:5862-5866
Publication Date(Web):2017/05/15
DOI:10.1002/anie.201701159
AbstractA regioselective cyclization of 1,n-diynes under rhenium catalysis was developed on the basis of a rare type of 1,1-difunctionalization of terminal alkynes with carbon nucleophiles, followed by sequential addition reactions of the resulting alkenylrhenium species. The reaction provides an efficient approach to the synthesis of complex cyclopentane-fused bi- and tricycles and spirocycles, which are useful building blocks for the construction of essential frameworks of biologically active compounds as well as functional materials, from simple starting materials by the formation of up to six new carbon–carbon bonds in a single step. The reaction proceeds under neutral conditions and does not require external ligands or additives. The key to this reactivity is the unique activation mode of the rhenium carbonyl complex, which prefers to interact with heteroatoms in polar carbon–heteroatom bonds as well as nonpolar carbon–carbon unsaturated π bonds.
Co-reporter:Sobi Asako, Sae Ishikawa, and Kazuhiko Takai
ACS Catalysis 2016 Volume 6(Issue 5) pp:3387
Publication Date(Web):April 14, 2016
DOI:10.1021/acscatal.6b00627
A simple molybdenum-based catalytic system for hydrosilylation of allenes has been developed. The reactions of mono- and disubstituted allenes with secondary and tertiary silanes proceeded smoothly and selectively to afford linear allylsilanes. The origin of the unprecedented linear selectivity was investigated by density functional theory studies to reveal that the reaction consists of the following steps: (1) concerted hydromolybdation/Si–H oxidative addition from a Mo(CO)4/allene/silane adduct to form (π-allyl)molybdenum, (2) allyl rotation from the initially formed (π-allyl)molybdenum to a thermodynamically more stable isomer, and (3) reductive elimination at the less-hindered allyl carbon to afford a linear allylsilane.Keywords: allenes; allyl rotation; density functional theory; hydrosilylation; linear allylsilanes; molybdenum
Co-reporter:Masahito Murai, Ryo Okada, Atsushi Nishiyama, and Kazuhiko Takai
Organic Letters 2016 Volume 18(Issue 17) pp:4380-4383
Publication Date(Web):August 11, 2016
DOI:10.1021/acs.orglett.6b02134
Use of a rhodium catalyst with (R)-(S)-BPPFA ligand allows efficient synthesis of sila[n]helicenes via dehydrogenative silylation of C–H bonds. By selecting the proper ligands, the current method provides the ability to prepare unsymmetrical sila[n]helicene derivatives without any oxidants. The resulting sila[6]helicene is a rare example of a five-membered ring-fused [6]helicene, which was isolated as a single pure enantiomer without substituents on the terminal benzene rings.
Co-reporter:Shunsuke Hori; Masahito Murai
Journal of the American Chemical Society 2015 Volume 137(Issue 4) pp:1452-1457
Publication Date(Web):January 6, 2015
DOI:10.1021/ja5090755
A novel anti-Markovnikov addition reaction of methanetricarboxylates with terminal acetylenes under neutral conditions was achieved using a rhenium complex. This transformation represents a rare example of intermolecular anti-Markovnikov addition of carbon nucleophiles to unactivated terminal acetylenes. 1,3-Diesters having bulky substituents at the active methylene carbon are also applicable as substrates to provide anti-Markovnikov adducts as single regio- and stereoisomers. Preliminary mechanistic studies imply that the rhenium vinylidene species is the key intermediate in the current catalytic cycle.
Co-reporter:Masahito Murai, Hiroyuki Maekawa, Shino Hamao, Yoshihiro Kubozono, David Roy, and Kazuhiko Takai
Organic Letters 2015 Volume 17(Issue 3) pp:708-711
Publication Date(Web):January 29, 2015
DOI:10.1021/ol503723j
Novel [6]phenacenes (fulminenes) with two long alkyl chains at the axis positions were synthesized. This short synthesis comprises the following three steps: (1) ruthenium-catalyzed direct C–H bond arylation; (2) conversion of directing groups by Wittig reaction; and (3) bismuth- or gold-catalyzed cyclization of vinyl ether. Organic field-effect transistor devices fabricated with a thin film of 3,11-di(tetradecyl)fulminene exhibited typical p-channel normally-off properties.
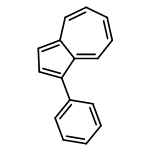
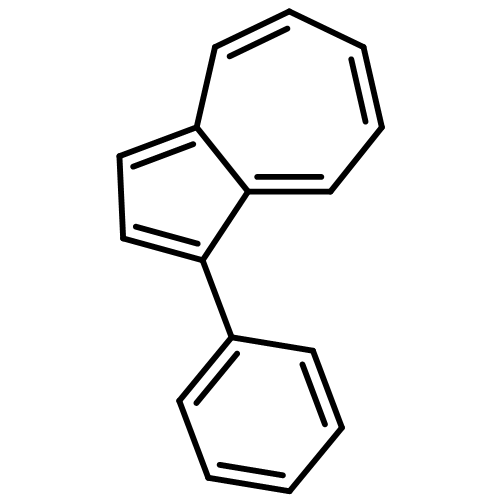
Co-reporter:Masahito Murai, Keishi Takami, Hirotaka Takeshima, and Kazuhiko Takai
Organic Letters 2015 Volume 17(Issue 7) pp:1798-1801
Publication Date(Web):March 24, 2015
DOI:10.1021/acs.orglett.5b00575
Use of an iridium catalyst allowed the efficient dehydrogenative functionalization of C–H bonds of azulenes with the production of hydrogen as the sole byproduct. The reaction occurred with excellent chemo- and regioselectivities to provide 2-silylazulenes even without any directing groups. Effective conjugation through the 2-position of the azulene ring was demonstrated by the unique stimuli-responsiveness against an acid–base reaction.
Co-reporter:Masahito Murai, Koji Matsumoto, Yutaro Takeuchi, and Kazuhiko Takai
Organic Letters 2015 Volume 17(Issue 12) pp:3102-3105
Publication Date(Web):June 10, 2015
DOI:10.1021/acs.orglett.5b01373
Use of a rhodium catalyst with electron-rich and bulky chiral diphosphine ligands having C2-symmetry allowed efficient dehydrogenative silylation of the C(sp2)–H bond of ferrocenes leading to chiral benzosiloloferrocenes. The substrate scope was expanded to hydrogermane and hydrosilanes having a ruthenocene backbone, which resulted in a new approach to benzosilole- and benzogermole-fused metallocenes.
Co-reporter:Takuya Nakagiri, Masahito Murai, and Kazuhiko Takai
Organic Letters 2015 Volume 17(Issue 13) pp:3346-3349
Publication Date(Web):June 12, 2015
DOI:10.1021/acs.orglett.5b01583
The combination of a catalytic amount of Re2O7 and triphenyl phosphite as a reductant is effective for the deoxygenation of unactivated aliphatic epoxides to alkenes. The reaction proceeds stereospecifically with variously substituted epoxides under neutral conditions and is compatible with various functional groups. Protection and deprotection of a double bond functionality using an epoxide are shown as an example of the current rhenium-catalyzed deoxygenation protocol. The effect of reductants for the stereoselectivity has also been studied, indicating that the use of electron-deficient phosphines or phosphites is the key for the stereospecific deoxygenation.
Co-reporter:Dr. Masahito Murai;Keishi Takami ;Dr. Kazuhiko Takai
Chemistry - A European Journal 2015 Volume 21( Issue 12) pp:4566-4570
Publication Date(Web):
DOI:10.1002/chem.201406508
Abstract
This study describes the iridium-catalyzed intermolecular dehydrogenative silylation of C(sp2)H bonds of polycyclic aromatic compounds without directing groups. The reaction produced various arylsilanes through both SiH and CH bond activation, with hydrogen as the sole byproduct. Reactivity was affected by the electronic nature of the aromatic compounds, and silylation of electron-deficient and polycyclic aromatic compounds proceeded efficiently. Site-selectivity was controlled predominantly by steric factors. Therefore, the current functionalization proceeded with opposite chemo- and site-selectivity compared to that observed for general electrophilic functionalization of aromatic compounds.
Co-reporter:Masahito Murai, Hirotaka Takeshima, Haruka Morita, Yoichiro Kuninobu, and Kazuhiko Takai
The Journal of Organic Chemistry 2015 Volume 80(Issue 11) pp:5407-5414
Publication Date(Web):May 11, 2015
DOI:10.1021/acs.joc.5b00920
The current work describes the marked rate of acceleration caused by phosphine ligands on the rhodium-catalyzed dehydrogenative silylation and germylation of unactivated C(sp3)–H bonds. The reactivity was affected by the steric and electronic nature of the phosphine ligands. The use of the bulky and electron-rich diphosphine ligand (R)-DTBM-SEGPHOS was highly effective to yield the dehydrogenative silylation products selectively in the presence of a hydrogen acceptor. An appropriate choice of C2-symmetric chiral diphosphine ligand enables the asymmetric dehydrogenative silylation via the enantioselective desymmetrization of the C(sp3)–H bond. The unprecedented catalytic germylation of C(sp3)–H bonds with dehydrogenation was also examined with the combination of the rhodium complex and a wide bite angle diphosphine ligand to provide the corresponding 2,3-dihydrobenzo[b]germoles in good yield.
Co-reporter:Yusuke Nishida; Naoki Hosokawa; Masahito Murai
Journal of the American Chemical Society 2014 Volume 137(Issue 1) pp:114-117
Publication Date(Web):December 24, 2014
DOI:10.1021/ja5114535
Treatment of gem-di(iodozincio)methane with pyridine or diamine derivatives resulted in the isolation of a storable gem-di(iodozincio)methane species. Use of the sterically bulky bipyridine ligand gave a gem-di(iodozincio)methane complex, which allowed the first X-ray structural analysis of such species. This work represents a rare example of the isolation of an organometallic reactive species in Schlenk equilibrium and thus provides new insight into the design of efficient and storable organometallic reagents. The isolated gem-di(iodozincio)methane complexes serve as effective methylene dianion synthons for olefination of carbonyl compounds.
Co-reporter:Masahito Murai, Kazuki Origuchi, and Kazuhiko Takai
Organic Letters 2014 Volume 16(Issue 14) pp:3828-3831
Publication Date(Web):July 9, 2014
DOI:10.1021/ol501744g
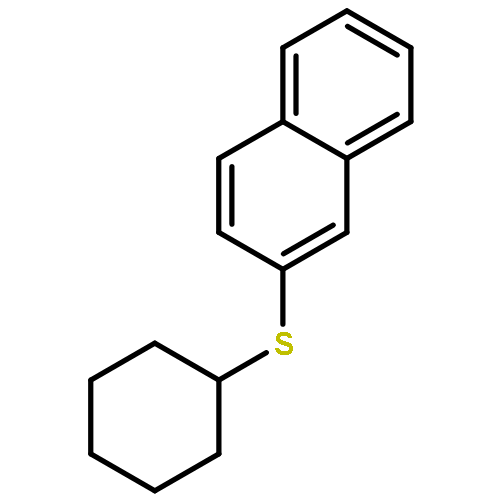
Use of a bismuth catalyst allowed efficient dehydrative substitution of phenolic hydroxy groups with alcohols and thiols to form C–O and C–S bonds. The reaction required equimolar amounts of two readily available substrates that generated H2O as the only byproduct. The relatively mild reaction conditions were compatible with the functional groups selected, and provided excellent chemoselectivity.
Co-reporter:Masahito Murai, Naoki Hosokawa, David Roy, and Kazuhiko Takai
Organic Letters 2014 Volume 16(Issue 16) pp:4134-4137
Publication Date(Web):July 30, 2014
DOI:10.1021/ol5018273
The reaction of 2-(2-arylphenyl)vinyl ethers in the presence of a catalytic amount of bismuth(III) triflate gave substituted phenanthrenes in excellent yields under mild reaction conditions. The reaction was also applied to the construction of other polycyclic aromatic hydrocarbons (PAHs), such as chrysene, helicene, and pyrene having a phenanthrene backbone, via regioselective cyclization. This method has the advantages of easy availability of the cyclization precursors, operational simplicity, and high reaction efficiency.
Co-reporter:Masahito Murai, Koji Matsumoto, Ryo Okada, and Kazuhiko Takai
Organic Letters 2014 Volume 16(Issue 24) pp:6492-6495
Publication Date(Web):December 10, 2014
DOI:10.1021/ol503355q
Rhodium-catalyzed dehydrogenative germylation leading to unsymmetrically functionalized 9-germafluorenes via Ge–H and C–H bond activation is described. Despite the significant achievements made in dehydrogenative functionalization of C–H bonds, only a limited number of examples with the fourth-row atom–H bonds have been reported. The current method enabled the synthesis of various 9-germafluorene derivatives, including tetracyclic as well as donor–acceptor substituted germoles, which may be useful for electronic device applications.
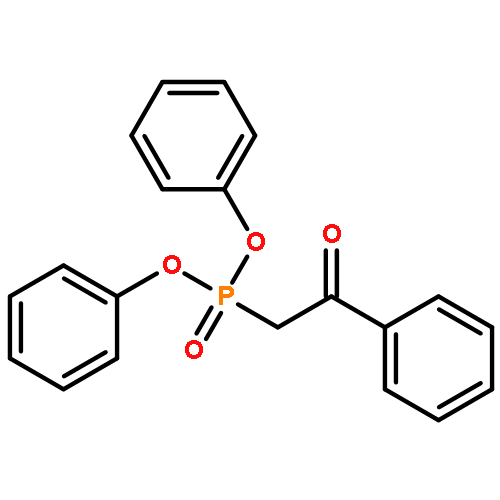
Co-reporter:Masahito Murai, Masahiro Nakamura, and Kazuhiko Takai
Organic Letters 2014 Volume 16(Issue 21) pp:5784-5787
Publication Date(Web):October 24, 2014
DOI:10.1021/ol502859w
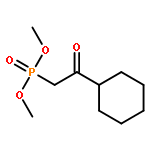
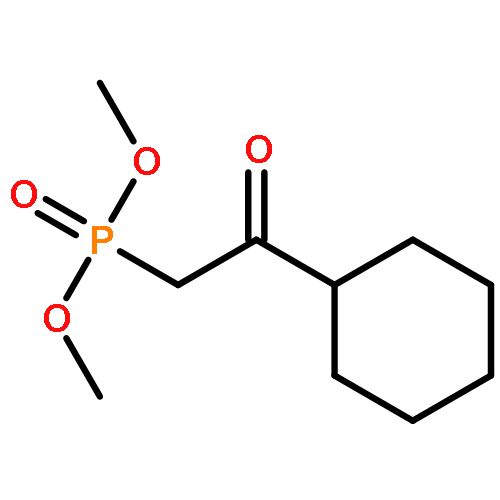
Treatment of β-keto phosphonates (Horner–Wadsworth–Emmons reagents) with terminal alkynes in the presence of a rhenium catalyst gave 2H-1,2-oxaphosphorin 2-oxides with various substitution patterns. The reaction proceeds via two consecutive processes: cleavage of a carbon–carbon σ-bond of the β-keto phosphonate with insertion of the alkyne in a regio- and stereoselective manner, followed by cyclization of the resulting δ-phosphonyl α,β-unsaturated ketone yielding the 2H-1,2-oxaphosphorin 2-oxide. Horner–Wadsworth–Emmons reagents were found to add to nonpolar unsaturated compounds under neutral conditions.
Co-reporter:Yoichiro Kuninobu, Takahiro Nakahara, Hirotaka Takeshima, and Kazuhiko Takai
Organic Letters 2013 Volume 15(Issue 2) pp:426-428
Publication Date(Web):January 10, 2013
DOI:10.1021/ol303353m
The treatment of a variety of hydrosilanes, each incorporating a benzylic C(sp3)–H bond, with a rhodium catalyst resulted in intramolecular dehydrogenative silylation. This silylation reaction was found to occur at typically unreactive C(sp3)–H bonds located at terminal positions on alkyl chains. Interestingly, the rhodium catalyst also promoted regioselective silylation at a site internal to an alkyl chain.
Co-reporter:Dr. Yoichiro Kuninobu;Kanae Yamauchi;Naoya Tamura;Takayuki Seiki;Dr. Kazuhiko Takai
Angewandte Chemie International Edition 2013 Volume 52( Issue 5) pp:1520-1522
Publication Date(Web):
DOI:10.1002/anie.201207723
Co-reporter:Dr. Yoichiro Kuninobu;Takashi Iwanaga;Tetsuya Omura;Dr. Kazuhiko Takai
Angewandte Chemie International Edition 2013 Volume 52( Issue 16) pp:4431-4434
Publication Date(Web):
DOI:10.1002/anie.201210328
Co-reporter:Mitsumi Nishi, Yoichiro Kuninobu, and Kazuhiko Takai
Organic Letters 2012 Volume 14(Issue 23) pp:6116-6118
Publication Date(Web):November 12, 2012
DOI:10.1021/ol302810u
The reaction of α-thioketones and alkynes in the presence of a rhenium catalyst, [HRe(CO)4]n, gave γ-thio-α,β-unsaturated ketones in excellent yields. The alkynes were inserted into the carbon–sulfur bond of the α-thioketones, and isomerization of a double bond provided the products with high regio- and stereoselectivities. This reaction also proceeded in an intramolecular fashion.
Co-reporter:Shun-ichi Yamamoto, Kana Okamoto, Makiko Murakoso, Yoichiro Kuninobu, and Kazuhiko Takai
Organic Letters 2012 Volume 14(Issue 12) pp:3182-3185
Publication Date(Web):2017-2-22
DOI:10.1021/ol301273j
A new method is described for the regioselective synthesis of multisubstituted pyridine derivatives. Treatment of N-acetyl β-enamino ketones with alkynes in the presence of the rhenium catalyst, Re2(CO)10, gives multisubstituted pyridines regioselectively. In this reaction, the N-acetyl moieties are important for the selective formation of the multisubstituted pyridines. This reaction proceeds via insertion of alkynes into a carbon–carbon single bond of β-enamino ketones, intramolecular nucleophilic cyclization, and elimination of acetic acid.
Co-reporter:Yoichiro Kuninobu and Kazuhiko Takai
Chemical Reviews 2011 Volume 111(Issue 3) pp:1938
Publication Date(Web):November 23, 2010
DOI:10.1021/cr100241u
Co-reporter:Yoichiro Kuninobu, Hironori Matsuzaki, Mitsumi Nishi, and Kazuhiko Takai
Organic Letters 2011 Volume 13(Issue 11) pp:2959-2961
Publication Date(Web):May 9, 2011
DOI:10.1021/ol2008507
Treatment of β-keto sulfones with terminal alkynes gave unsaturated δ-keto sulfones in good to excellent yields under rhenium catalysis. In this reaction, the insertion of the alkynes into the nonstrained carbon–carbon single bond between the α- and β-positions of the β-keto sulfones proceeded smoothly, and (Z)-isomers were produced with high regio- and stereoselectivities.
Co-reporter:Yoichiro Kuninobu, Kazuhiro Ohta and Kazuhiko Takai
Chemical Communications 2011 vol. 47(Issue 38) pp:10791-10793
Publication Date(Web):05 Sep 2011
DOI:10.1039/C1CC12359A
We have succeeded in the allylation of aromatic and olefinic C–H bonds of benzoic and acrylic acids using a rhenium catalyst, Re2(CO)10. In this reaction, isomerization of the introduced allyl group to the 1-propenyl group did not occur.
Co-reporter:Yoichiro Kuninobu, Takahiro Nakahara, Peng Yu, Kazuhiko Takai
Journal of Organometallic Chemistry 2011 696(1) pp: 348-351
Publication Date(Web):
DOI:10.1016/j.jorganchem.2010.09.064
Co-reporter:Dr. Yoichiro Kuninobu;Tadamasa Uesugi;Dr. Atsushi Kawata ;Dr. Kazuhiko Takai
Angewandte Chemie International Edition 2011 Volume 50( Issue 44) pp:10406-10408
Publication Date(Web):
DOI:10.1002/anie.201104704
Co-reporter:Yoichiro Kuninobu, Tomohiro Tatsuzaki, Takashi Matsuki, and Kazuhiko Takai
The Journal of Organic Chemistry 2011 Volume 76(Issue 17) pp:7005-7009
Publication Date(Web):July 18, 2011
DOI:10.1021/jo200861s
Polycyclic aromatic compounds can be synthesized from 2-benzylic- or 2-allylbenzaldehydes using a catalytic amount of In(III) or Re(I) complexes. By using this method, polycyclic aza-aromatic compounds can also be prepared efficiently. In these reactions, only water is formed as a side product.
Co-reporter:Yoichiro Kuninobu, Takuya Yoshida, and Kazuhiko Takai
The Journal of Organic Chemistry 2011 Volume 76(Issue 18) pp:7370-7376
Publication Date(Web):August 5, 2011
DOI:10.1021/jo201030j
Dibenzophosphole oxides were obtained from secondary hydrophosphine oxides with a biphenyl group by dehydrogenation via phosphine–hydrogen and carbon–hydrogen bond cleavage in the presence of a catalytic amount of palladium(II) acetate, Pd(OAc)2. By using this reaction, a ladder-type dibenzophosphole oxide could also be synthesized by double intramolecular dehydrogenative cyclization.
Co-reporter:Tomonari Ureshino ; Takuya Yoshida ; Yoichiro Kuninobu
Journal of the American Chemical Society 2010 Volume 132(Issue 41) pp:14324-14326
Publication Date(Web):September 24, 2010
DOI:10.1021/ja107698p
The rhodium-catalyzed synthesis of silafluorenes from biphenylhydrosilanes is described. This highly efficient reaction proceeds via both Si−H and C−H bond activation, producing only H2 as a side product. Using this method, a ladder-type bis-silicon-bridged p-terphenyl could also be synthesized.
Co-reporter:Yoichiro Kuninobu, Takashi Matsuki and Kazuhiko Takai
Organic Letters 2010 Volume 12(Issue 13) pp:2948-2950
Publication Date(Web):June 7, 2010
DOI:10.1021/ol100947p
By heating aryl aldehydes with catalytic amounts of a rhenium complex, ReBr(CO)5, and N-phenylacetamide in toluene, indenone derivatives are obtained in good to excellent yields. This reaction proceeds via (1) the formation of an isobenzofuran derivative by the insertion of an aldehyde into the C−H bond of another aldehyde (C−H bond activation) and successive intramolecular nucleophilic cyclization, (2) nucleophilic addition of the formed isobenzofuran derivative to the third aldehyde, (3) isomerization, and (4) intramolecular aldol condensation.
Co-reporter:Yoichiro Kuninobu, Tomonari Ureshino, Shun-ichi Yamamoto and Kazuhiko Takai
Chemical Communications 2010 vol. 46(Issue 29) pp:5310-5312
Publication Date(Web):14 Jun 2010
DOI:10.1039/C0CC00243G
We have succeeded in formal regioselective functionalization of alkanes by iridium-catalyzed dehydrogenation, hydrozirconation of the resulting alkenes, and electrophilic reaction of the generated alkylzirconium intermediate.
Co-reporter:Yoichiro Kuninobu, Peng Yu, and Kazuhiko Takai
Organic Letters 2010 Volume 12(Issue 19) pp:4274-4276
Publication Date(Web):August 30, 2010
DOI:10.1021/ol101627x
Aminoindane derivatives were synthesized diastereoselectively by the treatment of aromatic imines with allenes in the presence of a catalytic amount of a rhenium complex, [HRe(CO)4]n. The allenes inserted into the aromatic C−H bonds.
Co-reporter:Yoichiro Kuninobu, Takayuki Seiki, Shunsuke Kanamaru, Yuta Nishina, and Kazuhiko Takai
Organic Letters 2010 Volume 12(Issue 22) pp:5287-5289
Publication Date(Web):October 20, 2010
DOI:10.1021/ol102349r
The synthesis of unsymmetric functionalized pentacenes from 1,4-anthraquinones and functionalized isobenzofurans, which were prepared by transformation via C−H bond activation, was successfully accomplished. Examples of the synthesis of pentacenes with functional groups at the 5-position are still rare. These obtained functionalized pentacenes are highly soluble in hexane, toluene, and THF.
Co-reporter:Yoichiro Kuninobu, Mitsumi Nishi and Kazuhiko Takai
Chemical Communications 2010 vol. 46(Issue 46) pp:8860-8862
Publication Date(Web):21 Oct 2010
DOI:10.1039/C0CC03781H
Treatment of tertiary amines with diazoacetate in the presence of a catalytic amount of an iron salt, FeCl3, in ethanol gave glycine derivatives. In this reaction, a carbon–nitrogen single bond of the amine was cleaved.
Co-reporter:Yoichiro Kuninobu, Mitsumi Nishi, Atsushi Kawata, Hisatsugu Takata, Yumi Hanatani, Yudha S. Salprima, Aya Iwai and Kazuhiko Takai
The Journal of Organic Chemistry 2010 Volume 75(Issue 2) pp:334-341
Publication Date(Web):December 9, 2009
DOI:10.1021/jo902072q
We have succeeded in the development of three approaches to the synthesis of aromatic compounds from 1,3-dicarbonyl compounds and alkynes. The first approach is a manganese-catalyzed [2+2+2] cycloaddition between 1,3-dicarbonyl compounds, which have no substituents at the active methylene moiety, and terminal alkynes. This reaction proceeds with high regioselectivity when aryl acetylenes are employed as the alkyne component. The second approach is a rhenium- or manganese-catalyzed formal [2+1+2+1] cycloaddition between β-keto esters and two kinds of alkynes. In this reaction, the aromatic compounds are obtained by the following reaction sequence: (1) insertion of the first alkyne into a carbon−carbon single bond of a β-keto ester, (2) formation of 2-pyranones via intramolecular cyclization with the elimination of ethanol, and (3) Diels−Alder reaction between the formed 2-pyranone and the second alkyne. This reaction provides multisubstituted aromatic compounds in a regioselective manner. The third approach is a rhenium-catalyzed formal [2+2+1+1] cycloaddition reaction from two 1,3-diketones and one alkyne. In this reaction, the aromatic skeleton is constructed from three carbons of the first 1,3-diketone, two carbons of the alkyne, and one carbon of the second 1,3-diketone.
Co-reporter:Yoichiro Kuninobu ; Takashi Matsuki
Journal of the American Chemical Society 2009 Volume 131(Issue 29) pp:9914-9915
Publication Date(Web):July 1, 2009
DOI:10.1021/ja904360k
Treatment of phenol derivatives with terminal alkenes in the presence of a catalytic amount of a rhenium complex, Re2(CO)10, gave monoalkylated phenol derivatives in good to excellent yields. This reaction proceeds at the ortho- or para-position of phenols regioselectively.
Co-reporter:Yoichiro Kuninobu, Yasuo Fujii, Takashi Matsuki, Yuta Nishina and Kazuhiko Takai
Organic Letters 2009 Volume 11(Issue 12) pp:2711-2714
Publication Date(Web):May 26, 2009
DOI:10.1021/ol900962v
Treatment of olefins bearing a directing group with α,β-unsaturated carbonyl compounds, alkynes, or aldehydes in the presence of a catalytic amount of a rhenium complex, [ReBr(CO)3(thf)]2 gave γ,δ-unsaturated carbonyl compounds, dienes, and allyl silyl ethers, respectively. This reaction proceeds via C−H bond activation, insertion of unsaturated molecules into the formed rhenium−carbon bond, and then reductive elimination (or transmetalation in the case of aldehydes).
Co-reporter:Yoichiro Kuninobu, Junya Morita, Mitsumi Nishi, Atsushi Kawata and Kazuhiko Takai
Organic Letters 2009 Volume 11(Issue 12) pp:2535-2537
Publication Date(Web):May 20, 2009
DOI:10.1021/ol900772h
Treatment of cyclic β-keto esters with terminal alkynes in the presence of a catalytic amount of a rhenium complex, [ReBr(CO)3(thf)]2, gave bicyclo[3.3.1]nonene derivatives. The reaction conditions and yields of the bicyclo[3.3.1]nonenes were improved by the sequential use of tetrabutylammonium fluoride (TBAF) after the rhenium-catalyzed reactions.
Co-reporter:Yoichiro Kuninobu ; Yuta Nishina ; Takashi Matsuki
Journal of the American Chemical Society 2008 Volume 130(Issue 43) pp:14062-14063
Publication Date(Web):October 1, 2008
DOI:10.1021/ja805921f
Treatment of an α,β-unsaturated ketimine with an α,β-unsaturated carbonyl compound in the presence of a rhenium complex, Re2(CO)10, gave a cyclopentadienyl−rhenium complex. This reaction proceeds via rhenium-catalyzed C−H bond activation of an olefinic C−H bond, insertion of an α,β-unsaturated carbonyl compound into a Re−C bond of the alkenylrhenium intermediate, intramolecular nucleophilic cyclization, reductive elimination, elimination of aniline to give a cyclopentadiene derivative, followed by the formation of a cyclopentadienyl−rhenium complex from the cyclopentadiene derivative and the rhenium complex.
Co-reporter:Yoichiro Kuninobu, Atsushi Kawata, Mitsumi Nishi, Hisatsugu Takata and Kazuhiko Takai
Chemical Communications 2008 (Issue 47) pp:6360-6362
Publication Date(Web):24 Oct 2008
DOI:10.1039/B814694B
Rhenium- and manganese-catalyzed reactions between β-keto esters and acetylenes, followed by treatment with tetrabutylammonium fluoride, gave 2-pyranone derivatives regioselectively.
Co-reporter:Salprima YudhaS. Dr.;Yoichiro Kuninobu Dr. Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 48) pp:9318-9321
Publication Date(Web):
DOI:10.1002/anie.200803350
Co-reporter:Yoichiro Kuninobu Dr.;Yuta Nishina;Takahiro Takeuchi Dr.
Angewandte Chemie 2007 Volume 119(Issue 34) pp:
Publication Date(Web):24 JUL 2007
DOI:10.1002/ange.200702256
Mn kommt ins Spiel: In Gegenwart eines Mangankatalysators und einer stöchiometrischen Menge Hydrosilan inserieren Aldehyde in Aryl-C-H-Bindungen von Verbindungen mit dirigierenden Gruppen (siehe Schema). Diese erste mangankatalysierte C-H-Aktivierung liefert Silylether in guten bis hervorragenden Ausbeuten und kann auch auf asymmetrische Umwandlungen angewendet werden.
Co-reporter:Atsushi Kawata;Kazumi Takata;Yoichiro Kuninobu Dr. Dr.
Angewandte Chemie 2007 Volume 119(Issue 41) pp:
Publication Date(Web):4 SEP 2007
DOI:10.1002/ange.200702798
Retroaldolreaktion: Die Indium-katalysierte Reaktion eines 1,3-Diketons mit einem Alkohol verläuft lösungsmittelfrei über einen nucleophilen Angriff des Alkohols an die Carbonylgruppe des 1,3-Diketons sowie eine C-C-Bindungsspaltung durch eine Retro-Claisen-Kondensation und liefert einen Ester in hoher Ausbeute (siehe Schema). Mit Wasser oder einem Amin als Nucleophil erhält man eine Carbonsäure bzw. ein Amid.
Co-reporter:Yoichiro Kuninobu Dr.;Yuta Nishina;Takahiro Takeuchi Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 34) pp:
Publication Date(Web):24 JUL 2007
DOI:10.1002/anie.200702256
Mn gets in the game: In the presence of a manganese catalyst and a stoichiometric amount of hydrosilane, aldehydes insert into CH bonds of aromatic rings of compounds with directing groups (see scheme). This first example of a manganese-catalyzed chemical transformation through CH bond activation gives silyl ethers in good to excellent yields and can also be applied to asymmetric transformation.
Co-reporter:Yoichiro Kuninobu Dr.;Eri Ishii Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 18) pp:
Publication Date(Web):23 MAR 2007
DOI:10.1002/anie.200700183
Very couply! Coupling reactions of propargyl or benzyl alcohols with allyl- or alkynylsilanes proceed efficiently using [{ReBr(CO)3(thf)}2] as catalyst. Additionally, diethynylmethane derivatives were obtained by the reaction of aromatic aldehydes with trimethyl(phenylethynyl)silane in the presence of both the rhenium complex and AuCl (see scheme).
Co-reporter:Atsushi Kawata;Kazumi Takata;Yoichiro Kuninobu Dr. Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 41) pp:
Publication Date(Web):4 SEP 2007
DOI:10.1002/anie.200702798
Retro-aldol reaction: Indium-catalyzed reaction of a 1,3-diketone with an alcohol proceeds under solvent-free conditions by nucleophilic attack of the alcohol on a carbonyl group of the 1,3-diketone and carbon–carbon bond cleavage by a retro-Claisen condensation to give an ester in high yield (see scheme). Using water and an amine as nucleophiles instead of an alcohol gave the corresponding carboxylic acid and amide.
Co-reporter:Yoichiro Kuninobu Dr.;Eri Ishii Dr.
Angewandte Chemie 2007 Volume 119(Issue 18) pp:
Publication Date(Web):23 MAR 2007
DOI:10.1002/ange.200700183
Kupplungskünstler: Katalytische Kupplungen zwischen Propargyl- oder Benzylalkoholen und Allyl- oder Alkinylsilanen gelingen effizient mit [{ReBr(CO)3(thf)}2]. In Gegenwart dieses Rheniumkomplexes und von AuCl wurden durch die Reaktion von Arylaldehyden mit Trimethyl(phenylethinyl)silan auch Diethinylmethan-Derivate erhalten (siehe Schema).
Co-reporter:Yoichiro Kuninobu, Shuhei Nishimura and Kazuhiko Takai
Organic & Biomolecular Chemistry 2006 vol. 4(Issue 2) pp:203-205
Publication Date(Web):13 Dec 2005
DOI:10.1039/B516916J
By the reactions of ketimines bearing a pyridyl or a picolyl group on a nitrogen atom of the imine moiety with tosylisocyanate, 4H-pyrido[1,2-a]pyrimidin-4-one derivatives could be obtained in quantitative yields. In these reactions, tosylisocyanate acts as a carbonyl precursor. The pyridyl or picolyl group is a key functional group because it is not only the constituent structure of the 4H-pyrido[1,2-a]pyrimidin-4-one framework but also the promoter of the formation of a ketene intermediate.
Co-reporter:Yoichiro Kuninobu Dr.;Yuta Nishina;Makoto Shouho Dr.
Angewandte Chemie 2006 Volume 118(Issue 17) pp:
Publication Date(Web):23 MAR 2006
DOI:10.1002/ange.200503627
Einer der Reaktionsschritte der rheniumkatalysierten Umsetzung von aromatischen Ketiminen mit Ethylacrylat, die in guten Ausbeuten die Indenderivate ergibt, ist eine C-H-Aktivierung. Die Indenderivate sind auch aus aromatischen Ketonen und α,β-ungesättigten Estern in Gegenwart eines Rheniumkomplex-Katalysators und p-Anisidin erhältlich (siehe Schema).
Co-reporter:Yoichiro Kuninobu Dr.;Yuta Nishina;Makoto Shouho Dr.
Angewandte Chemie International Edition 2006 Volume 45(Issue 17) pp:
Publication Date(Web):23 MAR 2006
DOI:10.1002/anie.200503627
Several reaction steps, including CH activation, lead to indene derivatives in good yields in the rhenium-catalyzed reaction of aromatic ketimines and ethyl acrylate. Indene derivatives can also be obtained by the reactions of aromatic ketones with α,β-unsaturated esters in the presence of a catalytic rhenium complex and p-anisidine (see scheme).
Co-reporter:Ryotaro Morita;Hiroshi Matsushita;Chika Toratsu
Chirality 2003 Volume 15(Issue 1) pp:17-23
Publication Date(Web):20 NOV 2002
DOI:10.1002/chir.10148
Treatment of an α,β-unsaturated ketone and an aldehyde with chromium(II) chloride and R3SiCl in DMF gives cross-pinacol-type coupling products, 1,2-diols selectively. The anti/syn ratios of the produced 1,2-diols are shown to depend on the reaction temperature: At lower temperatures, the anti adduct is produced selectively, but at higher temperatures, the anti/syn ratios gradually decrease. When a combination of manganese and a catalytic amount of lead are used instead of chromium(II), 1,6-diketone, a dimer of the α,β-unsaturated ketone, is produced selectively. Chirality 15:17–23, 2003. © 2002 Wiley-Liss, Inc.
Co-reporter:Kazuhiko Takai, Ryo Kokumai and Takahumi Nobunaka
Chemical Communications 2001 (Issue 12) pp:1128-1129
Publication Date(Web):31 May 2001
DOI:10.1039/B102387J
Treatment of a carbonate ester of 2,2,2-trichloroethanol
derivative with CrCl2–DMF in THF gives a
β-carbonate-coordinated geminal dichromium species, which adds to an
aldehyde and eliminates an acyloxychromium group to afford a
(Z)-2-chloroalk-2-en-1-ol stereoselectively.
Co-reporter:Kazuhiko Takai Dr.;Ryotaro Morita;Chika Toratsu
Angewandte Chemie 2001 Volume 113(Issue 6) pp:
Publication Date(Web):14 MAR 2001
DOI:10.1002/1521-3757(20010316)113:6<1150::AID-ANGE11500>3.0.CO;2-V
Co-reporter:Chika Toratsu;Takafumi Fujii;Takuma Suzuki Dr.
Angewandte Chemie 2000 Volume 112(Issue 15) pp:
Publication Date(Web):2 AUG 2000
DOI:10.1002/1521-3757(20000804)112:15<2837::AID-ANGE2837>3.0.CO;2-6
Co-reporter:Kazuhiko Takai;Naoto Matsukawa;Akira Takahashi;Takafumi Fujii
Angewandte Chemie International Edition 1998 Volume 37(Issue 1‐2) pp:
Publication Date(Web):17 DEC 1998
DOI:10.1002/(SICI)1521-3773(19980202)37:1/2<152::AID-ANIE152>3.0.CO;2-8
The mild reductant chromium(II) chloride can discriminate between alkyl iodides, alkyl radicals, and allylic radicals. In the presence of CrII the alkyl radical is so long-lived that it can undergo intermolecular addition to a 1,3-diene. Rapid one-electron reduction of the formed allylic radical results in an allylic chromium(II) species, which adds to an aldehyde to afford a three-component coupling product [see, for example, Eq. (a)].
Co-reporter:Kazuhiko Takai;Naoto Matsukawa;Akira Takahashi;Takafumi Fujii
Angewandte Chemie 1998 Volume 110(Issue 1‐2) pp:
Publication Date(Web):12 MAR 1999
DOI:10.1002/(SICI)1521-3757(19980116)110:1/2<160::AID-ANGE160>3.0.CO;2-7
Als mildes Reduktionsmittel ist Chrom(II) in der Lage, zwischen Alkyliodiden, Alkylradikalen und Allylradikalen zu diskriminieren: Das Alkylradikal ist in Gegenwart von CrII so langlebig, daß es mit einem 1,3-Dien reagieren kann, und das dabei entstehende Allylradikal bildet nach rascher Einelektronenreduktion eine Allylchrom(II)-Spezies, die an einen Aldehyd unter Bildung des Drei-Komponenten-Produkts addiert [siehe z. B. Gl. (a)].
Co-reporter:Yoichiro Kuninobu, Mitsumi Nishi and Kazuhiko Takai
Chemical Communications 2010 - vol. 46(Issue 46) pp:NaN8862-8862
Publication Date(Web):2010/10/21
DOI:10.1039/C0CC03781H
Treatment of tertiary amines with diazoacetate in the presence of a catalytic amount of an iron salt, FeCl3, in ethanol gave glycine derivatives. In this reaction, a carbon–nitrogen single bond of the amine was cleaved.
Co-reporter:Yoichiro Kuninobu, Tomonari Ureshino, Shun-ichi Yamamoto and Kazuhiko Takai
Chemical Communications 2010 - vol. 46(Issue 29) pp:NaN5312-5312
Publication Date(Web):2010/06/14
DOI:10.1039/C0CC00243G
We have succeeded in formal regioselective functionalization of alkanes by iridium-catalyzed dehydrogenation, hydrozirconation of the resulting alkenes, and electrophilic reaction of the generated alkylzirconium intermediate.
Co-reporter:Kazuhiko Takai, Noriko Katsura and Yuji Kunisada
Chemical Communications 2001(Issue 18) pp:NaN1725-1725
Publication Date(Web):2001/08/13
DOI:10.1039/B105357B
Chromioenamines can be generated by treatment of O-acetyloximes with chromium(II) via two steps of one-electron reduction and successive isomerization, and the species react with aldehydes to give γ-amino alcohols after reduction with LiAlH4.
Co-reporter:Yoichiro Kuninobu, Kazuhiro Ohta and Kazuhiko Takai
Chemical Communications 2011 - vol. 47(Issue 38) pp:NaN10793-10793
Publication Date(Web):2011/09/05
DOI:10.1039/C1CC12359A
We have succeeded in the allylation of aromatic and olefinic C–H bonds of benzoic and acrylic acids using a rhenium catalyst, Re2(CO)10. In this reaction, isomerization of the introduced allyl group to the 1-propenyl group did not occur.
Co-reporter:Yoichiro Kuninobu, Atsushi Kawata, Mitsumi Nishi, Hisatsugu Takata and Kazuhiko Takai
Chemical Communications 2008(Issue 47) pp:NaN6362-6362
Publication Date(Web):2008/10/24
DOI:10.1039/B814694B
Rhenium- and manganese-catalyzed reactions between β-keto esters and acetylenes, followed by treatment with tetrabutylammonium fluoride, gave 2-pyranone derivatives regioselectively.

![Benzene, [(3-butyl-2-hepten-1-yl)dimethylsilyl]-](/data/chemimg/3722300/1903772-82-6.png)
![Benzene, [(3-butyl-2-hepten-1-yl)dimethylsilyl]-](/data/chemimg/3722300/1903772-82-6_b.png)
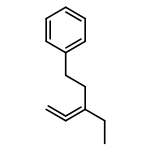

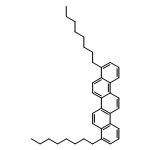
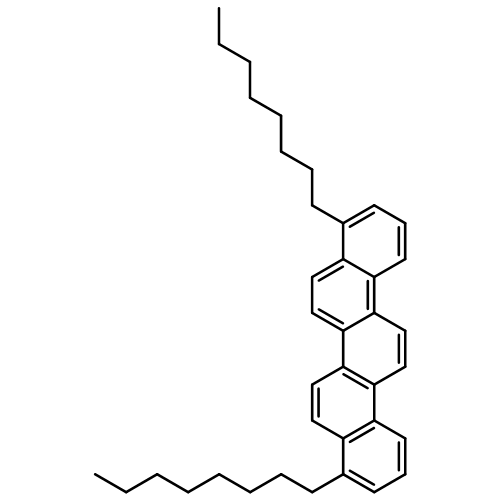
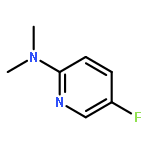
![Benzenamine, 4-methyl-N-[(2-octylphenyl)methylene]-](/data/chemimg/3744000/1648892-69-6.png)
![Benzenamine, 4-methyl-N-[(2-octylphenyl)methylene]-](/data/chemimg/3744000/1648892-69-6_b.png)


![Silane, [(2Z)-3-cyclohexyl-2-propenyl]dimethylphenyl-](http://img.cochemist.com/ccimg/868200/868169-40-8.png)
![Silane, [(2Z)-3-cyclohexyl-2-propenyl]dimethylphenyl-](http://img.cochemist.com/ccimg/868200/868169-40-8_b.png)


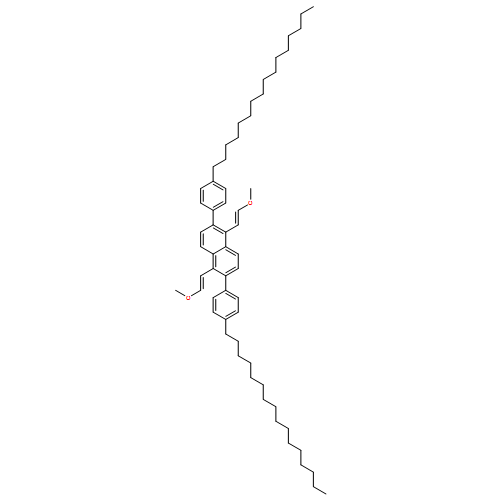
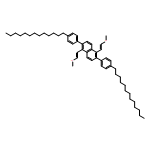
![Naphthalene, 2-[2,6-bis[(1Z)-2-methoxyethenyl]phenyl]-](http://img.cochemist.com/ccimg/653000/652977-01-0.png)
![Naphthalene, 2-[2,6-bis[(1Z)-2-methoxyethenyl]phenyl]-](http://img.cochemist.com/ccimg/653000/652977-01-0_b.png)
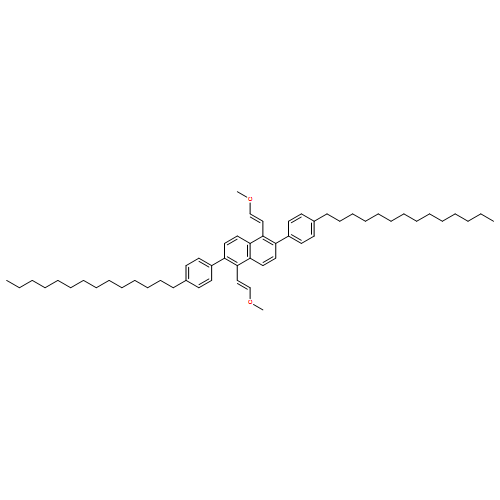
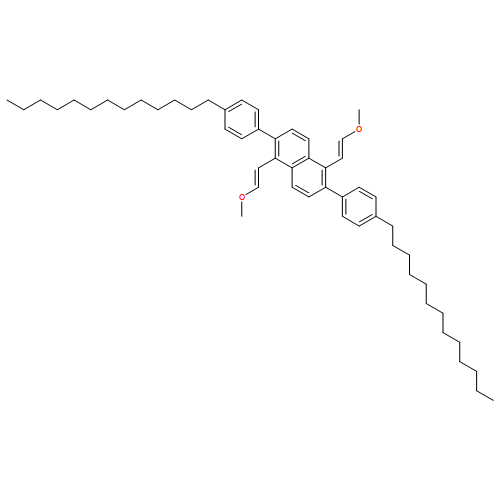
![Naphthalene, 2-[2,6-bis[(1E)-2-methoxyethenyl]phenyl]-](http://img.cochemist.com/ccimg/653000/652977-00-9.png)
![Naphthalene, 2-[2,6-bis[(1E)-2-methoxyethenyl]phenyl]-](http://img.cochemist.com/ccimg/653000/652977-00-9_b.png)

![Phosphine,1,1'-[(1R)-6,6'-dimethoxy[1,1'-biphenyl]-2,2'-diyl]bis[1,1-bis(3,4,5-trimethoxyphenyl)-](/data/chemimg/478300/256390-47-3.png)
![Phosphine,1,1'-[(1R)-6,6'-dimethoxy[1,1'-biphenyl]-2,2'-diyl]bis[1,1-bis(3,4,5-trimethoxyphenyl)-](/data/chemimg/478300/256390-47-3_b.png)

![[1,1':3',1''-Terphenyl]-2'-carboxaldehyde](http://img.cochemist.com/ccimg/169700/169618-84-2.png)
![[1,1':3',1''-Terphenyl]-2'-carboxaldehyde](http://img.cochemist.com/ccimg/169700/169618-84-2_b.png)
![(R)-1-[(1S)-2-(DIPHENYLPHOSPHINO)FERROCENYL]ETHYLDI-TERT-BUTYLPHOSPHINE](http://img.cochemist.com/ccimg/155900/155830-69-6.png)
![(R)-1-[(1S)-2-(DIPHENYLPHOSPHINO)FERROCENYL]ETHYLDI-TERT-BUTYLPHOSPHINE](http://img.cochemist.com/ccimg/155900/155830-69-6_b.png)







![Benzene, [(3E)-5-methyl-3-hexenyl]-](http://img.cochemist.com/ccimg/96100/96025-23-9.png)
![Benzene, [(3E)-5-methyl-3-hexenyl]-](http://img.cochemist.com/ccimg/96100/96025-23-9_b.png)
![[1,1'-Biphenyl]-2-acetaldehyde](http://img.cochemist.com/ccimg/90500/90401-64-2.png)
![[1,1'-Biphenyl]-2-acetaldehyde](http://img.cochemist.com/ccimg/90500/90401-64-2_b.png)















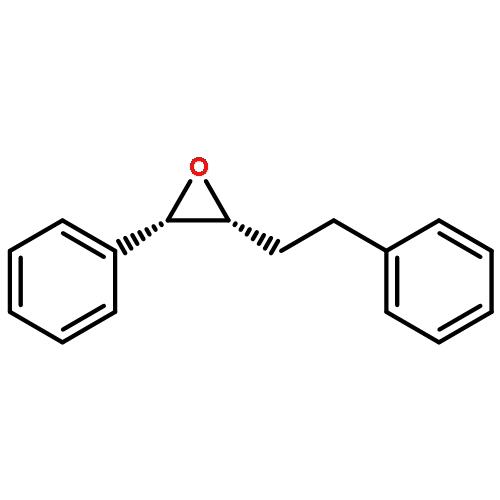
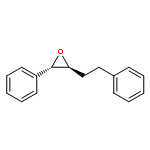
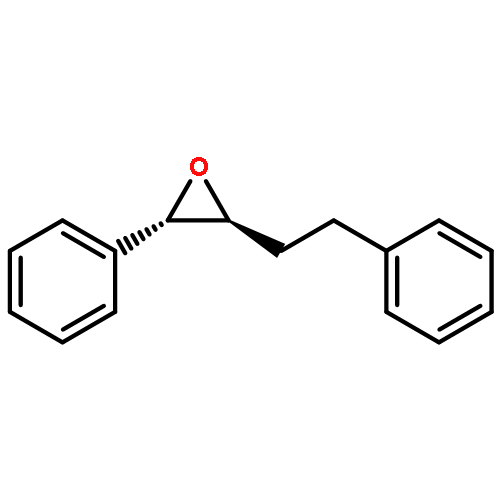

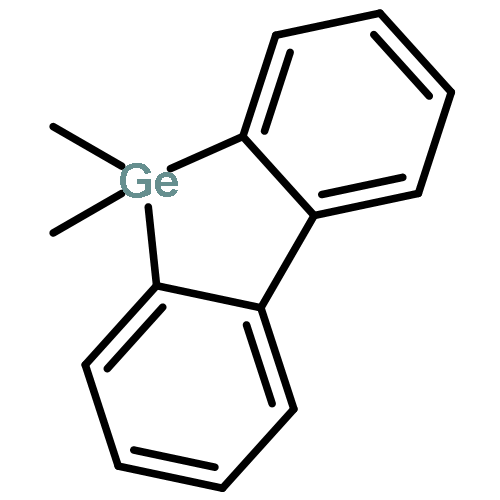

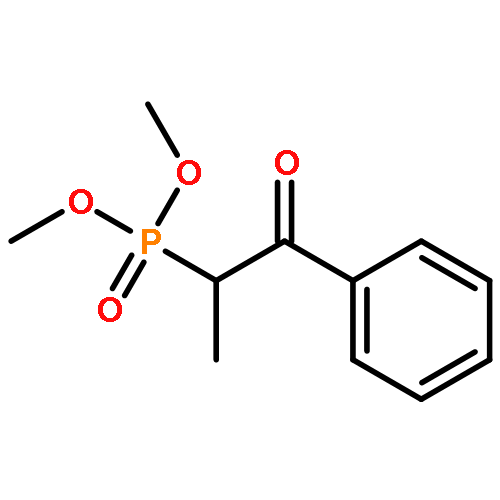



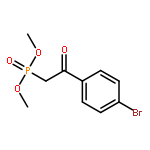
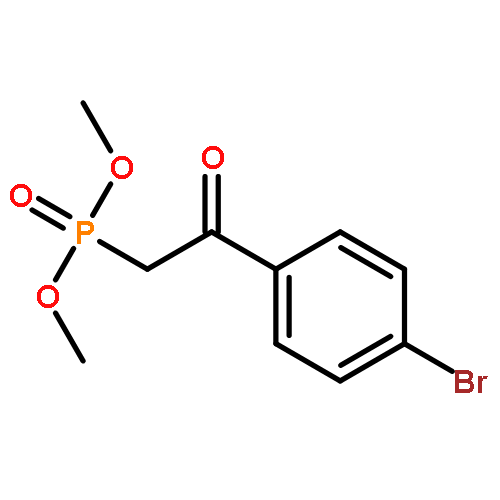
![PHOSPHONIC ACID, [2-(4-METHOXYPHENYL)-2-OXOETHYL]-, DIMETHYL ESTER](http://img.cochemist.com/ccimg/57100/57057-03-1.png)
![PHOSPHONIC ACID, [2-(4-METHOXYPHENYL)-2-OXOETHYL]-, DIMETHYL ESTER](http://img.cochemist.com/ccimg/57100/57057-03-1_b.png)




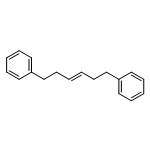
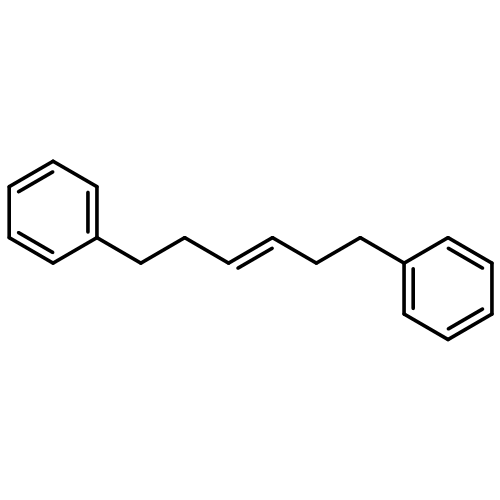


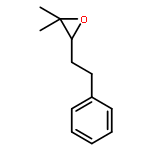
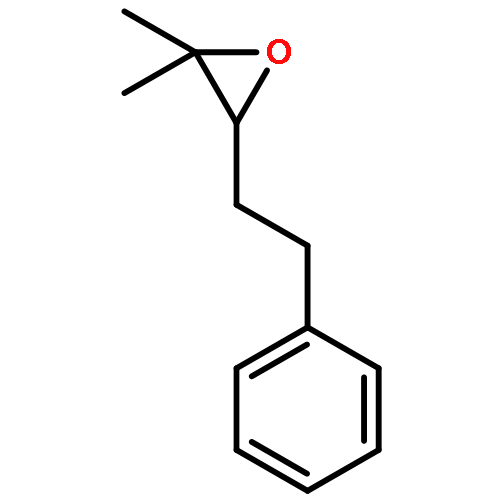
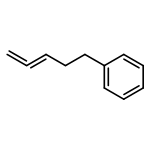
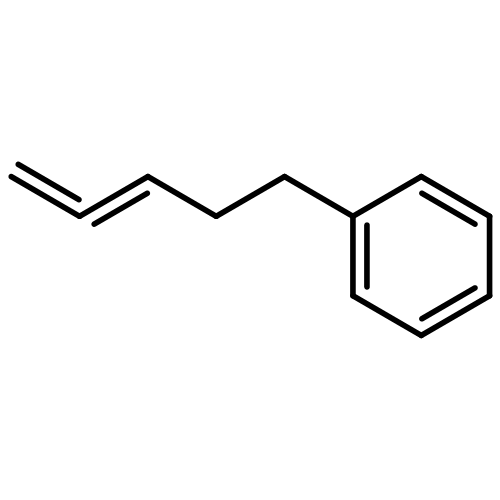

![Benzene, 1,1'-[(1E)-1-butene-1,4-diyl]bis-](/data/chemimg/1171700/27066-35-9.png)
![Benzene, 1,1'-[(1E)-1-butene-1,4-diyl]bis-](/data/chemimg/1171700/27066-35-9_b.png)









![Benzo[b]thiophen-5-ol](http://img.cochemist.com/ccimg/19400/19301-35-0.png)
![Benzo[b]thiophen-5-ol](http://img.cochemist.com/ccimg/19400/19301-35-0_b.png)
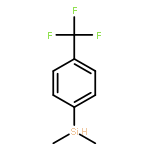

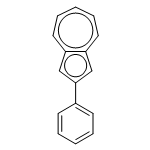
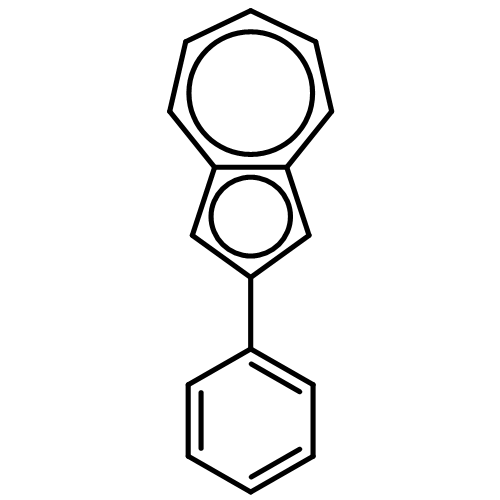
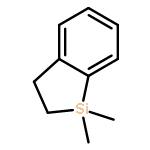
![[1,1'-Biphenyl]-2-carboxaldehyde,4'-methoxy-](http://img.cochemist.com/ccimg/16100/16064-04-3.png)
![[1,1'-Biphenyl]-2-carboxaldehyde,4'-methoxy-](http://img.cochemist.com/ccimg/16100/16064-04-3_b.png)






![Rhodium, bis[(1,2,5,6-h)-1,5-cyclooctadiene]di-m-methoxydi-](http://img.cochemist.com/ccimg/12200/12148-72-0.png)
![Rhodium, bis[(1,2,5,6-h)-1,5-cyclooctadiene]di-m-methoxydi-](http://img.cochemist.com/ccimg/12200/12148-72-0_b.png)

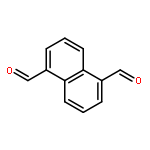
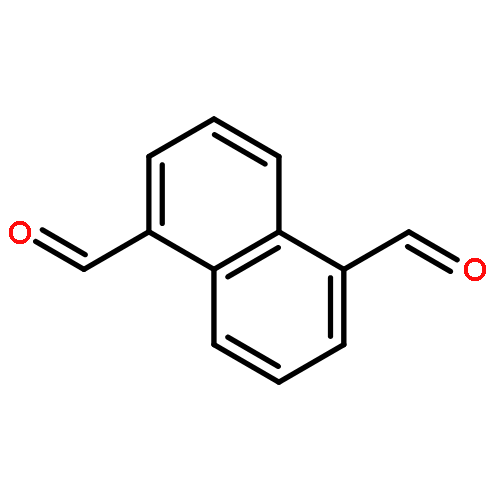


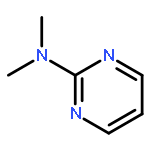
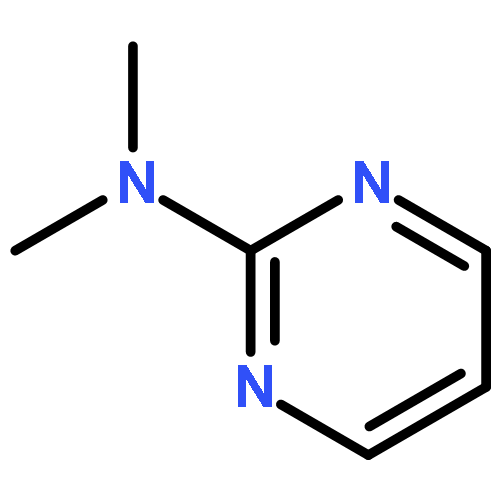
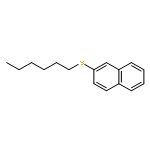
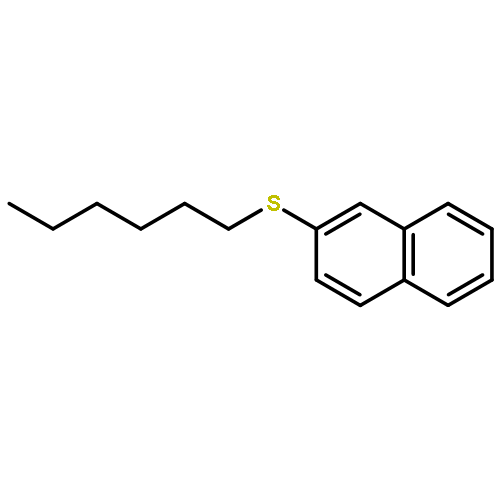
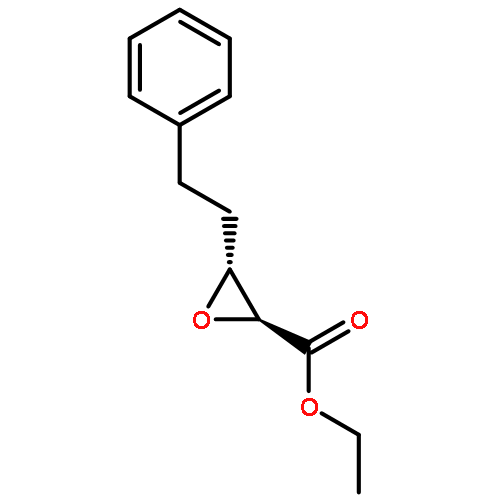
![13-Oxabicyclo[10.1.0]tridecane, (1R,12R)-rel-](http://img.cochemist.com/ccimg/4700/4683-60-7.png)
![13-Oxabicyclo[10.1.0]tridecane, (1R,12R)-rel-](http://img.cochemist.com/ccimg/4700/4683-60-7_b.png)
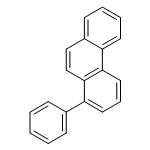
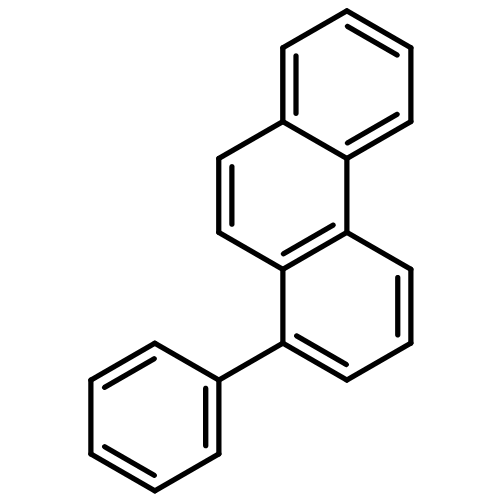


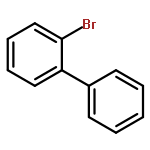
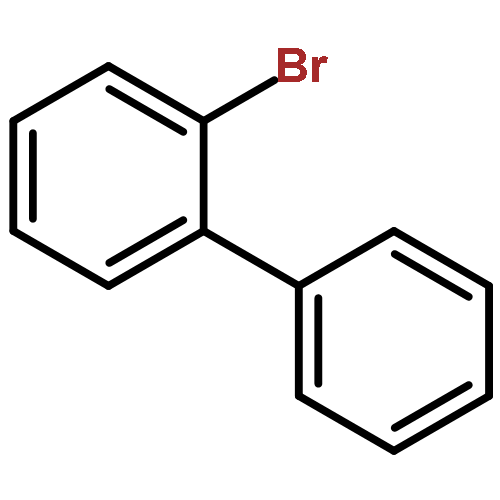




![13-Oxabicyclo[10.1.0]tridecane, (1R,12S)-rel-](http://img.cochemist.com/ccimg/1600/1502-29-0.png)
![13-Oxabicyclo[10.1.0]tridecane, (1R,12S)-rel-](http://img.cochemist.com/ccimg/1600/1502-29-0_b.png)
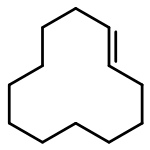
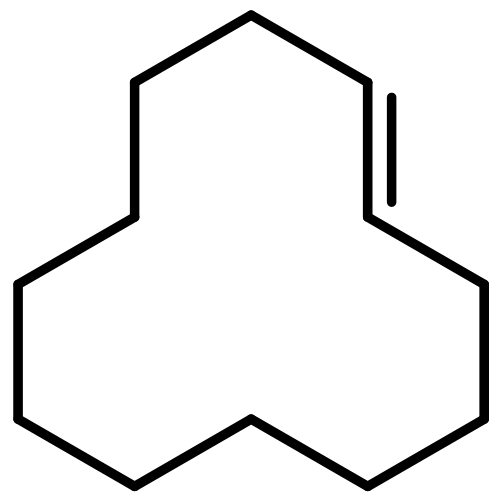

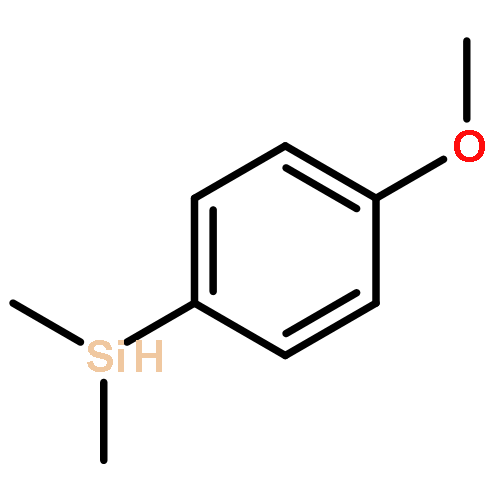

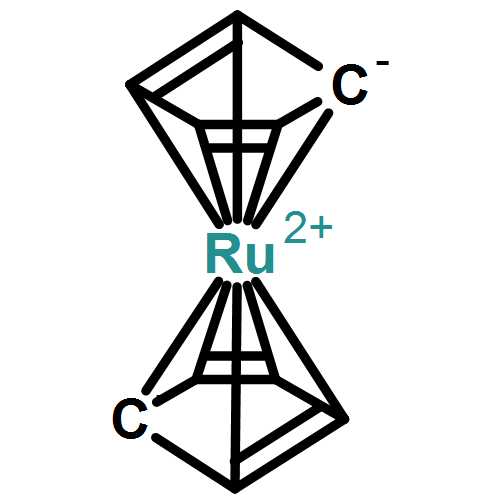
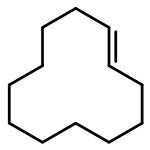
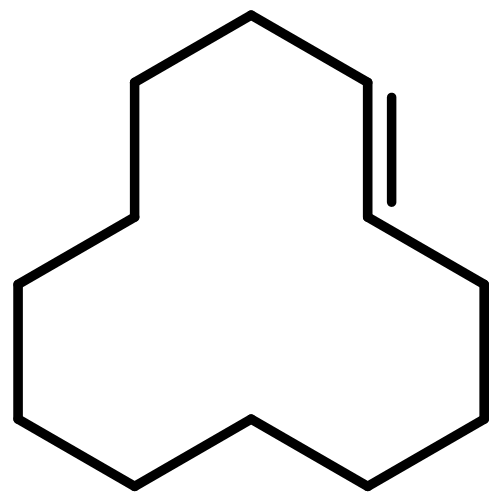









![Naphtho[1,2-b]thiophene](http://img.cochemist.com/ccimg/300/234-41-3.png)
![Naphtho[1,2-b]thiophene](http://img.cochemist.com/ccimg/300/234-41-3_b.png)
![Naphtho[2,1-b]thiophene](http://img.cochemist.com/ccimg/300/233-02-3.png)
![Naphtho[2,1-b]thiophene](http://img.cochemist.com/ccimg/300/233-02-3_b.png)
![Azulene, 1-[4-(trifluoromethyl)phenyl]-](/data/chemimg/3769300/1683535-30-9.png)
![Azulene, 1-[4-(trifluoromethyl)phenyl]-](/data/chemimg/3769300/1683535-30-9_b.png)
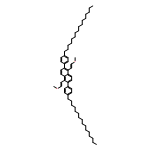
![Benzo[c]picene, 2,10-dihexadecyl-](/data/chemimg/3744000/1648892-77-6.png)
![Benzo[c]picene, 2,10-dihexadecyl-](/data/chemimg/3744000/1648892-77-6_b.png)
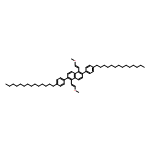
![Benzo[c]picene, 2,10-ditetradecyl-](/data/chemimg/3744000/1648892-76-5.png)
![Benzo[c]picene, 2,10-ditetradecyl-](/data/chemimg/3744000/1648892-76-5_b.png)
![Benzo[c]picene, 2,10-ditridecyl-](/data/chemimg/3744000/1648892-75-4.png)
![Benzo[c]picene, 2,10-ditridecyl-](/data/chemimg/3744000/1648892-75-4_b.png)