Co-reporter:Rizvan C. Uluisik, Neval Akbas, Gudrun S. Lukat-Rodgers, Seth A. Adrian, Courtni E. Allen, Michael P. Schmitt, Kenton R. Rodgers, Dabney W. Dixon
Journal of Inorganic Biochemistry 2017 Volume 167() pp:124-133
Publication Date(Web):February 2017
DOI:10.1016/j.jinorgbio.2016.11.027
•The heme in HtaA-CR2 is coordinated by an axial tyrosine.•Mutation of the axial ligand and a potential hydrogen-bonding partner His reduced heme binding.•Reconstitution of the protein after full unfolding significantly altered the heme binding pocket.•HtaA-CR2 is very stable toward heat and treatment with guanidinium hydrochloride.•Bound heme plays an essential role in the stability of HtaA-CR2.HtaA is a heme-binding protein that is part of the heme uptake system in Corynebacterium diphtheriae. HtaA contains two conserved regions (CR1 and CR2). It has been previously reported that both domains can bind heme; the CR2 domain binds hemoglobin more strongly than the CR1 domain. In this study, we report the biophysical characteristics of HtaA-CR2. UV–visible spectroscopy and resonance Raman experiments are consistent with this domain containing a single heme that is bound to the protein through an axial tyrosine ligand. Mutants of conserved tyrosine and histidine residues (Y361, H412, and Y490) have been studied. These mutants are isolated with very little heme (≤ 5%) in comparison to the wild-type protein (~ 20%). Reconstitution after removal of the heme with butanone gave an alternative form of the protein. The HtaA-CR2 fold is very stable; it was necessary to perform thermal denaturation experiments in the presence of guanidinium hydrochloride. HtaA-CR2 unfolds extremely slowly; even in 6.8 M GdnHCl at 37 °C, the half-life was 5 h. In contrast, the apo forms of WT HtaA-CR2 and the aforementioned mutants unfolded at much lower concentrations of GdnHCl, indicating the role of heme in stabilizing the structure and implying that heme transfer is effected only to a partner protein in vivo.The heme in the HtaA-CR2 heme uptake protein is coordinated by an axial tyrosine.The C. diphtheriae heme uptake protein HtaA-CR2 has an axial tyrosine. Reconstitution with hemin after unfolding gave a different form of the protein, as revealed by UV–visible and Raman spectroscopy. Thermal and chemical unfolding experiments showed that hemin plays a significant role in the stability of the protein.
Co-reporter:Neval Akbas, Elizabeth B. Draganova, Darci R. Block, Brian R. Sook, Yau Fong Chan, Joy Zhuo, Zehava Eichenbaum, Kenton R. Rodgers, Dabney W. Dixon
Journal of Inorganic Biochemistry 2016 Volume 158() pp:99-109
Publication Date(Web):May 2016
DOI:10.1016/j.jinorgbio.2015.10.016
•Residues near the heme have significant effects on heme binding.•The SiaA protein unfolds very slowly over time.•Slow unfolding may be an intrinsic characteristic of proteins in heme uptake pathways.•SiaA has little thermodynamic preference for folding around ferric over ferrous heme.The protein SiaA (HtsA) is part of a heme uptake pathway in Streptococcus pyogenes. In this report, we present the heme binding of the alanine mutants of the axial histidine (H229A) and methionine (M79A) ligands, as well as a lysine (K61A) and cysteine (C58A) located near the heme propionates (based on homology modeling) and a control mutant (C47A). pH titrations gave pKa values ranging from 9.0 to 9.5, close to the value of 9.7 for WT SiaA. Resonance Raman spectra of the mutants suggested that the ferric heme environment may be distinct from the wild-type; spectra of the ferrous states were similar. The midpoint reduction potential of the K61A mutant was determined by spectroelectrochemical titration to be 61 ± 3 mV vs. SHE, similar to the wild-type protein (68 ± 3 mV). The addition of guanidine hydrochloride showed two processes for protein denaturation, consistent with heme loss from protein forms differing by the orientation of the heme in the binding pocket (the half-life for the slower process ranged from less than half a day to two days). The ease of protein unfolding was related to the strength of interaction of the residues with the heme. We hypothesize that kinetically facile but only partial unfolding, followed by a very slow approach to the completely unfolded state, may be a fundamental attribute of heme trafficking proteins. Small motions to release/transfer the heme accompanied by resistance to extensive unfolding may preserve the three dimensional form of the protein for further uptake and release.SiaA is the lipoprotein component of an ABC transporter in Streptococcus pyogenes, playing an important role in heme uptake. The axial ligands and residues close to the heme propionates have a significant effect on heme release.
Co-reporter:Elizabeth B. Draganova;Seth A. Adrian
JBIC Journal of Biological Inorganic Chemistry 2016 Volume 21( Issue 7) pp:875-886
Publication Date(Web):2016 October
DOI:10.1007/s00775-016-1386-3
The heme-binding protein HmuT is part of the Corynebacterium diphtheriae heme uptake pathway and is responsible for the delivery of heme to the HmuUV ABC transporter. HmuT binds heme with a conserved His/Tyr heme axial ligation motif. Sequence alignment revealed additional conserved residues of potential importance for heme binding: R237, Y272 and M292. In this study, site-directed mutations at these three positions provided insight into the nature of axial heme binding to the protein and its effect on the thermal stability of the heme-loaded protein fold. UV–visible absorbance, resonance Raman (rR) and thermal unfolding experiments, along with collision-induced dissociation electrospray ionization mass spectrometry, were used to probe the contributions of each mutated residue to the stability of ϖ HmuT. Thermal unfolding and rR experiments revealed that R237 and M292 are important residues for heme binding. Arginine 237 is a hydrogen-bond donor to the phenol side chain of Y235, which serves as an axial heme ligand. Methionine 292 serves a supporting structural role, favoring the R237 hydrogen-bond donation, which elicits a, heretofore, unobserved modulating influence on π donation by the axial tyrosine ligand in the heme carbonyl complex, HmuT–CO.
Co-reporter:Elizabeth B. Draganova, Neval Akbas, Seth A. Adrian, Gudrun S. Lukat-Rodgers, Daniel P. Collins, John H. Dawson, Courtni E. Allen, Michael P. Schmitt, Kenton R. Rodgers, and Dabney W. Dixon
Biochemistry 2015 Volume 54(Issue 43) pp:6598-6609
Publication Date(Web):October 17, 2015
DOI:10.1021/acs.biochem.5b00666
The heme uptake pathway (hmu) of Corynebacterium diphtheriae utilizes multiple proteins to bind and transport heme into the cell. One of these proteins, HmuT, delivers heme to the ABC transporter HmuUV. In this study, the axial ligation of the heme in ferric HmuT is probed by examination of wild-type (WT) HmuT and a series of conserved heme pocket residue mutants, H136A, Y235A, and M292A. Characterization by UV–visible, resonance Raman, and magnetic circular dichroism spectroscopies indicates that H136 and Y235 are the axial ligands in ferric HmuT. Consistent with this assignment of axial ligands, ferric WT and H136A HmuT are difficult to reduce while Y235A is reduced readily in the presence of dithionite. The FeCO Raman shifts in WT, H136A, and Y235A HmuT–CO complexes provide further evidence of the axial ligand assignments. Additionally, these frequencies provide insight into the nonbonding environment of the heme pocket. Ferrous Y235A and the Y235A–CO complex reveal that the imidazole of H136 exists in two forms, one neutral and one with imidazolate character, consistent with a hydrogen bond acceptor on the H136 side of the heme. The ferric fluoride complex of Y235A reveals the presence of at least one hydrogen bond donor on the Y235 side of the heme. Hemoglobin utilization assays showed that the axial Y235 ligand is required for heme uptake in HmuT.
Co-reporter:Reham A. I. Abou-Elkhair, Dabney W. Dixon and Thomas L. Netzel
The Journal of Organic Chemistry 2009 Volume 74(Issue 13) pp:4712-4719
Publication Date(Web):June 9, 2009
DOI:10.1021/jo900306g
Photoexcitation of anthraquinones (AQ) in association with DNA results in DNA damage mainly at guanine residues, with products from thymine oxidation also observed. Studies of adenine oxidation will be aided by systems with an increased driving force for charge transfer, achieved by adding electron-withdrawing groups to the AQ ring. Attaching AQ derivatives to adenine via a bridge with two carbon atoms should enable the intended regiocontrol within the DNA duplex structure. Herein we report the synthesis of conjugates between AQ and adenine in which the AQ moieties have been modified with a formyl, a trifluoroacetyl, and two methyl ester groups. These have been synthesized by palladium coupling of tert-butyldiphenylsilyl 5′-protected 8-ethynyl-2′-deoxyadenosine with the corresponding bromoanthraquinone intermediates. Bromo intermediates bearing formyl or trifluoroacetyl were prepared by monolithiation of 2,6-dibromoanthraquinone, a step that required protection of the anthraquinone carbonyls. A bromo intermediate bearing two methyl ester groups was obtained from 1,2,4-trimethylbenzene by Friedel−Crafts acylation with 4-bromobenzoyl chloride followed by oxidation to the tricarboxylic acid, cyclization to form the anthraquinone ring, and finally esterification. Hydrogenation of the ethynyl linker gave the ethanyl linker. Cyclic voltammetry showed that the conjugate with the two ester groups and ethynyl linker was the most easily reduced of the derivatives synthesized. These derivatives, with reduction potentials favorable for electron transfer, can be used in studies of adenine oxidation in DNA.
Co-reporter:Yu Cao, Anila Fiaz Gill, Dabney W. Dixon
Tetrahedron Letters 2009 50(30) pp: 4358-4360
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.05.039
Co-reporter:Michelle B. Kim
Journal of Physical Organic Chemistry 2008 Volume 21( Issue 9) pp:731-737
Publication Date(Web):
DOI:10.1002/poc.1334
Abstract
Water-soluble naphthalene diimides (NDIs) have found uses in a wide variety of applications including as electron acceptors in electron transfer reactions and as molecules that undergo spontaneous organization in aqueous solution. Many studies have looked at their interaction with nucleic acids including work with DNA duplexes, triplexes, quadruplexes, hairpins, and DNA–RNA heteroduplexes. In many of these interactions the NDIs serve as threading intercalators. Herein we describe the reversible hydroxide-catalyzed hydrolysis of NDIs bearing aliphatic side chains, with ring opening first to the monoimide and then to the diamide. Examples with N-methylpyrrolidinium groups placed two (1) and three (5) atoms from the central core were studied. The Ka values for the first and second hydrolyses for 1 were 2.5 ± 0.2 × 105 and 2.0 ± 0.1 × 102 M−1, respectively; for 5 they were 1.4 ± 0.1 × 105 and 44 ± 2 M−1, respectively. NDI 1 hydrolyzed 6.8 times faster than 5. The rates for the first and second ring opening of 1 in 100 mM hydroxide measured by stopped-flow were 17.0 ± 0.2 and 0.53 ± 0.01 s−1, respectively. Capillary electrophoresis in borate buffer showed separation of the diimide and monoimide with the former eluting first. Nuclear magnetic resonance (NMR) showed both the syn and anti isomers of the diamide species. Overall, the rate of hydrolysis of the NDI is increased when the cationic charge is moved closer to the NDI core. Copyright © 2008 John Wiley & Sons, Ltd.
Co-reporter:Dabney W. Dixon, Anila F. Gill, Lingamallu Giribabu, Andrei N. Vzorov, Atia B. Alam, Richard W. Compans
Journal of Inorganic Biochemistry 2005 Volume 99(Issue 3) pp:813-821
Publication Date(Web):March 2005
DOI:10.1016/j.jinorgbio.2004.12.013
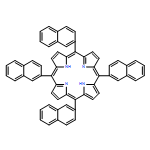
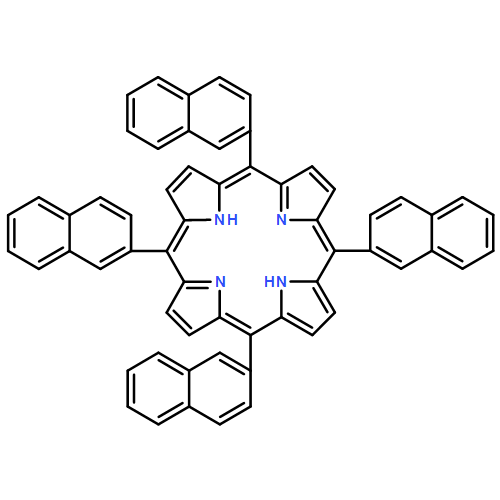
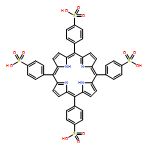
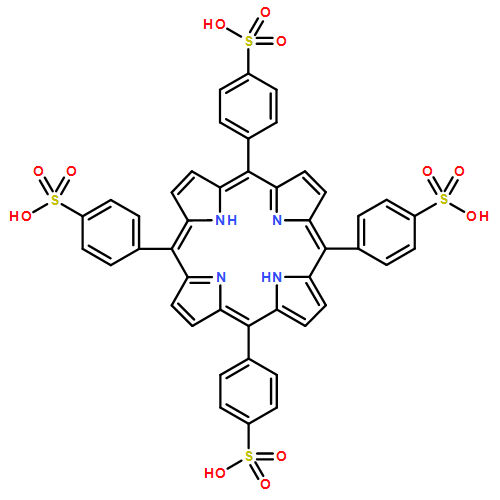
Sulfonated 5,10,15,20-tetra(1-naphthyl)porphyrin (T1NapS) and 5,10,15,20-tetra(2-naphthyl)porphyrin (T2NapS) and their copper and iron chelates show activity against the human immunodeficiency virus (HIV-1). The porphyrins were prepared by sulfonation of the parent structures with sulfuric acid. More highly sulfonated structures were prepared by sulfonation for longer times. Matrix-assisted laser desorption/ionization (MALDI) mass spectrometry showed species with as many as eight sulfonates. Some of the mass spectral peaks for the copper chelates were consistent with loss of water, apparently from intramolecular sulfone formation between two adjacent naphthalene rings that took place during copper insertion. The compounds could be separated using capillary electrophoresis; addition of β- or γ-cyclodextrin gave substantially better separation of the components. Activity against HIV was evaluated using an epithelial HeLa-CD4-CCR5 cell line; EC50 values for HIV-1 IIIB and HIV-1 JR-FL ranged from 1 to 15 μg/ml. The compounds exhibit low toxicity for human epithelial cells and have potential as microbicides which might be used to provide protection against sexual transmission of HIV.
.jpg)
![1-Propanaminium, 3,3',3'',3'''-[21H,23H-porphine-5,10,15,20-tetrayltetrakis(4,1-phenyleneoxy)]tetrakis[N,N,N-trimethyl- (9CI)](/data/chemimg/3723800/143266-26-6.png)
![1-Propanaminium, 3,3',3'',3'''-[21H,23H-porphine-5,10,15,20-tetrayltetrakis(4,1-phenyleneoxy)]tetrakis[N,N,N-trimethyl- (9CI)](/data/chemimg/3723800/143266-26-6_b.png)
![Vanadium(4+),oxo[[4,4',4'',4'''-(21H,23H-porphine-5,10,15,20-tetrayl-kN21,kN22,kN23,kN24)tetrakis[1-methylpyridiniumato]](2-)]-,(SP-5-12)-](http://img.cochemist.com/ccimg/106100/106049-21-2.png)
![Vanadium(4+),oxo[[4,4',4'',4'''-(21H,23H-porphine-5,10,15,20-tetrayl-kN21,kN22,kN23,kN24)tetrakis[1-methylpyridiniumato]](2-)]-,(SP-5-12)-](http://img.cochemist.com/ccimg/106100/106049-21-2_b.png)
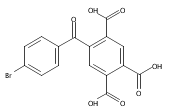
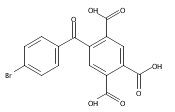
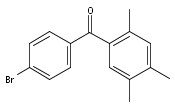
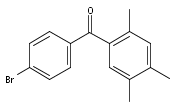


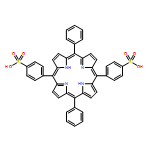
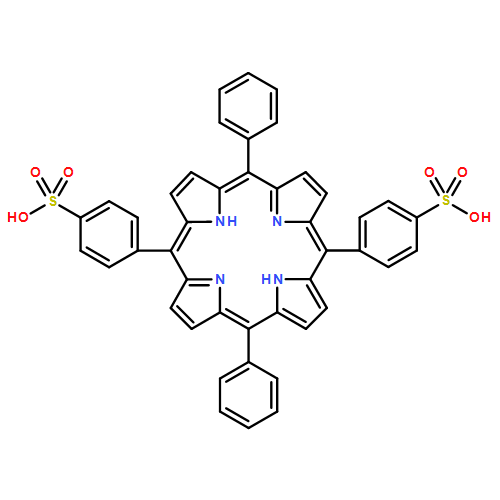
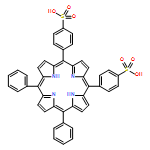

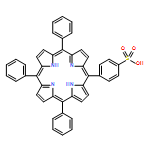
![Carbamic acid, [3-[(3-aminopropyl)methylamino]propyl]-,1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/87600/87530-14-1.png)
![Carbamic acid, [3-[(3-aminopropyl)methylamino]propyl]-,1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/87600/87530-14-1_b.png)
![Manganese(5 ), [[4,4',4'',4'''-(21H,23H-porphine-5,10,15,20-tetrayl-κN21,κN22,κN23,κN24)tetrakis[1-methylpyridiniumato]](2-)]-, (SP-4-1)-](/data/chemimg/1563200/70649-54-6.png)
![Manganese(5 ), [[4,4',4'',4'''-(21H,23H-porphine-5,10,15,20-tetrayl-κN21,κN22,κN23,κN24)tetrakis[1-methylpyridiniumato]](2-)]-, (SP-4-1)-](/data/chemimg/1563200/70649-54-6_b.png)
![Zincate(4-),[29H,31H-phthalocyanine-C,C,C,C-tetrasulfonato(6-)-kN29,kN30,kN31,kN32]-, hydrogen (1:4)](http://img.cochemist.com/ccimg/61600/61586-86-5.png)
![Zincate(4-),[29H,31H-phthalocyanine-C,C,C,C-tetrasulfonato(6-)-kN29,kN30,kN31,kN32]-, hydrogen (1:4)](http://img.cochemist.com/ccimg/61600/61586-86-5_b.png)
![Iron(5+),[[4,4',4'',4'''-(21H,23H-porphine-5,10,15,20-tetrayl-kN21,kN22,kN23,kN24)tetrakis[1-methylpyridiniumato]](2-)]-,(SP-4-1)-](http://img.cochemist.com/ccimg/60500/60489-13-6.png)
![Iron(5+),[[4,4',4'',4'''-(21H,23H-porphine-5,10,15,20-tetrayl-kN21,kN22,kN23,kN24)tetrakis[1-methylpyridiniumato]](2-)]-,(SP-4-1)-](http://img.cochemist.com/ccimg/60500/60489-13-6_b.png)
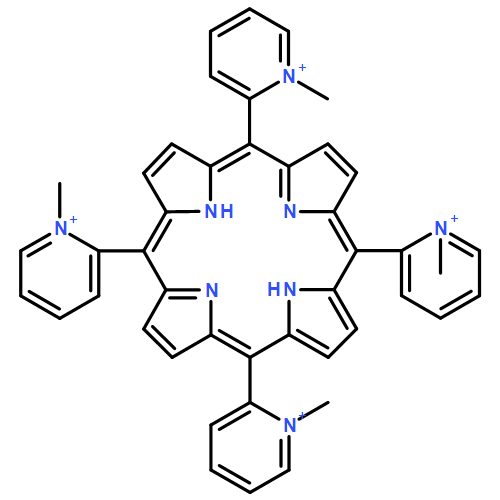
![Cobalt(5+),[[4,4',4'',4'''-(21H,23H-porphine-5,10,15,20-tetrayl-kN21,kN22,kN23,kN24)tetrakis[1-methylpyridiniumato]](2-)]-,(SP-4-1)-](http://img.cochemist.com/ccimg/51400/51329-41-0.png)
![Cobalt(5+),[[4,4',4'',4'''-(21H,23H-porphine-5,10,15,20-tetrayl-kN21,kN22,kN23,kN24)tetrakis[1-methylpyridiniumato]](2-)]-,(SP-4-1)-](http://img.cochemist.com/ccimg/51400/51329-41-0_b.png)
![Copper(4 ), [[4,4',4'',4'''-(21H,23H-porphine-5,10,15,20-tetrayl-κN21,κN22,κN23,κN24)tetrakis[1-methylpyridiniumato]](2-)]-, (SP-4-1)-](/data/chemimg/2567900/48242-70-2.png)
![Copper(4 ), [[4,4',4'',4'''-(21H,23H-porphine-5,10,15,20-tetrayl-κN21,κN22,κN23,κN24)tetrakis[1-methylpyridiniumato]](2-)]-, (SP-4-1)-](/data/chemimg/2567900/48242-70-2_b.png)
![Zinc(4+),[[4,4',4'',4'''-(21H,23H-porphine-5,10,15,20-tetrayl-kN21,kN22,kN23,kN24)tetrakis[1-methylpyridiniumato]](2-)]-,(SP-4-1)-](/data/chemimg/59500/40603-58-5.png)
![Zinc(4+),[[4,4',4'',4'''-(21H,23H-porphine-5,10,15,20-tetrayl-kN21,kN22,kN23,kN24)tetrakis[1-methylpyridiniumato]](2-)]-,(SP-4-1)-](/data/chemimg/59500/40603-58-5_b.png)


![Iron, chloro[5,10,15,20-tetrakis(4-methoxyphenyl)-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-5-12)-](/data/chemimg/27600/36995-20-7.png)
![Iron, chloro[5,10,15,20-tetrakis(4-methoxyphenyl)-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-5-12)-](/data/chemimg/27600/36995-20-7_b.png)
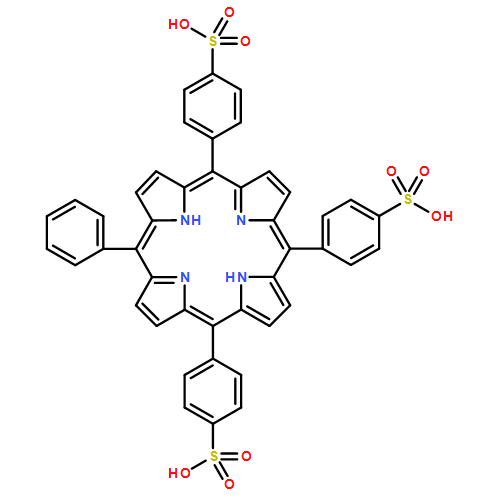

![Iron, chloro[dimethyl7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(2-)-kN21,kN22,kN23,kN24]-, (SP-5-13)-](http://img.cochemist.com/ccimg/15800/15741-03-4.png)
![Iron, chloro[dimethyl7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(2-)-kN21,kN22,kN23,kN24]-, (SP-5-13)-](http://img.cochemist.com/ccimg/15800/15741-03-4_b.png)
![Iron, μ-oxobis[5,10,15,20-tetraphenyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]di-](/data/chemimg/404500/12582-61-5.png)
![Iron, μ-oxobis[5,10,15,20-tetraphenyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]di-](/data/chemimg/404500/12582-61-5_b.png)




![anthra[2,3-c]furan-1,3,5,10-tetrone](http://img.cochemist.com/ccimg/6800/6705-73-3.png)
![anthra[2,3-c]furan-1,3,5,10-tetrone](http://img.cochemist.com/ccimg/6800/6705-73-3_b.png)
![1H-Naphth[2,3-f]isoindole-1,3,5,10(2H)-tetrone](http://img.cochemist.com/ccimg/6800/6705-70-0.png)
![1H-Naphth[2,3-f]isoindole-1,3,5,10(2H)-tetrone](http://img.cochemist.com/ccimg/6800/6705-70-0_b.png)
![1H,3H-Naphtho[2,3-c:6,7-c']difuran-1,3,6,8-tetrone](/data/chemimg/3154300/3711-01-1.png)
![1H,3H-Naphtho[2,3-c:6,7-c']difuran-1,3,6,8-tetrone](/data/chemimg/3154300/3711-01-1_b.png)
![[2,2'-Bipyridine]-5-carboxylic acid](/data/chemimg/1089300/1970-80-5.png)
![[2,2'-Bipyridine]-5-carboxylic acid](/data/chemimg/1089300/1970-80-5_b.png)








![2,6-Epoxy-2H-naphthaceno[1,2-b]oxocin-14-carboxylicacid, 11-[(6-deoxy-3-C-methyl-2,3,4-tri-O-methyl-a-L-mannopyranosyl)oxy]-4-(dimethylamino)-3,4,5,6,9,11,12,13,14,16-decahydro-3,5,8,10,13-pentahydroxy-6,13-dimethyl-9,16-dioxo-,methyl ester, (2R,3S,4R,5R,6R,11S,13S,14R)-](/data/chemimg/470500/1404-15-5.png)
![2,6-Epoxy-2H-naphthaceno[1,2-b]oxocin-14-carboxylicacid, 11-[(6-deoxy-3-C-methyl-2,3,4-tri-O-methyl-a-L-mannopyranosyl)oxy]-4-(dimethylamino)-3,4,5,6,9,11,12,13,14,16-decahydro-3,5,8,10,13-pentahydroxy-6,13-dimethyl-9,16-dioxo-,methyl ester, (2R,3S,4R,5R,6R,11S,13S,14R)-](/data/chemimg/470500/1404-15-5_b.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)