Co-reporter:Tanaji T. Talele
Journal of Medicinal Chemistry 2016 Volume 59(Issue 19) pp:8712-8756
Publication Date(Web):June 14, 2016
DOI:10.1021/acs.jmedchem.6b00472
Recently, there has been an increasing use of the cyclopropyl ring in drug development to transition drug candidates from the preclinical to clinical stage. Important features of the cyclopropane ring are, the (1) coplanarity of the three carbon atoms, (2) relatively shorter (1.51 Å) C–C bonds, (3) enhanced π-character of C–C bonds, and (4) C–H bonds are shorter and stronger than those in alkanes. The present review will focus on the contributions that a cyclopropyl ring makes to the properties of drugs containing it. Consequently, the cyclopropyl ring addresses multiple roadblocks that can occur during drug discovery such as (a) enhancing potency, (b) reducing off-target effects, (c) increasing metabolic stability, (d) increasing brain permeability, (e) decreasing plasma clearance, (f) contributing to an entropically more favorable binding to the receptor, (g) conformational restriction of peptides/peptidomimetics to prevent proteolytic hydrolysis, and (h) altering drug pKa to reduce its P-glycoprotein efflux ratio.
Co-reporter:Satyakam Singh ; Nagarajan Rajendra Prasad ; Eduardo E. Chufan ; Bhargav A. Patel ; Yi-Jun Wang ; Zhe-Sheng Chen ; Suresh V. Ambudkar ;Tanaji T. Talele
Journal of Medicinal Chemistry 2014 Volume 57(Issue 10) pp:4058-4072
Publication Date(Web):April 28, 2014
DOI:10.1021/jm401966m
P-glycoprotein (P-gp) serves as a therapeutic target for the development of multidrug resistance reversal agents. In this study, we synthesized 21 novel compounds by peptide coupling at corresponding carboxyl and amino termini of (S)-valine-based bis-thiazole and monothiazole derivatives with diverse chemical scaffolds. Using calcein-AM efflux assay, we identified compound 28 (IC50 = 1.0 μM) carrying 3,4,5-trimethoxybenzoyl and 2-aminobenzophenone groups, respectively, at the amino and carboxyl termini of the monothiazole zwitter-ion. Compound 28 inhibited the photolabeling of P-gp with [125I]-iodoarylazidoprazosin with IC50 = 0.75 μM and stimulated the basal ATP hydrolysis of P-gp in a concentration-dependent manner (EC50 ATPase = 0.027 μM). Compound 28 at 3 μM reduced resistance in cytotoxicity assay to paclitaxel in P-gp-expressing SW620/Ad300 and HEK/ABCB1 cell lines. Biochemical and docking studies showed site-1 to be the preferable binding site for 28 within the drug-binding pocket of human P-gp.
Co-reporter:Maulik R. Patel ; Aaditya Bhatt ; Jamin D. Steffen ; Adel Chergui ; Junko Murai ; Yves Pommier ; John M. Pascal ; Louis D. Trombetta ; Frank R. Fronczek ;Tanaji T. Talele
Journal of Medicinal Chemistry 2014 Volume 57(Issue 13) pp:5579-5601
Publication Date(Web):June 12, 2014
DOI:10.1021/jm5002502
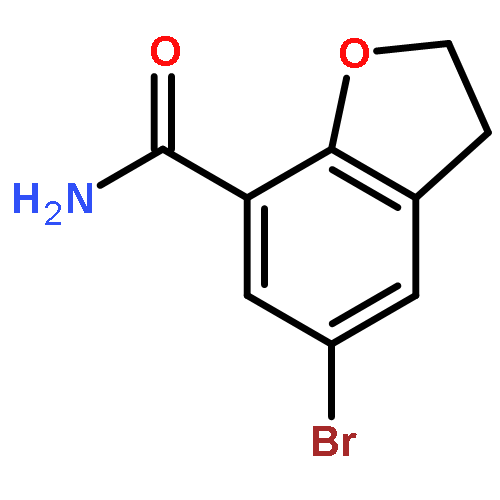
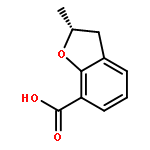
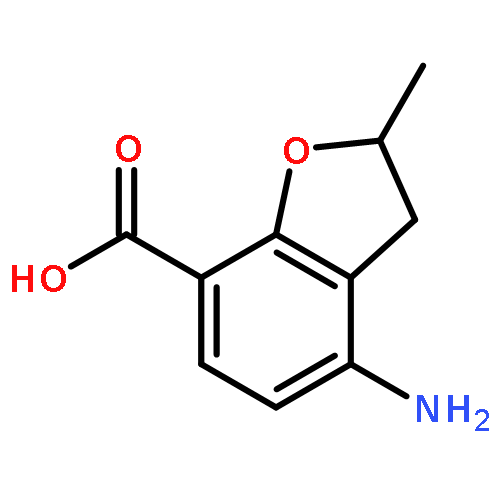
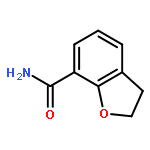
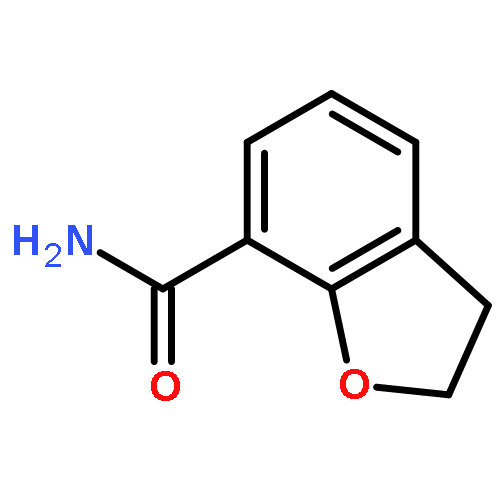
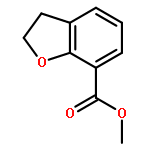
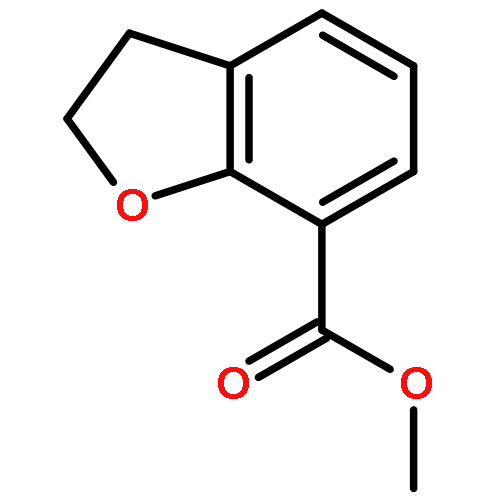
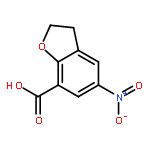
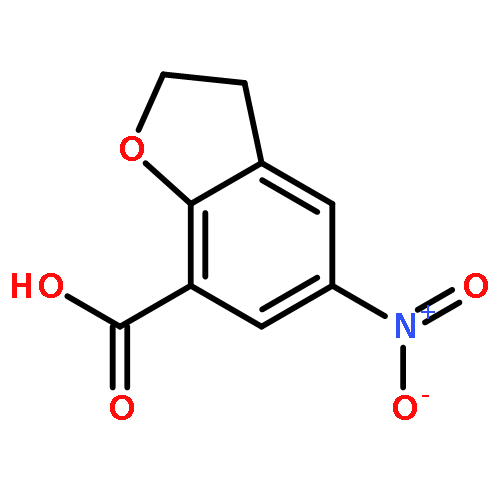
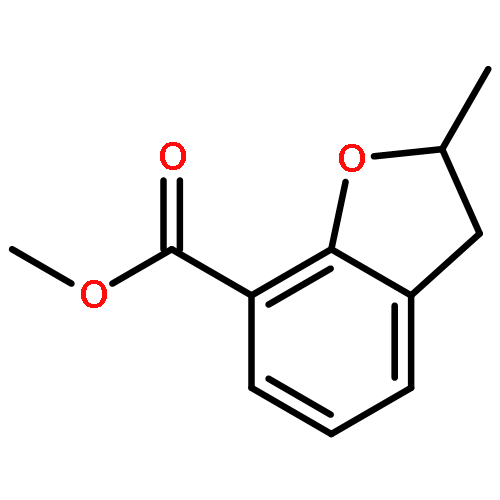
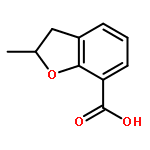
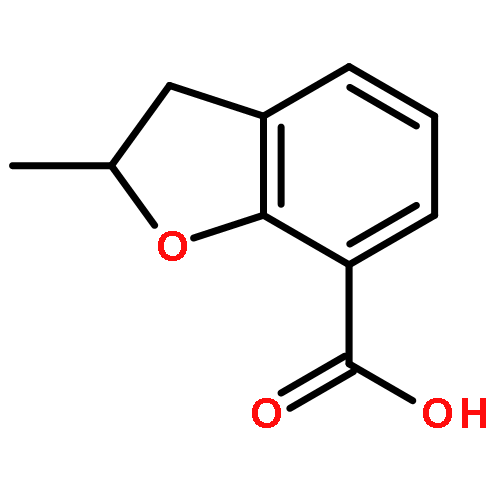
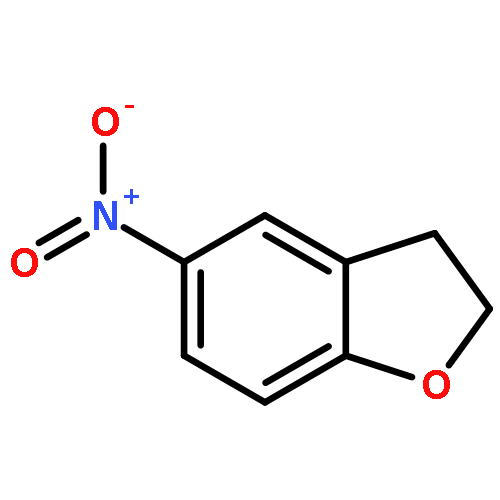
Novel substituted 2,3-dihydrobenzofuran-7-carboxamide (DHBF-7-carboxamide) and 2,3-dihydrobenzofuran-3(2H)-one-7-carboxamide (DHBF-3-one-7-carboxamide) derivatives were synthesized and evaluated as inhibitors of poly(ADP-ribose)polymerase-1 (PARP-1). A structure-based design strategy resulted in lead compound 3 (DHBF-7-carboxamide; IC50 = 9.45 μM). To facilitate synthetically feasible derivatives, an alternative core was designed, DHBF-3-one-7-carboxamide (36, IC50 = 16.2 μM). The electrophilic 2-position of this scaffold was accessible for extended modifications. Substituted benzylidene derivatives at the 2-position were found to be the most potent, with 3′,4′-dihydroxybenzylidene 58 (IC50 = 0.531 μM) showing a 30-fold improvement in potency. Various heterocycles attached at the 4′-hydroxyl/4′-amino of the benzylidene moiety resulted in significant improvement in inhibition of PARP-1 activity (e.g., compounds 66–68, 70, 72, and 73; IC50 values from 0.718 to 0.079 μM). Compound 66 showed selective cytotoxicity in BRCA2-deficient DT40 cells. Crystal structures of three inhibitors (compounds (−)-13c, 59, and 65) bound to a multidomain PARP-1 structure were obtained, providing insights into further development of these inhibitors.
Co-reporter:Dr. Satyakam Singh;Dr. Nagarajan Rajendra Prasad;Dr. Khyati Kapoor;Dr. Eduardo E. Chufan;Bhargav A. Patel;Dr. Suresh V. Ambudkar;Dr. Tanaji T. Talele
ChemBioChem 2014 Volume 15( Issue 1) pp:157-169
Publication Date(Web):
DOI:10.1002/cbic.201300565
Abstract
Multidrug resistance caused by ATP binding cassette transporter P-glycoprotein (P-gp) through extrusion of anticancer drugs from the cells is a major cause of failure in cancer chemotherapy. Previously, selenazole-containing cyclic peptides were reported as P-gp inhibitors and were also used for co-crystallization with mouse P-gp, which has 87 % homology to human P-gp. It has been reported that human P-gp can simultaneously accommodate two to three moderately sized molecules at the drug binding pocket. Our in silico analysis, based on the homology model of human P-gp, spurred our efforts to investigate the optimal size of (S)-valine-derived thiazole units that can be accommodated at the drug binding pocket. Towards this goal, we synthesized varying lengths of linear and cyclic derivatives of (S)-valine-derived thiazole units to investigate the optimal size, lipophilicity, and structural form (linear or cyclic) of valine-derived thiazole peptides that can be accommodated in the P-gp binding pocket and affects its activity, previously an unexplored concept. Among these oligomers, lipophilic linear (13) and cyclic trimer (17) derivatives of QZ59S-SSS were found to be the most and equally potent inhibitors of human P-gp (IC50=1.5 μM). As the cyclic trimer and linear trimer compounds are equipotent, future studies should focus on noncyclic counterparts of cyclic peptides maintaining linear trimer length. A binding model of the linear trimer 13 within the drug binding site on the homology model of human P-gp represents an opportunity for future optimization, specifically replacing valine and thiazole groups in the noncyclic form.
Co-reporter:Bhargav A. Patel, Ramalingam Krishnan, Nikhil Khadtare, K.R. Gurukumar, Amartya Basu, Payal Arora, Aaditya Bhatt, Maulik R. Patel, Dibyendu Dana, Sanjai Kumar, Neerja Kaushik-Basu, Tanaji T. Talele
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 11) pp:3262-3271
Publication Date(Web):1 June 2013
DOI:10.1016/j.bmc.2013.03.041
Hepatitis C virus (HCV) NS5B polymerase is a key target for anti-HCV therapeutics development. Herein, we report the synthesis and in vitro evaluation of anti-NS5B polymerase activity of a molecular hybrid of our previously reported lead compounds 1 (IC50 = 7.7 μM) and 2 (IC50 = 10.6 μM) as represented by hybrid compound 27 (IC50 = 6.7 μM). We have explored the optimal substituents on the terminal phenyl ring of the 3-phenoxybenzylidene moiety in 27, by generating a set of six analogs. This resulted in the identification of compound 34 with an IC50 of 2.6 μM. To probe the role of stereochemistry towards the observed biological activity, we synthesized and evaluated the d-isomers 41 (IC50 = 19.3 μM) and 45 (IC50 = 5.4 μM) as enantiomers of the l-isomers 27 and 34, respectively. The binding site of compounds 32 and 34 was mapped to palm pocket-I (PP-I) of NS5B. The docking models of 34 and 45 within the PP-I of NS5B were investigated to envisage the molecular mechanism of inhibition.
Co-reporter:Bhargav A. Patel, Charles R. Ashby Jr., Diane Hardej, Tanaji T. Talele
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 20) pp:5523-5527
Publication Date(Web):15 October 2013
DOI:10.1016/j.bmcl.2013.08.059
A focused library of rhodanine compounds containing novel substituents at the C5-position was synthesized and tested in vitro against a panel of clinically relevant MRSA strains. The present SAR study was based on our lead compound 1 (MIC = 1.95 μg/mL), with a focus on identifying optimal C5-arylidene substituents. In order to obtain this objective, we condensed several unique aromatic aldehydes with phenylalanine-derived rhodanine intermediates to obtain C5-substituted target rhodanine compounds for evaluation as anti-MRSA compounds. These efforts produced three compounds with significant efficacy: 23, 32 and 44, with MIC values ranging from 0.98 to 1.95 μg/mL against all tested MRSA strains as compared to the reference antibiotics penicillin G (MIC = 15.60–250.0 μg/mL) and ciprofloxacin (MIC = 7.80–62.50 μg/mL) and comparable to that of vancomycin (MIC = 0.48 μg/mL). In addition, compounds 24, 28, 37, 41, 46 and 48 (MIC = 1.95–3.90 μg/mL) were efficacious against all MRSA strains. The majority of the synthesized compounds had bactericidal activity at concentrations only two to fourfold higher than their MIC. Overall, the results suggest that compounds 23, 32 and 44 may be of potential use in the treatment of MRSA infections.Lead optimization strategy resulted in identification of several rhodanine compounds with anti-MRSA efficacy comparable to that of vancomycin.
Co-reporter:Kevin Hannigan, Shridhar S. Kulkarni, Volodymyr G. Bdzhola, Andriy G. Golub, Sergiy M. Yarmoluk, Tanaji T. Talele
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 21) pp:5790-5794
Publication Date(Web):1 November 2013
DOI:10.1016/j.bmcl.2013.09.007
Poly(ADP-ribose)polymerase-1 (PARP-1) is an abundant and ubiquitous chromatin-bound nuclear protein. PARP-1, a DNA repair enzyme, has been in the limelight as a chemotherapeutic target. In this study, we demonstrated the successful use of structure-based virtual screening to identify inhibitors of PARP-1 from Otava databases comprised of nearly 260,000 compounds. Five novel inhibitors belonging to thienopyrimidinone, isoquinolinoquinazolinone, pyrroloquinazolinone, and cyclopentenothienopyrimidinone scaffolds revealed inhibitory potencies with IC50 values ranged from 9.57 μM to 0.72 μM. Structural features relevant to the activity of these novel compounds within the active site of PARP-1 are discussed in detail and will guide future SAR investigation on these scaffolds.Structure-based virtual screening of nearly 260,000 Otava compounds resulted in five novel PARP-1 inhibitors.
Co-reporter:Maulik R. Patel;Kashyap G. Pya;Cesar A. Lau-Cam;Satyakam Singh;Maria A. Pino;Blase Billack;Kurt Degenhardt ;Tanaji T. Talele
Chemical Biology & Drug Design 2012 Volume 79( Issue 4) pp:488-496
Publication Date(Web):
DOI:10.1111/j.1747-0285.2011.01302.x
A group of novel N-1-substituted indazole-3-carboxamide derivatives were synthesized and evaluated as inhibitors of poly(ADP-ribose)polymerase-1 (PARP-1). A structure-based design strategy was applied to a weakly active unsubstituted 1H-indazole-3-carboxamide 2, by introducing a three carbon linker between 1H-indazole-3-carboxamide and different heterocycles, and led to compounds 4 [1-(3-(piperidine-1-yl)propyl)-1H-indazole-3-carboxamide, IC50 = 36 μm] and 5 [1-(3-(2,3-dioxoindolin-1-yl)propyl)-1H-indazole-3-carboxamide, IC50 = 6.8 μm]. Compound 5 was evaluated in rats for its protective action against diabetes induced by a treatment with streptozotocin, a known diabetogenic agent. In addition to preserving the ability of the pancreas to secrete insulin, compound 5 was also able to attenuate the ensuing hyperglycemic response to a significant extent.
Co-reporter:Shridhar S. Kulkarni, Satyakam Singh, Janki R. Shah, Woon-Kai Low, Tanaji T. Talele
European Journal of Medicinal Chemistry 2012 50() pp: 264-273
Publication Date(Web):
DOI:10.1016/j.ejmech.2012.02.001
Co-reporter:Aaditya Bhatt, K.R. Gurukumar, Amartya Basu, Maulik R. Patel, Neerja Kaushik-Basu, Tanaji T. Talele
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 10) pp:5138-5145
Publication Date(Web):October 2011
DOI:10.1016/j.ejmech.2011.08.028
Hepatitis C virus (HCV) NS5B polymerase is a key target for anti-HCV therapeutics development. Here we report the synthesis and biological evaluation of a new series of α,γ-diketo acids (DKAs) as NS5B polymerase inhibitors. We initiated structure–activity relationship (SAR) optimization around the furan moiety of compound 1a [IC50 = 21.8 μM] to achieve more active NS5B inhibitors. This yielded compound 3a [IC50 = 8.2 μM] bearing the 5-bromobenzofuran-2-yl moiety, the first promising lead compound of the series. Varying the furan moiety with thiophene, thiazole and indazole moieties resulted in compound 11a [IC50 = 7.5 μM] bearing 3-methylthiophen-2-yl moiety. Finally replacement of the thiophene ring with a bioisosteric phenyl ring further improved the inhibitory activity as seen in compounds 21a [IC50 = 5.2 μM] and 24a [IC50 = 2.4 μM]. Binding mode of compound 24a using glide docking within the active site of NS5B polymerase will form the basis for future SAR optimization.Optimization of specificity domain of α,γ-diketo acids (DKAs) resulted in identification of low micromolar NS5B polymerase inhibitor 24a [IC50 = 2.4 μM]. Binding mode of compound 24a using glide docking within the active site of NS5B polymerase will form the basis for future SAR optimization.Highlights► HCV NS5B polymerase is an attractive target for intervention of HCV infection. ► Specificity aryl/heteroaryl domain in α,γ-diketo acids influences NS5B inhibition. ► 2,4-difluorophenyl as the specificity domain resulted in potent inhibition of NS5B. ► Docking model of most active analog suggests clues for future SAR optimization.
Co-reporter:Aaditya Bhatt;Pallav D. Patel;Maulik R. Patel;Satyakam Singh;Cesar A. Lau-Cam ;Tanaji T. Talele
Chemical Biology & Drug Design 2011 Volume 77( Issue 5) pp:361-372
Publication Date(Web):
DOI:10.1111/j.1747-0285.2011.01112.x
GPR40, a G-protein-coupled receptor has been well established to play a crucial role in regulating blood glucose levels. Hence, GPR40 is a potential target for future antidiabetic agents. The present 3D QSAR study is aimed at delineating structural parameters governing GPR40 agonistic activity. To meet this objective, a comparative molecular similarity indices analysis for 63 different GPR40 agonists was performed using two methods; a ligand-based 3D QSAR model employing the atom fit alignment method and a receptor-based 3D QSAR model that was derived from the predicted binding conformations obtained by docking all the GPR40 agonists at the active site of GPR40. The results of these studies showed the ligand-based model to be superior ( value of 0.610) to the receptor-based model ( value of 0.519) in terms of statistical data. The predictive ability of these models was evaluated using a test set of 15 compounds not included in the preliminary training set of 48 compounds. The predictive r2 values for the ligand- and the receptor-based models were found to be 0.863 and 0.599, respectively. Further, interpretation of the comparative molecular similarity indices analysis contour maps with reference to the active site of GPR40 provided an insight into GPR40-agonist interactions.
Co-reporter:Pallav D. Patel, Maulik R. Patel, Bela Kocsis, Erika Kocsis, Steven M. Graham, Andrew R. Warren, Stacia M. Nicholson, Blase Billack, Frank R. Fronczek, Tanaji T. Talele
European Journal of Medicinal Chemistry 2010 Volume 45(Issue 6) pp:2214-2222
Publication Date(Web):June 2010
DOI:10.1016/j.ejmech.2010.01.062
In an effort to find inhibitors that are effective against both Candida and Aspergillus spp., a series of 5(6)-(un)substituted benzotriazole analogs, represented by compounds 3a–3h and 3b′–3f′, were prepared using a crystalline oxirane intermediate 1 previously synthesized in our laboratory. All the compounds were evaluated for inhibitory activity against various species of Candida and Aspergillus. Compounds 3b′ (5,6-dimethylbenzotriazol-2-yl derivative), 3d (5-chlorobenzotriazol-1-yl derivative) and 3e′ (6-methylbenzotriazol-1-yl derivative) exhibited potent antifungal activity, with the MICs for Candida spp. and Aspergillus niger, ranging from 1.6 μg/mL to 25 μg/mL and 12.5 μg/mL to 25 μg/mL, respectively. The present work describes the design, synthesis, regioisomer characterization (through COSY and NOESY 2D-NMR spectroscopy and single molecule X-ray crystallography), antifungal evaluation, molecular docking, and structure-activity relationships of the various 5(6)-(un)substituted benzotriazole analogs.A series of 5(6)-(un)substituted benzotriazole analogs were identified as broad spectrum antifungal agents.
Co-reporter:Diane Hardej, Charles R. Ashby Jr., Nikhil S. Khadtare, Shridhar S. Kulkarni, Satyakam Singh, Tanaji T. Talele
European Journal of Medicinal Chemistry 2010 Volume 45(Issue 12) pp:5827-5832
Publication Date(Web):December 2010
DOI:10.1016/j.ejmech.2010.09.045
A series of rhodanine compounds containing various substituents at the N3- and C5-positions were synthesized and their in vitro activity against a panel of clinically relevant MRSA strains was determined. The anti-MRSA activity of compounds 21 (MIC = 3.9 μg/mL, MBC = 7.8 μg/mL) and 22 (MIC = 1.95 μg/mL, MBC = 7.8 μg/mL) was significantly greater than that of the lead compounds, 1–3 and reference antibiotics penicillin G (MIC = 31.25 μg/mL) and ciprofloxacin (MIC = 7.8 μg/mL) and comparable to that of vancomycin (MIC = 0.97 μg/mL). Compounds 21 and 22 were found to be bactericidal at only 2–4-fold higher than their MIC concentrations. In addition, their MIC values remained unchanged in the presence or absence of 10% serum. Overall, the results suggest that compounds 21 and 22 may be of potential use in the treatment of MRSA infections.The anti-MRSA activity of the phenylalanine-derived rhodanine analogs 21 (MIC = 3.9 μg/mL, MBC = 7.8 μg/mL) and 22 (MIC = 1.95 μg/mL, MBC = 7.8 μg/mL) was found to be comparable to the reference antibiotic vancomycin (MIC = 0.97 μg/mL).
Co-reporter:Tanaji T. Talele, Payal Arora, Shridhar S. Kulkarni, Maulik R. Patel, Satyakam Singh, Maksim Chudayeu, Neerja Kaushik-Basu
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 13) pp:4630-4638
Publication Date(Web):1 July 2010
DOI:10.1016/j.bmc.2010.05.030
Hepatitis C virus (HCV) NS5B polymerase is a key target for the development of therapeutic agents aimed at the treatment of HCV infections. Here we report on the identification of novel allosteric inhibitors of HCV NS5B through a combination of structure-based virtual screening, synthesis and structure–activity relationship (SAR) optimization approach. Virtual screening of 260,000 compounds from the ChemBridge database against the tetracyclic indole inhibitor binding pocket of NS5B (allosteric pocket-1, AP-1), sequentially down-sized the library by 4 orders of magnitude to yield 23 candidates. In vitro evaluation of the NS5B inhibitory activity of the in-silico selected compounds resulted in 17% hit rate, identifying two novel chemotypes. Of these, compound 3, bearing the rhodanine scaffold, proved amenable for productive SAR exploration and synthetic modification. As a result, 25 derivatives that exhibited IC50 values ranging from 7.7 to 68.0 μM were developed. Docking analysis of lead compound 28 within the tetracyclic indole- and benzylidene-binding allosteric pockets (AP-1 and AP-3, respectively) of NS5B revealed topological similarities between these two pockets. Compound 28, a novel rhodanine analog with NS5B inhibitory potency in the low micromolar level range may be a promising lead for future development of more potent NS5B inhibitors.A total of 25 inhibitors belonging to the rhodanine scaffold with IC50 values in the range of 7.7–68.0 μM were identified through a combined use of virtual screening, SAR analysis, synthesis and biological evaluation.
Co-reporter:Kirandeep Kaur;Tanaji T. Talele
Journal of Computer-Aided Molecular Design 2009 Volume 23( Issue 1) pp:25-36
Publication Date(Web):2009 January
DOI:10.1007/s10822-008-9235-2
Phosphoinositide-dependent protein kinase-1 (PDK1) is a Ser/Thr kinase which phosphorylates and activates members of the AGC kinase group known to control processes such as tumor cell growth, protection from apoptosis, and tumor angiogenesis. In this paper, CoMFA and CoMSIA studies were carried out on a training set of 56 conformationally rigid indolinone inhibitors of PDK1. Predictive 3D QSAR models, established using atom fit alignment rule based on crystallographic-bound conformation, had cross-validated (rcv2) values of 0.738 and 0.816 and non-cross-validated (rncv2) values of 0.912 and 0.949 for CoMFA and CoMSIA models, respectively. The predictive ability of the CoMFA and CoMSIA models was determined using a test set of 14 compounds, which gave predictive correlation coefficients (rpred2) of 0.865 and 0.837, respectively. Structure-based interpretation of the CoMFA and CoMSIA field properties provided further insights for the rational design of new PDK1 inhibitors.
Co-reporter:Pallav D. Patel;Tanaji T. Talele;Frank R. Fronczek
Journal of Chemical Crystallography 2009 Volume 39( Issue 12) pp:
Publication Date(Web):2009 December
DOI:10.1007/s10870-009-9593-1
Preparation of oxirane 3 was accomplished in two steps. 1H-1,2,4-triazole was reacted with 2,4-difluoro-α-chloroacetophenone 1 in presence of K2CO3 in refluxing toluene to provide compound 2. Compound 2 was treated with trimethylsulfoxonium iodide (TMSI) in aq. NaOH and toluene to provide oxirane 3. Oxirane 3, previously isolated as an oil, was crystallized from (DCM/MeOH) and characterized by X-ray crystallography: triclinic, space group P − 1, a = 7.3225 (15), b = 7.5833 (15), c = 9.856 (2) Å, α = 91.908 (12), β = 100.824 (11), γ = 103.800 (11)°, V = 520.28 (18) Å3, Z = 2. The molecule has a stepped conformation with nearly parallel triazole and phenyl rings. Lack of classical hydrogen-bond donors leads to packing dominated by weaker interactions, including C–H···N, C–H···F and F···F contacts.
Co-reporter:Kirandeep Kaur, Tanaji T. Talele
Journal of Molecular Graphics and Modelling 2008 Volume 27(Issue 4) pp:409-420
Publication Date(Web):November 2008
DOI:10.1016/j.jmgm.2008.07.003
A number of CCK2 antagonists have been reported to play an important role in controlling gastric acid-related conditions, nervous system related disorders and certain types of cancer. To obtain the helpful information for designing potent antagonists with novel structures and to investigate the quantitative structure–activity relationship of a group of 62 different CCK2 receptor antagonists with varying structures and potencies, CoMFA, CoMSIA, and HQSAR studies were carried out on a series of 1,3,4-benzotriazepine-based CCK2 receptor antagonists. QSAR models were derived from a training set of 49 compounds. By applying leave-one-out (LOO) cross-validation study, cross-validated (rcv2) values of 0.673 and 0.608 and non-cross-validated (rncv2) values of 0.966 and 0.969 were obtained for the CoMFA and CoMSIA models, respectively. The predictive ability of the CoMFA and CoMSIA models was determined using a test set of 13 compounds, which gave predictive correlation coefficients (rpred2) of 0.793 and 0.786, respectively. HQSAR was also carried out as a complementary study, and the best HQSAR model was generated using atoms, bonds, hydrogen atoms, and chirality as fragment distinction with fragment size (2–5) and six components showing rcv2 and rncv2 values of 0.744 and 0.918, respectively. CoMFA steric and electrostatic, CoMSIA hydrophobic and hydrogen bond acceptor fields, and HQSAR atomic contribution maps were used to analyze the structural features of the datasets that govern their antagonistic potency.
Co-reporter:Tanaji T. Talele, Mark L. McLaughlin
Journal of Molecular Graphics and Modelling 2008 Volume 26(Issue 8) pp:1213-1222
Publication Date(Web):June 2008
DOI:10.1016/j.jmgm.2007.11.003
The binding modes of a known 1,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole, quinazoline, pyrimidine and indolinone series of Aurora A kinase inhibitors have been studied using molecular docking and molecular dynamics (MD) simulations. Crystallographic bound compound 8 was precisely predicted by our docking procedure as evident from 0.43 Å root mean square (rms) deviations. In addition compound 25 (AZ_68) has been successfully cross-docked within the Aurora A kinase active site, which was pre-organized for inhibitor 8. We found four key sites (A: solvent-exposed front pocket, B: hinge region, C: selectivity pocket and D: solvent-exposed phosphate binding region) of the Aurora A kinase contributing towards the binding of these compounds. We suggest that the small hydrophobic substituents at C-6 position of pyrrolopyrazole nucleus (in compounds 1–8); C-6 and C-7 positions of the quinazoline moiety (in compounds 9–23); C-2 position of the quinazoline and C-4 position of the pyrimidine (in compound 25) could be more effective and selective through increased hydrophobic contacts and selectivity pocket interactions with these modifications of Aurora A kinase inhibitors. Five representative complexes were subjected to 1000 ps of MD simulation to determine the stability of the predicted binding conformations. The low value of the root mean square deviations (ranging from 0.725 to 1.820 Å) between the starting complex structure and the energy minimized final average complex structure suggests that the Glide Extra Precision (XP) derived docked complexes are in a state of near equilibrium. The structure-based drug design strategy described in this study will be highly useful for the development of new inhibitors with high potency and selectivity.
Co-reporter:Shang-jun Tang, Li-kun Chen, Fang Wang, Yun-kai Zhang, Zhen-cong Huang, Kenneth Kin Wah To, Xiao-kun Wang, Tanaji T. Talele, Zhe-sheng Chen, Wei-qiang Chen, Li-wu Fu
Biochemical Pharmacology (15 September 2014) Volume 91(Issue 2) pp:144-156
Publication Date(Web):15 September 2014
DOI:10.1016/j.bcp.2014.07.008

![3-[8-amino-1-(2-phenylquinolin-7-yl)imidazo[1,5-a]pyrazin-3-yl]-1-methylcyclobutan-1-ol](http://img.cochemist.com/ccimg/867200/867160-71-2.png)
![3-[8-amino-1-(2-phenylquinolin-7-yl)imidazo[1,5-a]pyrazin-3-yl]-1-methylcyclobutan-1-ol](http://img.cochemist.com/ccimg/867200/867160-71-2_b.png)


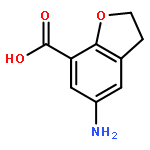
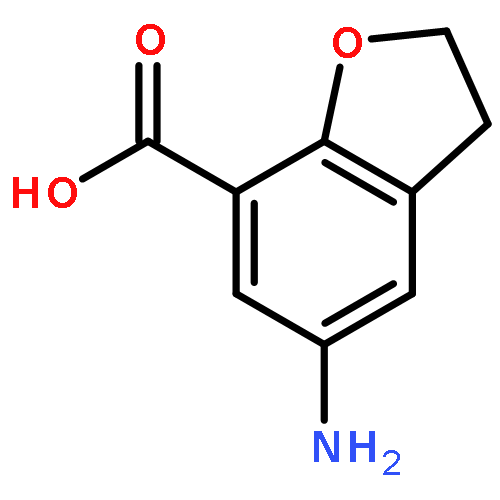
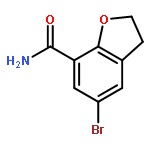


![(2R)-6-AMINO-2-[(2-METHYLPROPAN-2-YL)OXYCARBONYLAMINO]HEXANOIC ACID](http://img.cochemist.com/ccimg/121600/121584-18-7.png)
![(2R)-6-AMINO-2-[(2-METHYLPROPAN-2-YL)OXYCARBONYLAMINO]HEXANOIC ACID](http://img.cochemist.com/ccimg/121600/121584-18-7_b.png)
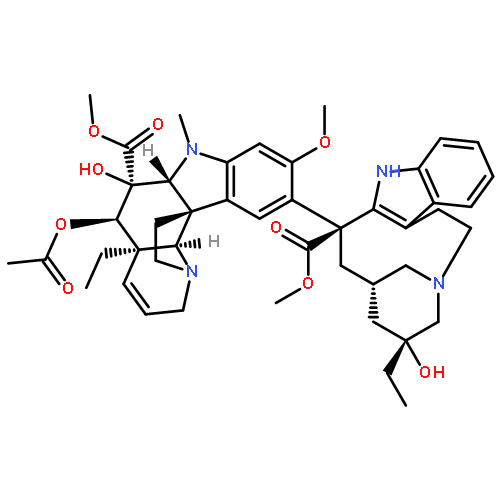
![5H,14H-Pyrrolo[1'',2'':4',5']pyrazino[1',2':1,6]pyrido[3,4-b]indole-5,14-dione,1,2,3,5a,6,11,12,14a-octahydro-9-methoxy-12-(2-methyl-1-propen-1-yl)-,(5aS,12S,14aS)-](http://img.cochemist.com/ccimg/119000/118974-02-0.png)
![5H,14H-Pyrrolo[1'',2'':4',5']pyrazino[1',2':1,6]pyrido[3,4-b]indole-5,14-dione,1,2,3,5a,6,11,12,14a-octahydro-9-methoxy-12-(2-methyl-1-propen-1-yl)-,(5aS,12S,14aS)-](http://img.cochemist.com/ccimg/119000/118974-02-0_b.png)
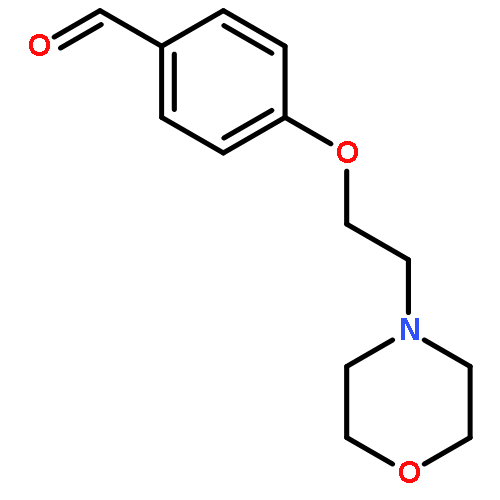
![4-[2-(4-Methyl-piperazin-1-yl)-ethoxy]-benzaldehyde](http://img.cochemist.com/ccimg/100900/100875-69-2.png)
![4-[2-(4-Methyl-piperazin-1-yl)-ethoxy]-benzaldehyde](http://img.cochemist.com/ccimg/100900/100875-69-2_b.png)
![Carbamic acid, [(1S)-1-(aminothioxomethyl)-2-methylpropyl]-,1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/97000/96929-02-1.png)
![Carbamic acid, [(1S)-1-(aminothioxomethyl)-2-methylpropyl]-,1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/97000/96929-02-1_b.png)
![3,6-diamino-9-[2-(methoxycarbonyl)phenyl]xanthylium chloride](/data/chemimg/622800/62669-70-9.png)
![3,6-diamino-9-[2-(methoxycarbonyl)phenyl]xanthylium chloride](/data/chemimg/622800/62669-70-9_b.png)







![(2E,6R)-6-[(3R,3AR,5AS,7Z,9AR,9BS)-6-(2-CARBOXYETHYL)-7-(1-CARBOX<WBR />YETHYLIDENE)-3A,5A,9B-TRIMETHYLDODECAHYDRO-1H-CYCLOPENTA[A]NAPHTH<WBR />ALEN-3-YL]-2-METHYL-2-HEPTENOIC ACID](http://img.cochemist.com/ccimg/332100/332012-40-5.png)
![(2E,6R)-6-[(3R,3AR,5AS,7Z,9AR,9BS)-6-(2-CARBOXYETHYL)-7-(1-CARBOX<WBR />YETHYLIDENE)-3A,5A,9B-TRIMETHYLDODECAHYDRO-1H-CYCLOPENTA[A]NAPHTH<WBR />ALEN-3-YL]-2-METHYL-2-HEPTENOIC ACID](http://img.cochemist.com/ccimg/332100/332012-40-5_b.png)
![4-Thiazolecarboxylic acid, 2-[(1S)-1-amino-2-methylpropyl]-, ethyl ester](http://img.cochemist.com/ccimg/220800/220717-59-9.png)
![4-Thiazolecarboxylic acid, 2-[(1S)-1-amino-2-methylpropyl]-, ethyl ester](http://img.cochemist.com/ccimg/220800/220717-59-9_b.png)
![4-Thiazolecarboxylic acid,2-[(1S)-1-[[(1,1-dimethylethoxy)carbonyl]amino]-2-methylpropyl]-](http://img.cochemist.com/ccimg/149400/149356-90-1.png)
![4-Thiazolecarboxylic acid,2-[(1S)-1-[[(1,1-dimethylethoxy)carbonyl]amino]-2-methylpropyl]-](http://img.cochemist.com/ccimg/149400/149356-90-1_b.png)
![4-Thiazolecarboxylicacid, 2-[(1R)-2-methyl-1-[[(phenylmethoxy)carbonyl]amino]propyl]-, ethyl ester](http://img.cochemist.com/ccimg/117100/117076-27-4.png)
![4-Thiazolecarboxylicacid, 2-[(1R)-2-methyl-1-[[(phenylmethoxy)carbonyl]amino]propyl]-, ethyl ester](http://img.cochemist.com/ccimg/117100/117076-27-4_b.png)