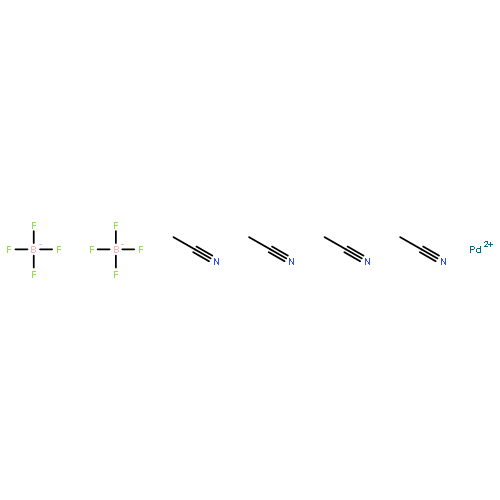Co-reporter:Valeska von Kiedrowski, Florian Quentin, and Martin Hiersemann
Organic Letters August 18, 2017 Volume 19(Issue 16) pp:
Publication Date(Web):August 1, 2017
DOI:10.1021/acs.orglett.7b02126
The first total synthesis of (+)-curvicollide C has been accomplished. Cross-metathesis and Julia–Kocienski olefination were instrumental in the synthesis of 1,3-diene segments and allowed for a ternary-convergent synthetic design. A full structural assignment is proposed for (−)-curvicollide C, a uniquely structured polyketide of fungal origin.
Co-reporter:David Tymann, André Klüppel, Wolf Hiller, and Martin Hiersemann
Organic Letters 2014 Volume 16(Issue 16) pp:4062-4065
Publication Date(Web):August 1, 2014
DOI:10.1021/ol501204m
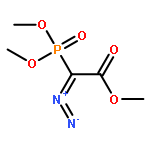
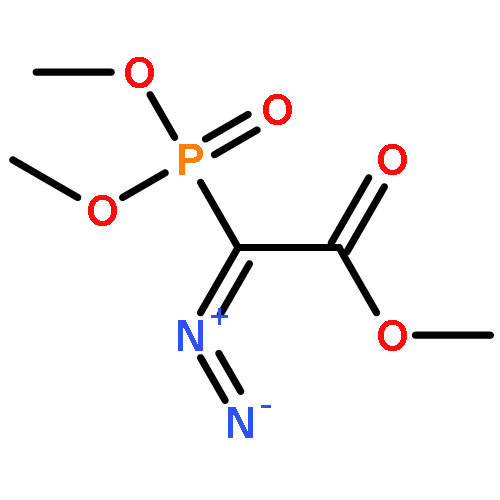
The uncatalyzed intramolecular carbonyl ene (ICE) reaction of substituted ε,ζ-unsaturated α-keto esters to terpenoid-related building blocks has been studied. We found a beneficial effect of a silyl substituent at the ene segment on the kinetics of the ICE reaction. A generalizable and scalable synthesis of ε,ζ-unsaturated α-keto esters from allylic alcohols was developed.
Co-reporter:Julia Becker, Lena Butt, Valeska von Kiedrowski, Elisabeth Mischler, Florian Quentin, and Martin Hiersemann
The Journal of Organic Chemistry 2014 Volume 79(Issue 7) pp:3040-3051
Publication Date(Web):March 12, 2014
DOI:10.1021/jo5001466
The enantioselective synthesis of (−)-9,10-dihydroecklonialactone B is described. The catalytic asymmetric Claisen rearrangement of a Gosteli-type allyl vinyl ether was utilized to afford an acyclic α-keto ester building block endowed with functionality amenable to the preparation of the carbocyclic target molecule by suitable postrearrangement transformations: A highly diastereoselective Corey–Bakshi–Shibata reduction of a β-chiral α-keto ester and a reductive homologation of an α-hydroxy ester. A transprotection tactic by a chemoselective intramolecular 6-exo-trig iodoetherification enabled regioselective ring-closing alkene metatheses to afford the 5- as well as the 14-membered ring, however, with mixed success in terms of E/Z selectivity.
Co-reporter:Björn Nelson, Sonja Herres-Pawlis, Wolf Hiller, Hans Preut, Carsten Strohmann, and Martin Hiersemann
The Journal of Organic Chemistry 2012 Volume 77(Issue 11) pp:4980-4995
Publication Date(Web):May 11, 2012
DOI:10.1021/jo3004088
The PdII-catalyzed cycloisomerization of 3-alkoxycarbonyl-3-hydroxy-substituted 1,5-hexadienes has been studied experimentally and computationally. Experimentally, the reaction is characterized by a rapid room temperature formation of monomeric as well as dimeric cycloisomerization products using the commercially available precatalyst [(CH3CN)4Pd](BF4)2. In situ NMR measurements indicate the initial kinetic advantage of the desired cycloisomerization pathway to methylene cyclopentanes; however, double bond isomerization, elimination, and dimer formation are competitive undesired pathways. Evaluation of the obtained product structures by NMR spectroscopy and X-ray crystallography indicates that the sole determinant for the monomer/dimer ratio is the regioselectivity of the initial hydropalladation in favor of the allylic (monomer formation) or the homoallylic double bond (dimer formation). In order to account for the experimental results, we propose the coexistence of two product-forming catalytic cycles, an open, monomer generating, as well as an interrupted and redirected, dimer generating, hydropalladation/carbopalladation/β-hydride elimination (HCHe) process. Results from computational studies of the proposed competing catalytic cycles are supportive to our mechanistic hypothesis and pinpoint the pivotal importance of PdII-hydroxo-chelate complexes for the reactivity–stability interplay of on- and off-pathway intermediates.
Co-reporter:Christoph Schnabel and Martin Hiersemann
Organic Letters 2009 Volume 11(Issue 12) pp:2555-2558
Publication Date(Web):May 19, 2009
DOI:10.1021/ol900819u
The enantioselective total synthesis of the jatrophane diterpene (−)-15-O-acetyl-3-O-propionylcharaciol is described. Starting from an advanced cyclopentane building block, a B-alkyl Suzuki−Miyaura cross-coupling and carbonyl addition were utilized to assemble a fully functionalized triene, and a ring-closing metathesis was then employed to construct the rigid 12-membered ring. Twenty-five years after the original report on the isolation of the natural product, our total synthesis unambiguously corroborates the original tentative structural assignment.



![7-Octenal, 3,3-dimethyl-4-[(triethylsilyl)oxy]-](http://img.cochemist.com/ccimg/886300/886221-05-2.png)
![7-Octenal, 3,3-dimethyl-4-[(triethylsilyl)oxy]-](http://img.cochemist.com/ccimg/886300/886221-05-2_b.png)
![7-OCTEN-4-OL, 3,3-DIMETHYL-1-[(TRIETHYLSILYL)OXY]-](http://img.cochemist.com/ccimg/886300/886221-02-9.png)
![7-OCTEN-4-OL, 3,3-DIMETHYL-1-[(TRIETHYLSILYL)OXY]-](http://img.cochemist.com/ccimg/886300/886221-02-9_b.png)
![7-Octen-4-ol, 3,3,7-trimethyl-1-[(triethylsilyl)oxy]-](http://img.cochemist.com/ccimg/886300/886221-01-8.png)
![7-Octen-4-ol, 3,3,7-trimethyl-1-[(triethylsilyl)oxy]-](http://img.cochemist.com/ccimg/886300/886221-01-8_b.png)
![BUTANAL, 2,2-DIMETHYL-4-[(TRIETHYLSILYL)OXY]-](http://img.cochemist.com/ccimg/886300/886221-00-7.png)
![BUTANAL, 2,2-DIMETHYL-4-[(TRIETHYLSILYL)OXY]-](http://img.cochemist.com/ccimg/886300/886221-00-7_b.png)
![7-OCTENAL, 3,3,7-TRIMETHYL-4-[(TRIETHYLSILYL)OXY]-](http://img.cochemist.com/ccimg/886300/886220-88-8.png)
![7-OCTENAL, 3,3,7-TRIMETHYL-4-[(TRIETHYLSILYL)OXY]-](http://img.cochemist.com/ccimg/886300/886220-88-8_b.png)
![Acetic acid, [[(2Z)-4-(phenylmethoxy)-2-butenyl]oxy]-, methyl ester](http://img.cochemist.com/ccimg/878100/878006-92-9.png)
![Acetic acid, [[(2Z)-4-(phenylmethoxy)-2-butenyl]oxy]-, methyl ester](http://img.cochemist.com/ccimg/878100/878006-92-9_b.png)


![4-Pentenal, 3-[(4-methoxyphenyl)methoxy]-2,2-dimethyl-, (3S)-](http://img.cochemist.com/ccimg/861200/861118-37-8.png)
![4-Pentenal, 3-[(4-methoxyphenyl)methoxy]-2,2-dimethyl-, (3S)-](http://img.cochemist.com/ccimg/861200/861118-37-8_b.png)


![2-Dodecenoic acid, 12-[(4-methoxyphenyl)methoxy]-, ethyl ester, (2E)-](http://img.cochemist.com/ccimg/691100/691006-71-0.png)
![2-Dodecenoic acid, 12-[(4-methoxyphenyl)methoxy]-, ethyl ester, (2E)-](http://img.cochemist.com/ccimg/691100/691006-71-0_b.png)


![2-Oxazolidinone, 4-(1-methylethyl)-3-[(2S)-2-methyl-1-oxohexyl]-, (4S)-](http://img.cochemist.com/ccimg/541500/541497-88-5.png)
![2-Oxazolidinone, 4-(1-methylethyl)-3-[(2S)-2-methyl-1-oxohexyl]-, (4S)-](http://img.cochemist.com/ccimg/541500/541497-88-5_b.png)
![Nonanal, 9-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/531600/531513-01-6.png)
![Nonanal, 9-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/531600/531513-01-6_b.png)
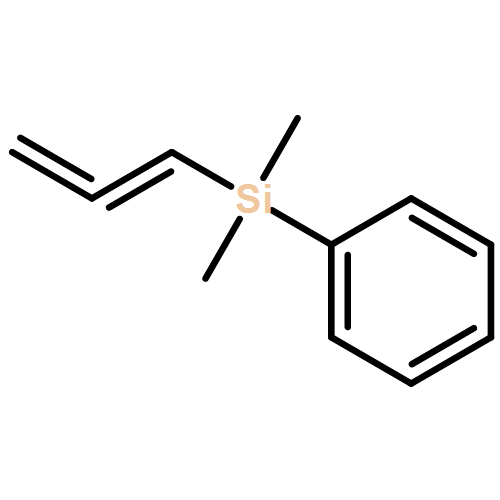
![Ruthenium, [1,3-bis(2,4,6-triMethylphenyl)-2-imidazolidinylidene]dichloro[[2-(1-Methylethoxy-κO)-5-nitrophenyl]Methylene-κC]-, (SP-5-41)-](/data/chemimg/1247800/502964-52-5.png)
![Ruthenium, [1,3-bis(2,4,6-triMethylphenyl)-2-imidazolidinylidene]dichloro[[2-(1-Methylethoxy-κO)-5-nitrophenyl]Methylene-κC]-, (SP-5-41)-](/data/chemimg/1247800/502964-52-5_b.png)
![2-BUTENOIC ACID, 2-[(3-METHYL-2-BUTENYL)OXY]-, 1-METHYLETHYL ESTER, (2E)-](http://img.cochemist.com/ccimg/413000/412966-99-5.png)
![2-BUTENOIC ACID, 2-[(3-METHYL-2-BUTENYL)OXY]-, 1-METHYLETHYL ESTER, (2E)-](http://img.cochemist.com/ccimg/413000/412966-99-5_b.png)


![1-Nonanol, 9-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/267100/267005-80-1.png)
![1-Nonanol, 9-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/267100/267005-80-1_b.png)
![1-Decanol, 10-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/239200/239115-48-1.png)
![1-Decanol, 10-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/239200/239115-48-1_b.png)


![Butanal,4-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-3-methyl-, (3R)-](http://img.cochemist.com/ccimg/186700/186641-79-2.png)
![Butanal,4-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-3-methyl-, (3R)-](http://img.cochemist.com/ccimg/186700/186641-79-2_b.png)




![(+)-2,2'-Isopropylidenebis[(4R)-4-phenyl-2-oxazoline]](/data/chemimg/116500/150529-93-4.png)
![(+)-2,2'-Isopropylidenebis[(4R)-4-phenyl-2-oxazoline]](/data/chemimg/116500/150529-93-4_b.png)







![Benzene, [(3-bromopropyl)seleno]-](http://img.cochemist.com/ccimg/118900/118824-31-0.png)
![Benzene, [(3-bromopropyl)seleno]-](http://img.cochemist.com/ccimg/118900/118824-31-0_b.png)


![Acetaldehyde, [[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/115900/115869-43-7.png)
![Acetaldehyde, [[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/115900/115869-43-7_b.png)
![2-Buten-1-ol, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (Z)-](http://img.cochemist.com/ccimg/113200/113123-37-8.png)
![2-Buten-1-ol, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (Z)-](http://img.cochemist.com/ccimg/113200/113123-37-8_b.png)





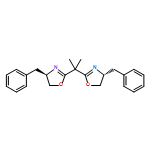
dimethyl-](http://img.cochemist.com/ccimg/96100/96045-13-5.png)
dimethyl-](http://img.cochemist.com/ccimg/96100/96045-13-5_b.png)




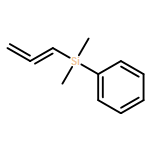


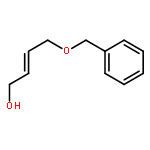













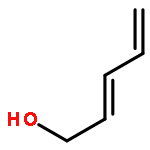
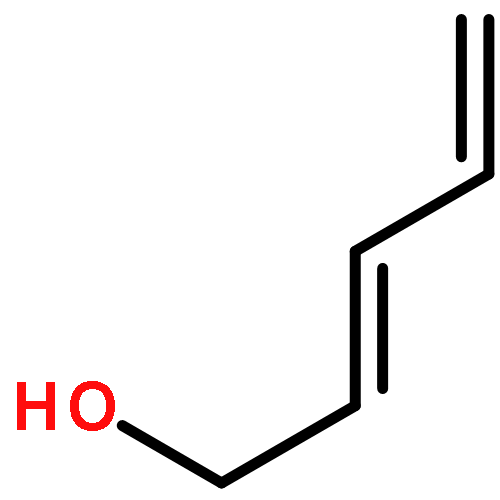




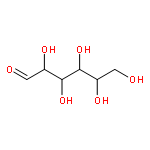
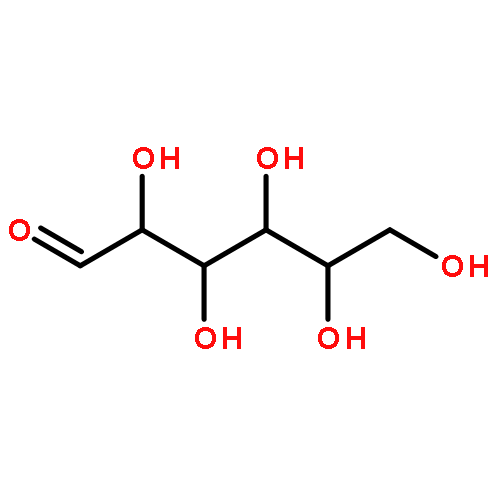










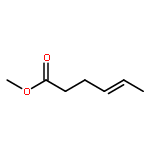

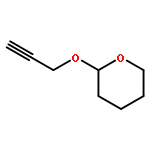
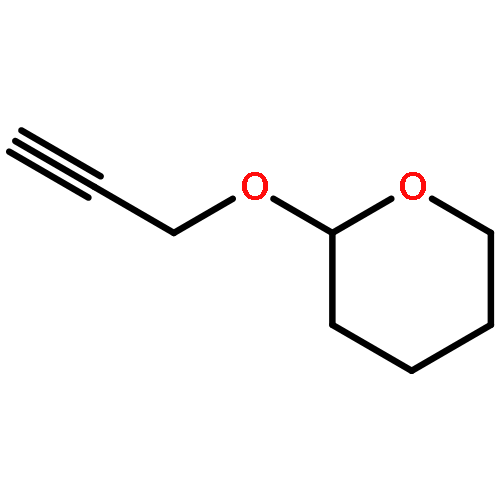
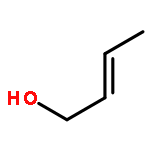
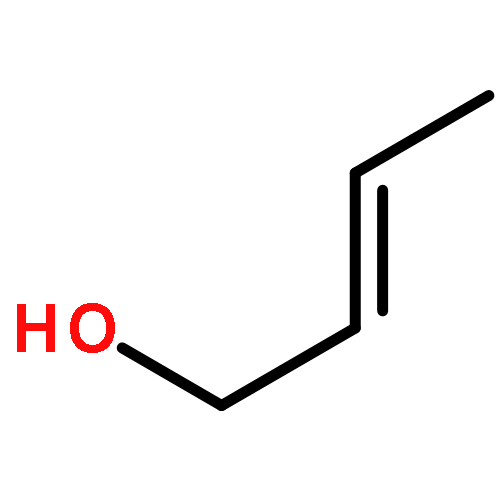


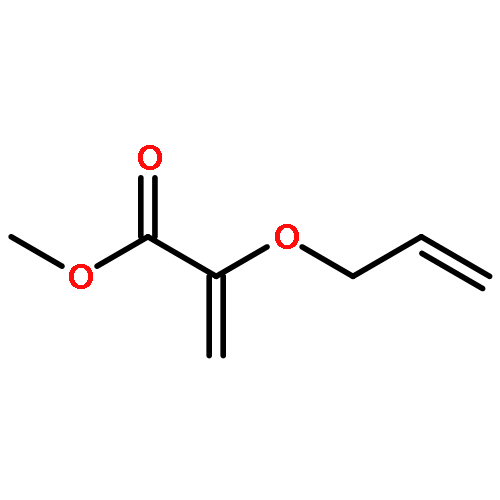
![5-(IODOMETHYL)-2,2,7,7-TETRAMETHYL-5,5A,8A,8B-TETRAHYDRO-3AH-DI[1,3]DIOXOLO[4,5-A:5',4'-D]PYRAN](http://img.cochemist.com/ccimg/4100/4026-28-2.png)
![5-(IODOMETHYL)-2,2,7,7-TETRAMETHYL-5,5A,8A,8B-TETRAHYDRO-3AH-DI[1,3]DIOXOLO[4,5-A:5',4'-D]PYRAN](http://img.cochemist.com/ccimg/4100/4026-28-2_b.png)
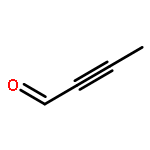
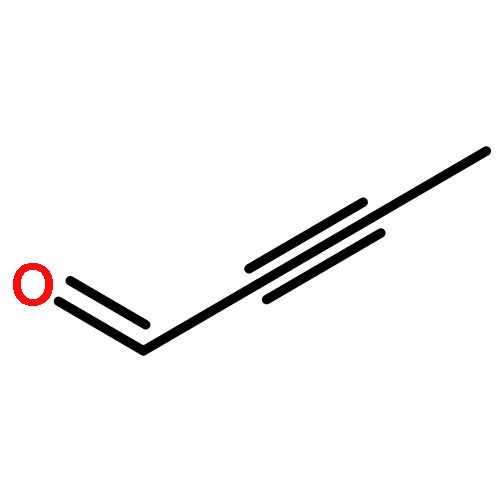




![Oxireno[3,4]cyclopent[1,2-c]oxacyclotetradecin-5(2H)-one,3-ethyl-1a,2a,3,6,7,8,9,10,11,12,14a,14b-dodecahydro-,(1aS,2aR,3S,13Z,14aS,14bR)-](http://img.cochemist.com/ccimg/122000/121923-96-4.png)
![Oxireno[3,4]cyclopent[1,2-c]oxacyclotetradecin-5(2H)-one,3-ethyl-1a,2a,3,6,7,8,9,10,11,12,14a,14b-dodecahydro-,(1aS,2aR,3S,13Z,14aS,14bR)-](http://img.cochemist.com/ccimg/122000/121923-96-4_b.png)



![Silane, [[1-(1-cyclopenten-1-yl)ethenyl]oxy](1,1-dimethylethyl)dimethyl-](http://img.cochemist.com/ccimg/86900/86891-78-3.png)
![Silane, [[1-(1-cyclopenten-1-yl)ethenyl]oxy](1,1-dimethylethyl)dimethyl-](http://img.cochemist.com/ccimg/86900/86891-78-3_b.png)