Co-reporter:Li-Cheng SongYu Lu, Liang Zhu, Qian-Li Li
Organometallics February 13, 2017 Volume 36(Issue 3) pp:
Publication Date(Web):February 1, 2017
DOI:10.1021/acs.organomet.6b00942
As the active site models of [NiFe]hydrogenases, three types of dichalcogenido E (E = S, Se)-bridged NiFe complexes (dppb)Ni[μ-E(CH2)3E-μ]Fe(CO)3 (dppb = bis(1,2-diphenylphosphino)benzene, 2a, E = S; 2b, E = Se), {(dppb)Ni[μ-E(CH2)3E-μ](μ-H)Fe(CO)3}BF4 (3a, E = S; 3b, E = Se), and {(dppb)Ni[μ-E(CH2)3E-μ](μ-D)Fe(CO)3}BF4 (4a, E = S; 4b, E = Se) were prepared by starting from the precursors (dppb)Ni[μ-E(CH2)3E-μ] (1a, E = S; 1b, E = Se) in satisfactory yields. While coordinations of 1a,b with Fe2(CO)9 gave the first-type dichalcogenido-bridged models 2a,b, the protonations of 2a,b with HBF4·Et2O afforded the second-type dichalcogenido-bridged μ-hydride models 3a,b the oxidative additions of 2a,b with DCl followed by anionic exchanges of the resulting intermediates with NaBF4 produced the third-type dichalcogenido-bridged μ-deuteride models 4a,b. Models 2a,b–4a,b were all fully characterized by elemental analysis, spectroscopy, and X-ray crystallography, and the H/D exchange reactions of μ-hydrides 3a,b with D2 and D2O were further studied under light irradiation by 1H and 2H NMR spectroscopy.
Co-reporter:Yuan-Qiang Guo, Yunchuang Wang, Li-Cheng Song, Feng Liu, Xiangjian Wan, Hongtao Zhang, and Yongsheng Chen
Chemistry of Materials April 25, 2017 Volume 29(Issue 8) pp:3694-3694
Publication Date(Web):April 7, 2017
DOI:10.1021/acs.chemmater.7b00642
A small molecule named DRBDTCO based on benzo[1,2-b:4,5-b′]dithiophene (BDT) with an asymmetric side chain and its dimer, dDRBDTCO, with the octamethylene connector were designed and synthesized as donor materials for solution-processed bulk heterojunction solar cells. The optimized power conversion efficiency of a DRBDTCO-based device was 8.18% under AM 1.5 G irradiation (100 mW cm–2), which is higher than that of its dDRBDTCO-based device. Both molecule-based devices exhibited high fill factors of >73%, which are attributed to their optimized morphologies with a feature size of 15–20 nm, highly efficient charge collection, less bimolecular recombination, and well-balanced charge transport properties. The results demonstrate that the asymmetric BDT unit could be a promising building block for donor materials with high efficiencies and high fill factors.
Co-reporter:Li-Cheng Song, Long-Duo Zhang, Bei-Bei Liu, Shu-Da Ding, Hao Chen, Xiu-Fang Xu, and Gui-Lan Fan
Organometallics April 10, 2017 Volume 36(Issue 7) pp:1419-1419
Publication Date(Web):March 23, 2017
DOI:10.1021/acs.organomet.7b00117
Three types of (diphosphine)Pd- or Pt-bridged butterfly Fe/S cluster complexes have been prepared by a simple and convenient one-pot synthetic method. The first type of such complexes involves the linear (diphosphine)Pd- or Pt-bridged double-butterfly Fe/S clusters [(μ-RS)(μ-S═CS)Fe2(CO)6]2[M(diphosphine)] (1–12; M = Pd and Pt; R = Et, t-Bu, Ph, and p-MeC6H4; diphosphine = dppe, dppv, and dppf), which were prepared by sequential reactions of monoanions [(μ-RS)(μ-CO)Fe2(CO)6]– (formed in situ from Fe3(CO)12, RSH, and Et3N) with excess CS2, followed by treatment of the resulting monoanions [(μ-RS)(μ-S═CS)Fe2(CO)6]– with (diphosphine)MCl2. The second type of complexes involves the macrocyclic (diphosphine)M-bridged double-butterfly Fe/S clusters [μ-S(CH2)4S-μ][(μ-S═CS)Fe2(CO)6]2[M(diphosphine)] (13–16; M = Pd and Pt; diphosphine = dppe and dppv), which were prepared by sequential reactions of dianion [{μ-S(CH2)4S-μ}{(μ-CO)Fe2(CO)6}2]2– (generated in situ from Fe3(CO)12, dithiol HS(CH2)4SH, and Et3N) with excess CS2, followed by treatment of the resultant dianion [{μ-S(CH2)4S-μ}{(μ-S═CS)Fe2(CO)6}2]2– with (diphosphine)MCl2. In contrast, when dithiol HS(CH2)4SH was replaced by HS(CH2)3SH (a dithiol with a shorter carbon chain), the aforementioned sequential reactions afforded the third type of macrocyclic complexes which involves the (diphosphine)M-bridged quadruple-butterfly Fe/S clusters [{μ-S(CH2)3S-μ}{(μ-S═CS)Fe2(CO)6}2]2[M(diphosphine)]2 (17–20; M = Pd and Pt; diphosphine = dppe and dppv). While the two possible pathways are suggested for production of the two types of novel macrocyclic Fe/S clusters 13–20, respectively, all new complexes 1–20 have been characterized by elemental analysis, spectroscopy, and, for some of them particularly, DFT calculations and X-ray crystallography.
Co-reporter:Li-Cheng Song;Xi-Yue Yang;Meng Cao;Xiu-Yun Gao;Bei-Bei Liu;Liang Zhu;Feng Jiang
Chemical Communications 2017 vol. 53(Issue 27) pp:3818-3821
Publication Date(Web):2017/03/30
DOI:10.1039/C7CC00149E
The structural and functional modeling of the active site of [NiFe]-hydrogenases has been proved to be challenging to a great extent. Herein, we report the synthesis, structures, and some properties of the NiFe-based dicarbonyl, terminal hydride, and μ-hydroxo models for the active site of [NiFe]-hydrogenases.
Co-reporter:Li-Cheng Song;Xiao-Feng Han;Wei Chen;Jia-Peng Li;Xu-Yong Wang
Dalton Transactions 2017 vol. 46(Issue 30) pp:10003-10013
Publication Date(Web):2017/08/01
DOI:10.1039/C7DT02203D
A new series of the structural and functional models for the active site of [NiFe]-H2ases has been prepared by a simple and convenient synthetic route. Thus, treatment of diphosphines RN(PPh2)2 (1a, R = p-MeC6H4CH2; 1b, R = EtO2CCH2) with an equimolar NiCl2·6H2O, NiBr2·3H2O, and NiI2 in refluxing CH2Cl2/MeOH or EtOH gave the mononuclear Ni complexes RN(PPh2)2NiX2 (2a, R = p-MeC6H4CH2, X = Cl; 2b, R = EtO2CCH2, X = Cl; 3a, R = p-MeC6H4CH2, X = Br; 3b, R = EtO2CCH2, X = Br; 4a, R = p-MeC6H4CH2, X = I; 4b, R = EtO2CCH2, X = I) in 67–97% yields. Further treatment of complexes 2a,b–4a,b with an equimolar mononuclear Fe complex (dppv)(CO)2Fe(pdt) and NaBF4 resulted in formation of the targeted model complexes [RN(PPh2)2Ni(μ-pdt)(μ-X)Fe(CO)(dppv)](BF4) (5a, R = p-MeC6H4CH2, X = Cl; 5b, R = EtO2CCH2, X = Cl; 6a, R = p-MeC6H4CH2, X = Br; 6b, R = EtO2CCH2, X = Br; 7a, R = p-MeC6H4CH2, X = I; 7b, R = EtO2CCH2, X = I) in 60–96% yields. All the new complexes 3a,b–4a,b and 5a,b–7a,b have been characterized by elemental analysis and spectroscopy, and particularly for some of them (3a,b/4a,b and 5b/6b) by X-ray crystallography. More interestingly, the electrochemical and electrocatalytic properties of such halogenido-bridged model complexes are first studied systematically and particularly they have been found to be pre-catalysts for proton reduction to H2 under CV conditions.
Co-reporter:Li-Cheng Song, Kai-Kai Xu, Xiao-Feng Han, and Ji-Wei Zhang
Inorganic Chemistry 2016 Volume 55(Issue 3) pp:1258-1269
Publication Date(Web):January 12, 2016
DOI:10.1021/acs.inorgchem.5b02490
As active site models of [Fe]-hydrogenase, tridentate 2-acylmethyl-6-methoxymethoxy-difunctionalized pyridine-containing complexes η3-(2-COCH2-6-MeOCH2OC5H3N)Fe(CO)2(L1) (4, L1 = I; 5, SCN; 6, PhCS2) were prepared via the following multistep reactions: (i) etherification of 2-MeO2C-6-HOC5H3N with ClCH2OMe to give 2-MeO2C-6-MeOCH2OC5H3N (1), (ii) reduction of 1 with NaBH4 to give 2-HOCH2-6-MeOCH2OC5H3N (2), (iii) esterification of 2 with 4-toluenesulfonyl chloride to give 2-TsOCH2-6-MeOCH2OC5H3N (3), (iv) nucleophilic substitution of 3 with Na2Fe(CO)4 followed by treatment of the resulting Fe(0) intermediate Na[(2-CH2-6-MeOCH2OC5H3N)Fe(CO)4] (M1) with I2 to give complex 4, and (v) condensation of 4 with KSCN and PhCS2K to give complexes 5 and 6, respectively. In contrast to the preparation of complexes 4–6, bidentate 2-acylmethyl-6-methoxymethoxy-difunctionalized pyridine-containing model complexes η2-(2-COCH2-6-MeOCH2OC5H3N)Fe(CO)2(I)(L2) (7, L2 = PPh3; 8, Cy-C6H11NC) and η2-(2-COCH2-6-MeOCH2OC5H3N)Fe(CO)2(L3) (9, L3 = 2-SC5H4N; 10, 8-SC9H6N) were prepared by ligand exchange reactions of 4 with PPh3, Cy-C6H11NC, 2-KSC5H4N, and 8-KSC9H6N, respectively. Particularly interesting is that the tridentate 2,6-bis(acylmethyl)pyridine- and 2-acylmethyl-6-arylthiomethylpyridine-containing model complexes η3-[2,6-(COCH2)2C5H3N]Fe(CO)2(L4) (11, L4 = PPh3; 12, CO) and η3-2-(COCH2-6-ArSCH2C5H3N)Fe(CO)2(ArS) (13, ArS = PhS; 14, 2-S-5-MeC4H2O) were obtained, unexpectedly, when 2,6-(TsOCH2)2C5H3N reacted with Na2Fe(CO)4 followed by treatment of the resulting mixture with ligands PPh3 and CO or disulfides (PhS)2 and (2-S-5-MeC4H2O)2. Reactions of ligand precursors 3 and 2,6-(TsOCH2)2C5H3N with Na2Fe(CO)4 were monitored by in situ IR spectroscopy, and the possible pathways for producing complexes 4 and 11–14 via intermediates Na[(2-CH2-6-MeOCH2OC5H3N)Fe(CO)4] (M1), Na[(2-CH2-6-TsOCH2C5H3N)Fe(CO)4] (M2), and (2-COCH2-6-CH2C5H3N)Fe(CO)3 (M3) are suggested. New compounds 1–14 were characterized by elemental analysis, spectroscopy, and, for some of them, X-ray crystallography.
Co-reporter:Li-Cheng Song, An-Guo Zhu and Yuan-Qiang Guo
Dalton Transactions 2016 vol. 45(Issue 12) pp:5021-5029
Publication Date(Web):04 Jan 2016
DOI:10.1039/C5DT04297F
As [FeFe]-hydrogenase models, the first thiodithiolate (TDT) ligand-containing μ-hydride complexes [(μ-TDT)Fe2(CO)4(PMe3)2(μ-H)]+Y− (2–7, Y = Cl, ClO4, PF6, BF4, CF3CO2, CF3SO3) have been prepared by protonation reactions of (μ-TDT)Fe2(CO)4(PMe3)2 (1) with the corresponding HY acids. While the protonation reactions are monitored by in situ1H and 31P{1H} NMR spectroscopy to show the isomer type and stability of 2–7, the structures of the isolated 2–7 are characterized by elemental analysis, spectroscopy and for some of them by X-ray crystallography. Although the H/D exchange of μ-hydride complex 7 (Y = CF3SO3) with D2 or D2O has been proved not to occur under the studied conditions, the H/D exchange of 7 with DCl gives the μ-deuterium complex [(μ-TDT)Fe2(CO)4(PMe3)2(μ-D)]+[CF3SO3]− (8) in a nearly quantitative yield. To our knowledge, 8 is the first crystallographically characterized μ-deuterium-containing butterfly [2Fe2S] complex produced by H/D exchange reaction.
Co-reporter:Li-Cheng Song, Yu Lu, Meng Cao and Xi-Yue Yang
RSC Advances 2016 vol. 6(Issue 45) pp:39225-39233
Publication Date(Web):13 Apr 2016
DOI:10.1039/C6RA07488J
While the parent Ni2 complex [Ni(RNPyS4)]2 (1a, R = H; RNPyS4 = 2,6-bis(2-mercaptophenylthiomethyl)-4-R-pyridine) cannot react with Fe(CO)3(BDA) in THF at room temperature, its substituted derivatives (1b, R = MeO; 1c, Cl; 1d, Br; 1e, i-Pr; 1f, BzO; 1g, MeS) have been found to react with Fe(CO)3(BDA) under the same conditions to give the first [RNPyS4] ligand-containing heterodinuclear [NiFe]-hydrogenase model complexes NiFe(RNPyS4)(CO)3 (2b, R = MeO; 2c, Cl; 2d, Br; 2e, i-Pr; 2f, BzO; 2g, MeS) along with the mononuclear [Fe]-hydrogenase model complexes Fe(RNPyS4)(CO) (3b, R = MeO; 3c, Cl; 3d, Br; 3e, i-Pr; 3f, BzO; 3g, MeS). However, when Ni2 complexes 1a–1g react with Fe(CO)3(BDA) in THF at the higher temperature of 40 °C, only the mononuclear complexes 3a–3g are obtained without the corresponding dinuclear complexes 2a–2g being isolated. While all the new complexes are characterized by elemental analysis and spectroscopy, the molecular structures of 1f, 2c, 3f and 3g have been further confirmed by X-ray crystallography. In addition, a possible pathway for the formation of 2a–2g and 3a–3g is suggested, which has been proved by monitoring the reaction course of dinuclear Ni2 complex 1e with Fe(CO)3(BDA) using in situ IR spectroscopy.
Co-reporter:Li-Cheng Song, Fei-Xian Luo, Bei-Bei Liu, Zhen-Chao Gu, and Hao Tan
Organometallics 2016 Volume 35(Issue 10) pp:1399-1408
Publication Date(Web):March 2, 2016
DOI:10.1021/acs.organomet.5b01040
The first phthalocyanine (Pc) macrocycle-containing [FeFe]-hydrogenase model complex, (i-BuO)8PcRu(CO)(μ-PDT)[(Ph2P)2NCH2C5H4N]Fe2(CO)4 (6), has been prepared by a multistep synthetic route. The treatment of 4-picolylamine with Ph2PCl/Et3N in CH2Cl2 at room temperature gave 4-picolyl-substituted azadiphosphine (Ph2P)2NCH2C5H4N (1), whereas further treatment of 1 with diiron complex (μ-PDT)Fe2(CO)6 in refluxing o-xylene produced the 4-picolylazadiphosphine-chelated diiron complex (μ-PDT)[(Ph2P)2NCH2C5H4N]Fe2(CO)4 (2). While 3,6-dihydroxyphthalonitrile reacted with i-BuBr/K2CO3 in DMF at 80 °C to give 3,6-diisobutoxyphthalonitrile 3, further reaction of 3 with lithium wire in refluxing i-BuOH afforded isobutoxyphthalocyanine (i-BuO)8PcH2 (4). Furthermore, 4 reacted with Ru3(CO)12 in refluxing PhCN to give ruthenium phthalocyanine (i-BuO)8PcRu(CO)2 (5). Finally, CO substitution reaction of 5 with diiron complex 2 in the presence of Me3NO in CH2Cl2 resulted in formation of the targeted model complex 6. While all the new compounds 1–6 were structurally characterized, complex 6 was found to be a catalyst for photoinduced H2 generation.
Co-reporter:Li-Cheng Song, Meng Cao and Yong-Xiang Wang
Dalton Transactions 2015 vol. 44(Issue 15) pp:6797-6808
Publication Date(Web):23 Feb 2015
DOI:10.1039/C5DT00067J
The homodinuclear complexes [Ni(RNPyS4)]2 (1a–1e; RNPyS4 = 2,6-bis(2-mercaptophenylthiomethyl)-4-R-pyridine; R = H, MeO, Cl, Br, i-Pr) were found to be prepared by reactions of the in situ generated Li2[Ni(1,2-S2C6H4)2] with 2,6-bis[(tosyloxy)methyl]pyridine and its substituted derivatives 2,6-bis[(tosyloxy)methyl]-4-R-pyridine. Further reactions of 1a–1e with Fe3(CO)12 gave both heterotrinuclear complexes NiFe2(RNPyS4)(CO)5 (2a–2e) and mononuclear complexes Fe(RNPyS4)(CO) (3a–3e), unexpectedly. Interestingly, complexes 2a–2e and 3a–3e could be regarded as models for the active sites of [NiFe]- and [Fe]-hydrogenases, respectively. All the prepared complexes were characterized by elemental analysis, spectroscopy, and particularly for some of them, by X-ray crystallography. In addition, the electrochemical properties of 2a–2e and 3a–3e as well as the electrocatalytic H2 production catalyzed by 2a–2e and 3a–3e were investigated by CV techniques.
Co-reporter:Li-Cheng Song, Hao Tan, An-Guo Zhu, Yuan-Yuan Hu, and Hao Chen
Organometallics 2015 Volume 34(Issue 9) pp:1730-1741
Publication Date(Web):April 28, 2015
DOI:10.1021/acs.organomet.5b00228
Interestingly, the intermediate salts A·[Et3NH] (A = (μ-RS)(μ-CO)Fe2(CO)6; R = i-Pr, sec-Bu, cy-C6H11, p-MeC6H4) prepared from Fe3(CO)12, RSH, and Et3N were found to react in situ with iodobenzene or its substituted derivatives in the presence of the catalyst precursor Pd(PPh3)4 to give the benzoyl type μ-acyl complexes (μ-RS)(μ-ArCO)Fe2(CO)6 (Ar = phenyl or substituted phenyl), whereas the in situ reactions of A·[Et3NH] with iodo-substituted aromatic heterocycles under the same conditions afforded the corresponding heterocyclic type μ-acyl complexes (μ-RS)(μ-ArCO)Fe2(CO)6 (Ar = heterocyclic or benzoheterocyclic group). Particularly worth noting is that such Pd-catalyzed C–C bond cross-coupling reactions are the first examples of catalytic reactions regarding A·[Et3NH] salts reported so far. In addition, all the prepared new μ-acyl complexes and the three new intermediate salts A·[Et3NH] (R = i-Pr, sec-Bu, cy-C6H11) were isolated and structurally characterized, while a possible pathway for such type Pd-catalyzed reactions regarding A·[Et3NH] salts is suggested.
Co-reporter:Li-Cheng Song, Zhen-Chao Gu, Wei-Wei Zhang, Qian-Li Li, Yong-Xiang Wang, and Hong-Fa Wang
Organometallics 2015 Volume 34(Issue 16) pp:4147-4157
Publication Date(Web):August 10, 2015
DOI:10.1021/acs.organomet.5b00560
The first [Fe2SP] model complexes for the active site of [FeFe]-hydrogenases have been prepared. Thus, the μ-CO-containing complex salt [Et3NH][(μ-CO)(μ-SCH2CH2OH)Fe2(CO)6] (m1) formed from Fe3(CO)12, HSCH2CH2OH, and Et3N was treated in situ with PCl3 and PPhCl2 followed by treatment with Et3N/DBU to give the all-carbonyl complexes (μ-SCH2CH2OPR-μ)Fe2(CO)6 (1, R = Cl; 3, Ph), whereas the functional transformation of 1 with PhOH/NaH and CO substitution of 3 with PPh3 or PPh2H in the presence of Me3NO afforded the corresponding complexes (μ-SCH2CH2OPOPh-μ)Fe2(CO)6 (2) and (μ-SCH2CH2OPPh-μ)Fe2(CO)5L (4, L = PPh3; 5, PPh2H), respectively. Similarly, when complex salt [Et3NH][(μ-CO)(μ-SC6H4OH)Fe2(CO)6] (m4) generated from Fe3(CO)12, 2-HOC6H4SH, and Et3N was treated in situ with PCl3 and PPhCl2 followed by treatment with Et3N/DBU, the expected all-carbonyl complexes (μ-SC6H4OPR-μ)Fe2(CO)6 (6, R = Cl; 8, Ph) were produced. In addition, further functional transformation of 6 with MeOH/Et3N and CO substitution of 8 with t-BuNC in the presence of Me3NO yielded the corresponding complexes (μ-SC6H4OPOMe-μ)Fe2(CO)6 (7) and (μ-SC6H4OPPh-μ)Fe2(CO)5(t-BuNC) (9), respectively. However, it should be noted that when complex salt m4 was treated with PBr3 under similar conditions, [(μ-SC6H4OPO(CH2)4Br-μ]Fe2(CO)6 (10) was unexpectedly obtained. While the possible pathways for formation of the unexpected 10 were suggested, all complexes 1–10 were characterized by elemental analysis and spectroscopy and for some of them by X-ray crystallography. Interestingly, model complexes 1, 4, and 8 have been found to be catalysts for HOAc proton reduction to H2 under CV conditions.
Co-reporter:Li-Cheng Song, Fu-Qiang Hu, Miao-Miao Wang, Zhao-Jun Xie, Kai-Kai Xu and Hai-Bin Song
Dalton Transactions 2014 vol. 43(Issue 21) pp:8062-8071
Publication Date(Web):14 Mar 2014
DOI:10.1039/C4DT00335G
As biomimetic models for [Fe]-hydrogenase, the 2-acylmethyl-6-ester group-difunctionalized pyridine-containing iron(II) complexes 1–4 have been successfully prepared via the following three separate steps. In the first step, the acylation or esterification of difunctionalized pyridine 2-(p-MeC6H4SO3CH2)-6-HOCH2C5H3N with acetyl chloride or benzoic acid gives the corresponding pyridine derivatives 2-(p-MeC6H4SO3CH2)-6-RCO2CH2C5H3N (A, R = Me; B, R = Ph). The second step involves reaction of A or B with Na2Fe(CO)4 followed by treatment of the intermediate Fe(0) complexes [Na(2-CH2-6-RCO2CH2C5H3N)Fe(CO)4] (M1, R = Me; M2, R = Ph) with iodine to afford 2-acylmethyl-6-acetoxymethyl or 6-benzoyloxymethyl-difunctionalized pyridine-containing Fe(II) iodide complexes [2-C(O)CH2-6-RCO2CH2C5H3N]Fe(CO)2I (1, R = Me; 3, R = Ph). Finally, when 1 or 3 is treated with sodium 2-mercaptopyridinate, the corresponding difunctionalized pyridine-containing Fe(II) mercaptopyridinate complexes [2-C(O)CH2-6-RCO2C5H3N]Fe(CO)2(2-SC5H4N) (2, R = Me; 4, R = Ph) are produced. While the structures of model complexes 1–4 are confirmed by X-ray crystallography, the electrochemical properties of 2 and 4 are compared with those of the two previously reported models. In addition, complexes 2 and 4 have been found to be catalysts for H2 production in the presence of TFA under CV conditions.
Co-reporter:Li-Cheng Song;Meng Cao;Zong-Qiang Du;Zhan-Heng Feng;Zhen Ma ;Hai-Bin Song
European Journal of Inorganic Chemistry 2014 Volume 2014( Issue 11) pp:1886-1895
Publication Date(Web):
DOI:10.1002/ejic.201301553
Abstract
Diiron carbonyl complex [{(μ-SCH2)2O}Fe2(CO)6] (A) reacted with Ph2PCl and Me3NO in MeCN at room temperature to give the expected and unexpected Ph2PCl-, Ph2PNMe2-, and Ph2PP(=O)Ph2-substituted complexes [{(μ-SCH2)2O}Fe2(CO)5L] [1, L = Ph2PCl; 2, L = Ph2PNMe2; 3, L = Ph2PP(=O)Ph2], whereas complex [{(μ-SCH2)2CH2}Fe2(CO)6] (B) reacted with Ph2PCl and Me3NO under similar conditions to afford only the unexpected complexes [{(μ-SCH2)2CH2}Fe2(CO)5L] [4, L = Ph2PNMe2; 5, L = Ph2PP(=O)Ph2]. Similarly, complex [{(μ-SeCH2)2O}Fe2(CO)6] (C) reacted with Ph2PCl and Me3NO in MeCN at room temperature to give the unexpected complexes [{(μ-SeCH2)2O}Fe2(CO)5L] [6, L = Ph2PNMe2; 7, L = Ph2PP(=O)Ph2], and the corresponding unexpected complexes [{(μ-SeCH2)2CH2}Fe2(CO)5L] [8, L = Ph2PNMe2; 9, L = Ph2PP(=O)Ph2] were produced by the reaction of complex [{(μ-SeCH2)2CH2}Fe2(CO)6] (D) with Ph2PCl and Me3NO under similar conditions. The structures of 1–9 were characterized by elemental analysis and spectroscopy, and for 1–5 and 7–9 by X-ray crystallography. Possible pathways for the formation of the functionalized phosphine-substituted complexes 1–9 have been suggested. Complexes 1–9 can be regarded as models for [FeFe] hydrogenases as their structures are similar to the active site of [FeFe] hydrogenases, and some have been found to be catalysts for proton reduction to hydrogen in the presence of HOAc.
Co-reporter:Li-Cheng Song;Meng Cao;Zong-Qiang Du;Zhan-Heng Feng;Zhen Ma ;Hai-Bin Song
European Journal of Inorganic Chemistry 2014 Volume 2014( Issue 11) pp:
Publication Date(Web):
DOI:10.1002/ejic.201400137
Abstract
Invited for the cover of this issue is the group of Li-Cheng Song at Nankai University, China. The cover image shows some of the [FeFe] hydrogenase models prepared, each with a butterfly 2Fe2S cluster core, and their catalytic function for H2 production.
Co-reporter:Li-Cheng Song;Meng Cao;Zong-Qiang Du;Zhan-Heng Feng;Zhen Ma ;Hai-Bin Song
European Journal of Inorganic Chemistry 2014 Volume 2014( Issue 11) pp:
Publication Date(Web):
DOI:10.1002/ejic.201490051
Co-reporter:Li-Cheng Song, Xiao-Jing Sun, Guo-Jun Jia, Miao-Miao Wang, Hai-Bin Song
Journal of Organometallic Chemistry 2014 Volume 761() pp:10-19
Publication Date(Web):1 July 2014
DOI:10.1016/j.jorganchem.2014.03.004
•The first four Ni-bridged butterfly Fe2Se2 complexes are prepared.•The first four Ni-bridged butterfly Fe2Te2 complexes are prepared.•Ten crystal structures for ten new complexes are reported.•The new complexes could be regarded as models for [NiFe]-hydrogenases.As the active site models of [NiFe]-hydrogenases, a series of trinuclear complexes (diphosphine)Ni(μ3-E)2Fe2(CO)6 (1−10, diphosphine = dppv, dppb, dppf, dppe, (Ph2PCH2)2NR (R = Me, t-Bu; E = S, Se, Te) has been prepared by the one-pot reactions of (μ-E2)Fe2(CO)6 with Et3BHLi, followed by treatment of the resultant intermediates (μ-LiE)2Fe2(CO)6 with the corresponding mononuclear complexes (diphosphine)NiCl2. All the new complexes 1−10 have been fully characterized by elemental analysis, spectroscopy, and X-ray crystallography. Electrochemical study reveals that reductions of the three representative complexes (dppv)Ni(μ3-E)2Fe2(CO)6 (1, E = S; 3, Se; 8, Te) become easier from the S to Se to Te complexes. Furthermore, electrocatalytic study demonstrates that complexes 1, 3, and 8 are catalysts for proton reduction to hydrogen in the presence of proton source HOTs.A series of new (diphosphine)Ni-bridged butterfly Fe2E2 (E = S, Se, Te) cluster complexes (1–10) was prepared and crystallographically characterized. Particularly interesting is that the three representative complexes (1, 3, and 8) were found to be electrocatalysts for proton reduction to hydrogen in the presence of HOTs.
Co-reporter:Li-Cheng Song, Liang-Xing Wang, Chang-Gong Li, Fengyu Li, Zhongfang Chen
Journal of Organometallic Chemistry 2014 749() pp: 120-128
Publication Date(Web):
DOI:10.1016/j.jorganchem.2013.09.007
Co-reporter:Li-Cheng Song, Hao Tan, Fei-Xian Luo, Yong-Xiang Wang, Zhen Ma, and Zheng Niu
Organometallics 2014 Volume 33(Issue 19) pp:5246-5253
Publication Date(Web):September 10, 2014
DOI:10.1021/om500571n
Two new ferrocenyl (Fc) group containing 1,5-diaza-3,7-diphosphacyclooctane ligands, PFc2NAr2 (1, Ar = Ph; 2, Ar = p-BrC6H4), were prepared by reaction of FcPH2 with 2 equiv of paraformaldehyde in glycol followed by treatment of the resulting FcP(CH2OH)2 with 1 equiv of aniline or p-bromoaniline in 79% and 67% yields, respectively; however, ligands 1 and 2 could be also prepared by treatment of the isolated and purified FcP(CH2OH)2 with 1 equiv of aniline or p-bromoaniline under similar conditions in 80% and 72% yields, respectively. Further treatment of 1 or 2 with the complex salt [Ni(MeCN)6](BF4)2 in MeCN at room temperature resulted in formation of the first ferrocenyl-containing complexes [Ni(PFc2NAr2)2](BF4)2 (3, Ar = Ph; 4, Ar = p-BrC6H4) in 95% and 90% yields, respectively. Compounds 1–4 were structurally characterized by elemental analysis and spectroscopy and particularly for 1, 2, and 4 by X-ray crystallography. When the cyclic voltammetric behavior of 1–4 was investigated, 3 and 4 were found to be electrocatalysts for proton reduction of TFA to give hydrogen under CV conditions.
Co-reporter:Peng-Hui Wei, Ling Xu, Li-Cheng Song, Wen-Xiong Zhang, and Zhenfeng Xi
Organometallics 2014 Volume 33(Issue 11) pp:2784-2789
Publication Date(Web):May 27, 2014
DOI:10.1021/om5002793
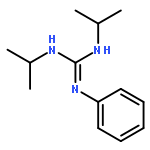
A series of mixed Cp′/bis(amidinato) (Cp′ = η5-C5Me4(SiMe3)) lanthanide complexes were synthesized by the 1:2 acid–base reaction between Cp′Ln(CH2SiMe3)2(THF) (Ln = Y, Dy, Er, Lu) and amidines. These Cp′/bis(amidinato) complexes showed excellent catalytic activity for the addition of amines to carbodiimides, yielding the corresponding guanidines. Isolation, structural characterization, and catalytic application of the binuclear lutetium amido complex showed clearly that the catalytic cycle was initiated by the dissociation of Cp′. These results demonstrated that Cp′, for the first time, acted as a reactive site to yield the active Ln–N species.
Co-reporter:Li-Cheng Song, Fu-Qiang Hu, Gao-Yu Zhao, Ji-Wei Zhang, and Wei-Wei Zhang
Organometallics 2014 Volume 33(Issue 22) pp:6614-6622
Publication Date(Web):November 4, 2014
DOI:10.1021/om5009296
We have developed two novel synthetic methods, by which two types of mononuclear Fe model complexes for the active site of [Fe]hydrogenase are successfully synthesized. The first type of 2-acylmethyl-6-hydroxymethylpyridine-containing complexes, [2-COCH2-6-HOCH2C5H3N]Fe(CO)2G (1, G = PhCO2; 2, PhCOS; 3, PhCS2; 4, 2-S-6-MeC5H3N), were prepared by a “one-pot” method involving reaction of 2-TsO-6-HOCH2C5H3N (Ts = 4-MeC6H4SO2) with Na2Fe(CO)4 followed by treatment of the resulting Fe(0) intermediate [Na(2-CH2-6-HOCH2C5H3N)Fe(CO)4] (M1) with (PhCO2)2, (PhCOS)2, (PhCS2)2, and (2-S-6-MeC5H3N)2 in 49–72% yields, respectively. The second type of 2-acylmethyl-6-hydroxypyridine-containing complexes, (2-COCH2-6-HOC5H3N)Fe(CO)2(2-SCO-6-RC5H3N) (9a, R = MeO; 9b, R = PhS), could be prepared via a multiple-step synthetic method. This method involves (i) treatment of 2-ClCO-6-RC5H3N (R = MeO, PhS) with NaSH followed by acidification with diluted HCl to give 2-HSCO-6-RC5H3N (5a, R = MeO; 5b, R = PhS); (ii) further treatment of 5a,b with KOH to afford 2-KSCO-6-RC5H3N (6a, R = MeO; 6b, R = PhS); (iii) treatment of 2-TsOCH2-6-PMBOC5H3N (PMB = 4-MeOC6H4CH2) with Na2Fe(CO)4 followed by treatment of the resulting Fe(0) intermediate [Na(2-CH2-6-PMBOC5H3N)Fe(CO)4] (M2) with Br2 or I2 to produce (2-COCH2-6-PMBOC5H3N)Fe(CO)3X (7a, X = Br; 7b, X = I); (iv) further treatment of 7a,b with 6a,b to yield (2-COCH2-6-PMBOC5H3N)Fe(CO)2(2-S-6-RC5H3N) (8a, R = MeO; 8b, R = PhS); and (v) finally, removal of the PMB groups from 8a,b under the action of deprotecting reagent CF3CO2H/EtSH to give complexes 9a,b. All compounds 1–4 and 5a,b–9a,b with the exception of 7b are new and have been characterized by elemental analysis, spectroscopy, and, particularly for 1, 4, and 7a–9a, X-ray crystallography.
Co-reporter:Li-Cheng Song, Jia-Peng Li, Zhao-Jun Xie, and Hai-Bin Song
Inorganic Chemistry 2013 Volume 52(Issue 19) pp:11618-11626
Publication Date(Web):September 24, 2013
DOI:10.1021/ic401978h
Four new dinuclear Ni/Mn model complexes RN(PPh2)2Ni(μ-SEt)2(μ-Cl)Mn(CO)3 (7, R = p-MeC6H4CH2; 8, R = EtO2CCH2) and RN(PPh2)2Ni(μ-SEt)2(μ-Br)Mn(CO)3 (9, R = p-MeC6H4CH2; 10, R = EtO2CCH2) have been prepared via the four separated step-reactions involving six new precursors RN(PPh2)2 (1, R = p-MeC6H4CH2; 2, R = EtO2CCH2), RN(PPh2)2NiCl2 (3, R = p-MeC6H4CH2; 4, R = EtO2CCH2), and RN(PPh2)2Ni(SEt)2 (5, R = p-MeC6H4CH2; 6, R = EtO2CCH2). The Et3N-assisted aminolysis of Ph2PCl with p-MeC6H4CH2NH2 or EtO2CCH2NH2·HCl in CH2Cl2 gave the azadiphosphine ligands 1 and 2 in 38% and 53% yields, whereas the coordination reaction of 1 or 2 with NiCl2·6H2O in CH2Cl2/MeOH afforded the mononuclear Ni dichloride complexes 3 and 4 in 59% and 78% yields, respectively. While thiolysis of 3 or 4 with EtSH under the assistance of Et3N in CH2Cl2 produced the mononuclear Ni dithiolate complexes 5 and 6 in 64% and 68% yields, further treatment of 5 and 6 with Mn(CO)5Cl or Mn(CO)5Br resulted in formation of the dinuclear Ni/Mn model complexes 7–10 in 31–73% yields. All the new compounds 1–10 have been structurally characterized, while model complexes 7 and 9 have been found to be catalysts for HOAc proton reduction to hydrogen under CV conditions.
Co-reporter:Li-Cheng Song, Qian-Li Li, Zhan-Heng Feng, Xiao-Jing Sun, Zhao-Jun Xie and Hai-Bin Song
Dalton Transactions 2013 vol. 42(Issue 5) pp:1612-1626
Publication Date(Web):16 Oct 2012
DOI:10.1039/C2DT31976D
Parent complex (μ-PDTe)Fe2(CO)6 (1, PDTe = μ-TeCH2CH2CH2Te-μ) is prepared via a new synthetic route involving the reaction of (μ-Te2)Fe2(CO)6 with Et3BHLi, followed by treatment of (μ-LiTe)2Fe2(CO)6 with Br(CH2)3Br in a 43% yield. Further reactions of 1 with 1 equiv of monophosphines in the presence of the decarbonylating agent Me3NO afford the corresponding monophosphine-substituted complexes (μ-PDTe)Fe2(CO)5(L) (2, L = PPh3; 3, PPh2H; 4, PMe3) in 37%–47% yields, whereas the N-heterocyclic carbene IMes-monosubstituted complex (μ-PDTe)Fe2(CO)5(IMes) (5) can be prepared in a 26% yield by treatment of 1 with the in situ generated IMes from the 1,3-bis(mesityl)imidazolium salt IMes·HCl and n-BuLi. While the diphosphine-bridged single-butterfly complexes (μ-PDTe)Fe2(CO)4(dppm) (6) and (μ-PDTe)Fe2(CO)4(dppn) (7) can be prepared in 28% and 21% yields by treatment of 1 with 1 equiv of the corresponding diphosphines in refluxing xylene, treatment of 1 with 0.5 equiv of diphosphines in the presence of Me3NO results in the formation of the corresponding diphosphine-bridged double-butterfly complexes [(μ-PDTe)Fe2(CO)5]2(dppp) (8), [(μ-PDTe)Fe2(CO)5]2(dppb) (9), and [(μ-PDTe)Fe2(CO)5]2(dppf) (10) in 25–37% yields. All the new substituted model complexes 2–10 are characterized by combustion analysis and spectroscopy, and particularly for 2, 3, 5, and 7–10, by X-ray crystallography. In addition, a comparative study on the electrochemical and electrocatalytic properties of the PDTe-type model complexes 1 and 7 with their corresponding selenium and sulfur analogs are reported.
Co-reporter:Li-Cheng Song;Fei-Xian Luo;Hao Tan;Xiao-Jing Sun;Zhao-Jun Xie ;Hai-Bin Song
European Journal of Inorganic Chemistry 2013 Volume 2013( Issue 14) pp:2549-2557
Publication Date(Web):
DOI:10.1002/ejic.201300038
Abstract
The first two model compounds for [FeFe]-hydrogenases that contain a subphthalocyanine (SubPc) macrocycle, namely, [{(μ-SCH2)2N(CH2)2CO2-3-C6H4S2C6H4-3′-O(SubPc)}Fe2(CO)6] (5) and [{(μ-SCH2)2NC6H4-4-O(SubPc)}Fe2(CO)6] (8), have been synthesized and structurally characterized. The treatment of chlorosubphthalocyanine (SubPc-Cl, 1) with (3-HOC6H4S)2 in toluene gave the corresponding phenoxy-substituted SubPc derivative 3-HOC6H4S2C6H4-3′-O(SubPc) (2) in 78 % yield, whereas the reaction of in-situ-generated [(μ-HOCH2S)2Fe2(CO)6] (3) with β-alanine afforded diiron complex [{(μ-CH2S)2N(CH2)2CO2H}Fe2(CO)6] (4) in 53 % yield. Further treatment of 2 with 4 in the presence of N,N′-dicyclocarbodiimide (DCC) and N,N-dimethyl-4-aminopyridine (DMAP) in CH2Cl2 resulted in the formation of model compound 5 in 86 % yield. Model compound 8 could be prepared by two methods. One method involves the reaction of in-situ-generated 3 with 4-aminophenol in tetrahydrofuran (THF) to give diiron complex [{(μ-CH2S)2NC6H4OH-4}Fe2(CO)6] (6) in 61 % yield and further treatment of SubPc-Cl (1) with 6 in toluene to give 8 in 13 % yield. The other method involves the reaction of SubPc-Cl (1) with silver triflate (AgOTf) followed by treatment of the resulting intermediate SubPc-OTf (7) with 6 in the presence of Et3N to produce 8 in a much higher yield (59 %). All the new precursors (2, 4, and 6) and the model compounds 5 and 8 have been fully characterized by elemental analysis and various spectroscopy techniques, as well by X-ray crystallography for 2, 4, 6, and 8. In addition, the photoinduced H2 production catalyzed by model 8 was preliminarily investigated.
Co-reporter:Li-Cheng Song, Gao-Yu Zhao, Zhao-Jun Xie, and Ji-Wei Zhang
Organometallics 2013 Volume 32(Issue 9) pp:2509-2512
Publication Date(Web):April 30, 2013
DOI:10.1021/om400240e
On synthesizing the PMB-protected mononuclear iron complexes [2-C(O)CH2-6-PMBOC5H3N]Fe(CO)3I (6) and [2-C(O)CH2-6-PMBOC5H3N]Fe(CO)2(2-SC5H4N) (7), the novel acylmethylpyridinol ligand containing dinuclear iron complex [2-C(O)CH2-6-HOC5H3N]Fe2(CO)4[2′-C(O)CH2-6′-OC5H3N](2-SC5H4N) (9), which is closely related to the active site of [Fe]-hydrogenase, has been prepared unexpectedly via removal of the PMB protecting group from 7 under the action of excess trifluoroacetic acid. The [Fe]-hydrogenase-related complex 9 and its precursor complexes 6 and 7 have been fully characterized by elemental analysis, spectroscopy, and X-ray crystallography.
Co-reporter:Li-Cheng Song, Bin Gai, Zhan-Heng Feng, Zong-Qiang Du, Zhao-Jun Xie, Xiao-Jing Sun, and Hai-Bin Song
Organometallics 2013 Volume 32(Issue 13) pp:3673-3684
Publication Date(Web):June 19, 2013
DOI:10.1021/om400309j
Parent complexes (μ-ODSe)Fe2(CO)6 (A, ODSe = SeCH2OCH2Se) and (μ-TDSe)Fe2(CO)6 (B, TDSe = SeCH2SCH2Se) could be prepared by oxidative addition of (HSeCH2)2X (X = O, S) with Fe3(CO)12. While reactions of A with 1 equiv of monophosphines in the presence of the decarbonylating agent Me3NO afforded the corresponding phosphine-monosubstituted complexes (μ-ODSe)Fe2(CO)5(L) (1, L = Ph3P; 2, L = Ph2POMe), the N-heterocyclic carbene (NHC)-monosubstituted complexes (μ-ODSe)Fe2(CO)5(L) (3, L = IMes; 4, L = IMes/Me) were prepared by reactions of the 1,3-bis(mesityl)imidazolium salt IMes·HCl and 1-mesityl-3-methylimidazolium salt IMes/Me·HI with n-BuLi, followed by treatment of the corresponding NHC intermediates with A. The phosphine-containing imidazolium salt IMes/CH2CH2PPh2·HCl reacted with A in the presence of Me3NO to give the imidazolium/phosphine-monosubstituted complex (μ-ODSe)Fe2(CO)5(IMes/CH2CH2PPh2·HCl) (5), whereas it reacted with t-BuOK or n-BuLi, followed by treatment of A with the resulting intermediate NHC/phosphine or both the resulting NHC/phosphine and phosphine Ph2PCH═CH2, to afford the corresponding NHC/phosphine-disubstituted complex [(μ-ODSe)Fe2(CO)5]2(IMes/CH2CH2PPh2) (6) and complex 6 along with (μ-ODSe)Fe2(CO)5(Ph2PCH═CH2) (7), respectively. In addition, 7 could also be produced simply by reaction of 6 with n-BuLi. The phosphine-monosubstituted complexes (μ-TDSe)Fe2(CO)5(L) (8, L = Ph3P; 9, L = Ph2PH) were similarly prepared by reactions of B with 1 equiv of the corresponding monophosphines in the presence of Me3NO, whereas reaction of B with m-chloroperoxybenzoic acid afforded the corresponding bridgehead S atom-oxidized complex (μ-TDSeO)Fe2(CO)6 (10, TDSeO = SeCH2S(O)CH2Se). While complexes A/B and 1–10 were structurally characterized, a comparative study on H2 production from HOAc catalyzed by parent complexes A/B and their sulfur analogues (μ-ODT)Fe2(CO)6/(μ-TDT)Fe2(CO)6 was carried out.
Co-reporter:Li-Cheng Song, Ling Li, Yuan-Yuan Hu, Hai-Bin Song
Journal of Organometallic Chemistry 2013 743() pp: 123-129
Publication Date(Web):
DOI:10.1016/j.jorganchem.2013.06.043
Co-reporter:Li-Cheng Song, Zhao-Jun Xie, Miao-Miao Wang, Gao-Yu Zhao, and Hai-Bin Song
Inorganic Chemistry 2012 Volume 51(Issue 14) pp:7466-7468
Publication Date(Web):July 3, 2012
DOI:10.1021/ic301146u
The first acylmethyl(hydroxymethyl)pyridine ligand-containing [Fe]hydrogenase model complexes 2–4 have been synthesized starting from the nucleophilic substitution reaction of 2-(4-MeC6H4SO3CH2)-6-HOCH2C5H3N with Na2Fe(CO)4. While the reaction course for producing complex 3 via the highly unstable intermediate complex 1 is monitored by in situ IR spectroscopy, the isolated model complexes 2–4 are fully characterized.
Co-reporter:Li-Cheng Song, Xiao-Jing Sun, Pei-Hua Zhao, Jia-Peng Li and Hai-Bin Song
Dalton Transactions 2012 vol. 41(Issue 29) pp:8941-8950
Publication Date(Web):22 May 2012
DOI:10.1039/C2DT30609C
The [N2S2]-type ligand 1,2-(2-C5H4NCH2S)2C6H4 (L) is prepared in 84% yield by a new method and its structure has been confirmed by X-ray crystallography. The new synthetic method involves sequential reaction of 1,2-phenylenedithiol with EtONa followed by treatment of the resulting disodium salt of 1,2-phenylenedithiol with in situ generated 2-(chloromethyl)pyridine from its HCl salt. Further treatment of ligand L with NiCl2·6H2O or NiI2 affords the expected new mononuclear Ni complexes Ni[1,2-(2-C5H4NCH2S)2C6H4]Cl2 (1) and Ni[1,2-(2-C5H4NCH2S)2C6H4]I2 (3) in 87–88% yields, whereas reaction of L with NiBr2 under similar conditions results in formation of the expected new mononuclear complex Ni[1,2-(2-C5H4NCH2S)2C6H4]Br2 (2) and one unexpected new mononuclear complex Ni[1-(2-C5H4NCH2S)-2-(2-C5H4NCH2SC6H4S)C6H4]Br2 (2*) in 82% and 5% yields, respectively. More interestingly, the ligand L-containing novel trinuclear NiFe2 complex Ni{[1,2-(2-C5H4NCH2S)2C6H4}Fe2(CO)6(μ3-S)2 (4) is found to be prepared by sequential reaction of (μ-S2)Fe2(CO)6 with Et3BHLi, followed by treatment of the resulting (μ-LiS)2Fe2(CO)6 with mononuclear complex 1, 2, or 3 in 12–20% yields. The new complexes 1–4 and 2* are fully characterized by elemental analysis and various spectroscopies, and the crystal structures of 1, 2* and 3 as well as some electrochemical properties of 1–4 are also reported.
Co-reporter:Li-Cheng Song, Wei Gao, Xiang Luo, Zhi-Xuan Wang, Xiao-Jing Sun, and Hai-Bin Song
Organometallics 2012 Volume 31(Issue 8) pp:3324-3332
Publication Date(Web):March 27, 2012
DOI:10.1021/om300136b
A series of new benzyloxy-functionalized 1,3-propanedithiolate (PDT)-type model complexes (A and 1–7) have been synthesized and structurally characterized. The benzyloxy-functionalized all-carbonyl complex [(μ-SCH2)2CH(OCH2Ph)]Fe2(CO)6 (A) can be prepared by condensation reaction of 2-benzyloxy-1,3-dibromopropane with the in situ generated (μ-LiS)2Fe2(CO)6, whereas it reacts with the in situ formed N-heterocyclic carbene 1-mesityl-3-methylimidazol-2-ylidene (IMes/Me) to give the corresponding carbene monosubstituted complex [(μ-SCH2)2CH(OCH2Ph)]Fe2(CO)5(IMes/Me) (1). The PMe3-monosubstituted complex [(μ-SCH2)2CH(OCH2Ph)]Fe2(CO)5(PMe3) (2) can be prepared by substitution of the CO ligand in parent complex A with 1 equiv of PMe3 in the presence of Me3NO·2H2O, whereas PPh3-monosubstituted and PPh3-disubstituted complexes [(μ-SCH2)2CH(OCH2Ph)]Fe2(CO)5(PPh3) (3) and [(μ-SCH2)2CH(OCH2Ph)]Fe2(CO)4(PPh3)2 (4) are prepared by reaction of A with 2 equiv of PPh3 under similar conditions. While the PPh3-disubstituted complex 4 can also be prepared by treatment of 3 in MeCN with 2 equiv of PPh3 in the presence of Me3NO·2H2O, treatment 4 with 2 equiv of PMe3 in refluxing toluene afforded unexpected PPh3/PMe3-disubstituted complex [(μ-SCH2)2CH(OCH2Ph)]Fe2(CO)4(PPh3)(PMe3) (5). Particularly interesting is that although the reaction of A with 1 equiv of diphosphine dppe in refluxing toluene affords the dppe-chelated single-butterfly complex [(μ-SCH2)2CH(OCH2Ph)]Fe2(CO)4(dppe) (6), treatment of A in MeCN with 1 equiv of dppe in the presence of Me3NO·2H2O results in formation of the dppe-bridged double-butterfly complex {[(μ-SCH2)2CH(OCH2Ph)]Fe2(CO)5}2(dppe) (7). All new model complexes have been chatacterized by elemental analysis, spectroscopy, and particularly for 1 and 4–7 X-ray crystallography. Furthermore, complexes A, 3, and 4 have been found to be catalysts for HOAc proton reduction to H2 under electrochemical conditions.
Co-reporter:Li-Cheng Song, Liang-Xing Wang, Guo-Jun Jia, Qian-Li Li, and Jiang-Bo Ming
Organometallics 2012 Volume 31(Issue 14) pp:5081-5088
Publication Date(Web):July 5, 2012
DOI:10.1021/om300418z
The first phosphinobenzaldehyde- and phosphinoporphyrin-functionalized model complexes (1–4) have been synthesized and structurally characterized. Thus, reaction of diiron complex [(μ-SCH2)2CH2]Fe2(CO)6 or [(μ-SCH2)2NC6H4CO2Me-p]Fe2(CO)6 with p-Ph2PC6H4CHO in the presence of Me3NO gave the phosphinobenzaldehyde-functionalized model complexes [(μ-SCH2)2CH2]Fe2(CO)5(p-Ph2PC6H4CHO) (1) and [(μ-SCH2)2NC6H4CO2Me-p]Fe2(CO)5(p-Ph2PC6H4CHO) (2) in 61% and 71% yields, respectively. Further reaction of 1 or 2 with PhCHO, pyrrole, and BF3·OEt2 followed by treatment with p-chloranil resulted in formation of the phosphinoporphyrin-functionalized model complexes 5-{[(μ-SCH2)2CH2]Fe2(CO)5(p-Ph2PC6H4)}-10,15,20-triphenylporphyrin (3) and 5-{[(μ-SCH2)2NC6H4CO2Me-p]Fe2(CO)5(p-Ph2PC6H4)}-10,15,20-triphenylporphyrin (4) in 19% and 18% yields, respectively. While the new complexes 1–4 were characterized by elemental analysis and spectroscopy, the structures of 1, 3, and 4 were confirmed by X-ray crystallography. Particularly interesting is that complex 4 was found to be a catalyst for the photoinduced H2 production in the presence of the electron donor EtSH and the proton source HOAc.
Co-reporter:Li-Cheng Song ; Chun-Hui Qi ; Hai-Lin Bao ; Xiao-Niu Fang ;Hai-Bin Song
Organometallics 2012 Volume 31(Issue 15) pp:5358-5370
Publication Date(Web):July 17, 2012
DOI:10.1021/om300395z
The 1,2,4,5-(CH2)4C6H2 moiety-containing butterfly Fe/S complexes [1,2-(CH2S2CH2)-4,5-(μ-SCH2)2C6H2][Fe2(CO)6] (1), [1,2,4,5-(μ-SCH2)4C6H2][Fe2(CO)6]2 (2), and [(1,2-Me2)-4,5-(μ-SCH2)2C6H2][Fe2(CO)6] (3) have been prepared by the reaction of tetrathiol 1,2,4,5-(HSCH2)4C6H2 with Fe3(CO)12 under different conditions. Treatment of complex 1 with PPh3 or PMe3 in the presence of Me3NO or with the in situ generated N-heterocyclic carbene IMes (IMes = 1,3-bis(mesityl)imidazol-2-ylidene) afforded the corresponding monosubstituted single-butterfly complexes [1,2-(CH2S2CH2)-4,5-(μ-SCH2)2C6H2][Fe2(CO)5L] (4, L = PPh3; 5, L = PMe3; 6, L = IMes). However, in contrast to the above-mentioned reaction of tetrathiol 1,2,4,5-(HSCH2)4C6H2 with Fe3(CO)12, the reactions of 1,2,4,5-(HSCH2)4C6H2 with Fe3(CO)12 in the presence of Et3N, followed by treatment with PhCOCl or PhC(Cl)═NPh, resulted in formation of the quadruple- and triple-butterfly complexes [(μ-PhC═O)Fe2(CO)6]4[1,2,4,5-(μ-SCH2)4C6H2] (7) and [(μ-PhC═NPh)Fe2(CO)6]2[Fe2(CO)6][1,2,4,5-(μ-SCH2)4C6H2] (8), whereas the same tetrathiol reaction system reacted with PhC≡CH to give quadruple- and triple-butterfly complexes [(μ-σ,π-PhCH═CH)Fe2(CO)6]4[1,2,4,5-(μ-SCH2)4C6H2] (9) and [(μ-σ,π-PhCH═CH)Fe2(CO)6]2[Fe2(CO)6][1,2,4,5-(μ-SCH2)4C6H2] (10). More interestingly, the dithioformato ligand-containing quadruple-butterfly complex [(μ-S═CSCH2Ph)Fe2(CO)6]4[1,2,4,5-(μ-SCH2)4C6H2] (11) could be prepared by the reaction of the tetrathiol system with CS2, followed by treatment with PhCH2Br, but the same tetrathiol/CS2 system reacted with CpFe(CO)2I to afford the triple-butterfly complex [(μ-S═CSFe(CO)2Cp)Fe2(CO)6]2[Fe2(CO)6][1,2,4,5-(μ-SCH2)4C6H2] (12). The possible pathways for production of complexes 1–3 and 7–12 are proposed, and the suggested intermediate [1,2-(HSCH2)2-4,5-(μ-SCH2)2C6H2][Fe2(CO)6] (M6) for formation of complex 1 has been successfully isolated under anaerobic conditions. The structures of complexes 1, 3, 4, 8, 9, and 11 have been confirmed by X-ray crystallography.
Co-reporter:Li-Cheng Song ; Xu-Feng Liu ; Zhao-Jun Xie ; Fei-Xian Luo ;Hai-Bin Song
Inorganic Chemistry 2011 Volume 50(Issue 21) pp:11162-11172
Publication Date(Web):October 14, 2011
DOI:10.1021/ic201713x
The [3 + 2] cycloaddition reaction of C60 with ethyl isonicotinoylacetate in the presence of piperidine in PhCl at room temperature or in the presence of Mn(OAc)3 in refluxing PhCl gave the pyridyl-containing dihydrofuran-fused C60 derivative (4-C5H4N)C(O)═C(C60)CO2Et (1), whereas the phenyl-containing C60 derivative PhC(O)═C(C60)CO2Et (2) was similarly prepared by [3 + 2] cycloaddition reaction of C60 with ethyl benzoylacetate in the presence of piperidine or Mn(OAc)3. More interestingly, one of the new porphyrin-fullerene dyads, i.e., [4-C5H4NC(O)═C(C60)CO2Et]·ZnTPPH (3, ZnTPPH = tetraphenylporphyrinozinc), could be prepared by coordination reaction of the pyridyl-containing C60 derivative 1 with equimolar ZnTPPH in CS2/hexane at room temperature. In addition, the β-keto ester-substituted porphyrin derivative H2TPPC(O)CH2CO2Et (4) was prepared by a sequential reaction of HO2CCH2CO2Et with n-BuLi in 1:2 molar ratio followed by treatment with H2TPPC(O)Cl in the presence of Et3N and then hydrolysis with diluted HCl, whereas the porphyrinozinc derivative ZnTPPC(O)CH2CO2Et (5) could be prepared by coordination reaction of 4 with Zn(OAc)2 in refluxing CHCl3/MeOH. Particularly interesting is that the second new porphyrin-fullerene dyad H2TPPC(O)═C(C60)CO2Et (6) could be prepared by [3 + 2] cycloaddition reaction of 4 with C60 in the presence of piperidine in PhCl at room temperature. In addition, treatment of 6 with Zn(OAc)2 in refluxing CHCl3/MeOH afforded the third new dyad ZnTPPC(O)═C(C60)CO2Et (7). All the new compounds 1–7 were characterized by elemental analysis and various spectroscopic methods and particularly for 2, 3, and 5 by X-ray crystallography. The five-component system consisting of an electron donor EDTA, dyad 3, an electron mediator methylviologen (MV2+), the catalyst colloidal Pt, and a proton source HOAc was proved to be effective for photoinduced H2 evolution. A possible pathway for such a type of H2 evolution was proposed.
Co-reporter:Li-Cheng Song, Zhao-Jun Xie, Xu-Feng Liu, Jiang-Bo Ming, Jian-Hua Ge, Xiao-Guang Zhang, Tian-Ying Yan and Peng Gao
Dalton Transactions 2011 vol. 40(Issue 4) pp:837-846
Publication Date(Web):09 Dec 2010
DOI:10.1039/C0DT00909A
A series of new diiron azadithiolate (ADT) complexes (1–8), which could be regarded as the active site models of [FeFe]hydrogenases, have been synthesized starting from parent complex [(μ-SCH2)2NCH2CH2OH]Fe2(CO)6 (A). Treatment of A with ethyl malonyl chloride or malonyl dichloride in the presence of pyridine afforded the malonyl-containing complexes [(μ-SCH2)2NCH2CH2O2CCH2CO2Et]Fe2(CO)6 (1) and [Fe2(CO)6(μ-SCH2)2NCH2CH2O2C]2CH2 (2). Further treatment of 1 and 2 with PPh3 under different conditions produced the PPh3-substituted complexes [(μ-SCH2)2NCH2CH2O2CCH2CO2Et]Fe2(CO)5(PPh3) (3), [(μ-SCH2)2NCH2CH2O2CCH2CO2Et]Fe2(CO)4(PPh3)2 (4), and [Fe2(CO)5(PPh3)(μ-SCH2)2NCH2CH2O2C]2CH2 (5). More interestingly, complexes 1–3 could react with C60 in the presence of CBr4 and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) via Bingel–Hirsch reaction to give the C60-containing complexes [(μ-SCH2)2NCH2CH2O2CC(C60)CO2Et]Fe2(CO)6 (6), [Fe2(CO)6(μ-SCH2)2NCH2CH2O2C]2C(C60) (7), and [(μ-SCH2)2NCH2CH2O2CC(C60)CO2Et]Fe2(CO)5(PPh3) (8). The new ADT-type models 1–8 were characterized by elemental analysis and spectroscopy, whereas 2–4 were further studied by X-ray crystallography and 6–8 investigated in detail by DFT methods.
Co-reporter:Wei Gao, Li-Cheng Song, Bang-Shao Yin, Hui-Ning Zan, De-Fu Wang, and Hai-Bin Song
Organometallics 2011 Volume 30(Issue 15) pp:4097-4107
Publication Date(Web):July 6, 2011
DOI:10.1021/om200395g
As the active site mimics of [FeFe]-hydrogenases, 14 new butterfly [2Fe2E] (E = Se, S) cluster complexes have been prepared by various synthetic routes. The N-substituted single-butterfly [2Fe2Se] complexes [(μ-SeCH2)2NC(O)R]Fe2(CO)6 (1, R = Me; 2, R = Ph; 3, R = PhCH2O) were prepared by reactions of the in situ formed (μ-LiSe)2Fe2(CO)6 with RC(O)N(CH2Cl)2, whereas the corresponding [2Fe2S] complexes [(μ-SCH2)2NC6H4R-p]Fe2(CO)6 (4, R = CO2Et; 5, R = CH2OH) were produced by reaction of the in situ generated (μ-HS)2Fe2(CO)6 with aqueous CH2O followed by treatment with p-RC6H4NH2. The parent single-butterfly [2Fe2Se] complex [(μ-SeCH2)2NH]Fe2(CO)6 (6) could be prepared by reaction of the N-substituted complex 3 with deprotecting reagent BBr3, BF3·OEt2/EtSH, or BF3·OEt2/Me2S, whereas the N-substituted single-butterfly [2Fe2Se] complexes [(μ-SeCH2)2NC(O)R]Fe2(CO)6 (7, R = Et; 8, R = PhCH2) were produced by reactions of 6 with acylating agents RC(O)Cl in the presence of Et3N. While the known parent single-butterfly [2Fe2S] complex [(μ-SCH2)2NH]Fe2(CO)6 reacted with 2,6-[ClC(O)]2C5H3N to afford double-butterfly [2Fe2S] complex [Fe2(CO)6(μ-SCH2)2NC(O)]2(2,6-C5H3N) (9), the new N-hydroxyethyl-substituted single-butterfly [2Fe2Se] complex [(μ-SeCH2)2N(CH2)2OH]Fe2(CO)6 (10) could be obtained by the in situ reaction of (μ-HSe)2Fe2(CO)6 with (HOCH2)2N(CH2)2OH. Interestingly, complex 10 could react with [ClC(O)]2CH2 or 1,3,5-[ClC(O)]3C6H3 in the presence of Et3N to give the corresponding double-butterfly [2Fe2Se] complex [Fe2(CO)6(μ-SeCH2)2N(CH2)2O2C]2CH2 (11) and triple-butterfly complex [Fe2(CO)6(μ-SeCH2)2N(CH2)2O2C]3(1,3,5-C6H3) (12), whereas the known single-butterfly [2Fe2S] complex [(μ-SCH2)2N(CH2)2OH]Fe2(CO)6 could react with 2,6-[ClC(O)]2C5H3N and 1,3,5-[ClC(O)]3C6H3 in the presence of Et3N to afford the corresponding double-butterfly [2Fe2S] complex [Fe2(CO)6(μ-SCH2)2N(CH2)2O2C]2(2,6-C5H3N) (13) and triple-butterfly complex [Fe2(CO)6(μ-SCH2)2N(CH2)2O2C]3(1,3,5-C6H3) (14), respectively. All the new complexes 1–14 have been characterized by elemental analysis and spectroscopy, as well as by X-ray crystallography for 1–4, 7–9, and 14. In addition, the electrochemical study indicated that complexes 1 and 2 can catalyze the proton reduction of HOAc to give hydrogen.
Co-reporter:Ling Li, Li-Cheng Song, Miao-Miao Wang, Qian-Li Li, and Hai-Bin Song
Organometallics 2011 Volume 30(Issue 18) pp:4899-4909
Publication Date(Web):August 25, 2011
DOI:10.1021/om2004767
Three new series of (diphosphine)Ni-bridged butterfly Fe/E (E = S, Se) cluster complexes have been prepared and structurally characterized. One such series of complexes includes the linear type of (diphosphine)Ni-bridged double-butterfly Fe/E complexes [(μ-RSe)(μ-S═CS)Fe2(CO)6]2[Ni(diphosphine)] (1–8, R = n-Bu, Ph; diphosphine = dppe, dppv, dppb, dppp, dppf). These complexes were prepared by one-pot reactions of the monoanion [(μ-RSe)(μ-CO)Fe2(CO)6]− (generated in situ from Fe3(CO)12, RSeH, and Et3N) with excess CS2 followed by treatment of the resulting monoanion [(μ-RSe)(μ-S═CS)Fe2(CO)6]− with (diphosphine)NiCl2. The second and third series include the macrocyclic type of (diphosphine)Ni-bridged quadruple- and double-butterfly Fe/E complexes [{μ-Se(CH2)3Se-μ}{(μ-S═CS)Fe2(CO)6}2]2[Ni(diphosphine)]2 (9a–11a, diphosphine = dppe, dppv, dppb) and [μ-Se(CH2)3Se(CH2)3Se-μ][(μ-S═CS)Fe2(CO)6]2[Ni(diphosphine)] (9b–11b, diphosphine = dppe, dppv, dppb), respectively. While macrocycles 9a–11a were produced by one-pot reactions of the dianion [{μ-Se(CH2)3Se-μ}{(μ-CO)Fe2(CO)6}2]2– (formed in situ from Fe3(CO)12, HSe(CH2)3SeH, and Et3N) with excess CS2 followed by treatment of the resulting dianion [{μ-Se(CH2)3Se-μ}{(μ-S═CS)Fe2(CO)6}2]2– with (diphosphine)NiCl2, 9b–11b were obtained by reactions of the dianion [{μ-Se(CH2)3Se(CH2)3Se-μ}{(μ-CO)Fe2(CO)6}2]2– (formed in situ from Fe3(CO)12, HSe(CH2)3Se(CH2)3SeH, and Et3N) with excess CS2 and subsequent treatment of the resultant [Et3NH]+ salt of the two-μ-CS2-containing dianion [{μ-Se(CH2)3Se(CH2)3Se-μ}{(μ-S═CS)Fe2(CO)6}2]2– with (diphosphine)NiCl2. A possible pathway for production of the two types of novel macrocycles 9a–11a and 9b–11b is suggested. In addition, the structures of all the linear and macrocyclic products were characterized by elemental analysis and spectroscopy and, for some of them, by X-ray crystallography.
Co-reporter:Li-Cheng Song ; Yu-Long Li ; Ling Li ; Zhen-Chao Gu ;Qing-Mei Hu
Inorganic Chemistry 2010 Volume 49(Issue 21) pp:10174-10182
Publication Date(Web):September 29, 2010
DOI:10.1021/ic101451y
Three series of new Ni/Fe/S cluster complexes have been prepared and structurally characterized. One series of such complexes includes the linear type of (diphosphine)Ni-bridged double-butterfly Fe/S complexes [(μ-RS)(μ-S═CS)Fe2(CO)6]2[Ni(diphosphine)] (1−6; R = Et, t-Bu, n-Bu, Ph; diphosphine = dppv, dppe, dppb), which were prepared by reactions of monoanions [(μ-RS)(μ-CO)Fe2(CO)6]− (generated in situ from Fe3(CO)12, Et3N, and RSH) with excess CS2, followed by treatment of the resulting monoanions [(μ-RS)(μ-S═CS)Fe2(CO)6]−with (diphosphine)NiCl2. The second series consists of the macrocyclic type of (diphosphine)Ni-bridged double-butterfly Fe/S complexes [μ-S(CH2)4S-μ][(μ-S═CS)Fe2(CO)6]2[Ni(diphosphine)] (7−9; diphosphine = dppv, dppe, dppb), which were produced by the reaction of dianion [{μ-S(CH2)4S-μ}{(μ-CO)Fe2(CO)6}2]2− (formed in situ from Fe3(CO)12, Et3N, and dithiol HS(CH2)4SH with excess CS2, followed by treatment of the resulting dianion [{μ-S(CH2)4S-μ}{(μ-S═CS)Fe2(CO)6}2]2− with (diphosphine)NiCl2. However, more interestingly, when dithiol HS(CH2)4SH (used for the production of 7−9) was replaced by HS(CH2)3SH (a dithiol with a shorter carbon chain), the sequential reactions afforded another type of macrocyclic Ni/Fe/S complex, namely, the (diphosphine)Ni-bridged quadruple-butterfly Fe/S complexes [{μ-S(CH2)3S-μ}{(μ-S═CS)Fe2(CO)6}2]2[Ni(diphosphine)]2 (10−12; diphosphine = dppv, dppe, dppb). While a possible pathway for the production of the two types of novel metallomacrocycles 7−12 is suggested, all of the new complexes 1−12 were characterized by elemental analysis and spectroscopy and some of them by X-ray crystallography.
Co-reporter:Li-Cheng Song;Qing-Shan Li;Zhi-Yong Yang;Yu-Juan Hua;Hong-Zhu Bian ;Qing-Mei Hu
European Journal of Inorganic Chemistry 2010 Volume 2010( Issue 7) pp:1119-1128
Publication Date(Web):
DOI:10.1002/ejic.200901023
Abstract
On the basis of preparation of the known complex [Fe2(μ-SCH2)2S(CO)6] (A) by a new method involving condensation of [(μ-LiS)2Fe2(CO)6] with excess S(CH2Br)2, twelve new diiron thiadithiolates as mimics of the [FeFe]-hydrogenase active site have been synthesized by substitution of the CO ligand and coordination at the central S atom of complex A with appropriate reagents. Treatment of A with 1 equiv. of the monodentate ligands PPh3 and [(η5-C5H5)(η5-Ph2PC5H4)Fe] in the presence of Me3NO and with 1 equiv. tBuNC and cyclohexyl isocyanide gave the single [2Fe3S]-cluster-containing monosubstituted complexes [Fe2(μ-SCH2)2S(CO)5(L1)] (1, L1 = PPh3; 2, L1 = (η5-C5H5)(η5-Ph2PC5H4)Fe; 3, L1 = tBuNC; 4, L1 = C6H11NC), whereas the double [2Fe3S]-cluster-containing disubstituted complexes [{Fe2(μ-SCH2)2S(CO)5}2(L2)] [5, L2 = 4,4′-(Ph2P)2(C6H4)2; 6, L2 = trans-Ph2PCH=CHPPh2; 7, L2 = 1,4-(CN)2C6H4; 8, L2 = (η5-Ph2PC5H4)2Fe; 9, L2 = (η5-Ph2PC5H4)2Ru] were produced by reaction of A with 0.5 equiv. of the corresponding bidentate ligands in the presence of Me3NO. In addition, the single [2Fe3S]-cluster-containing complexes in which the central S atom is coordinated, [{Fe2(μ-SCH2)2S(CO)6}{(η5-MeC5H4)(CO)2Fe}(BF4)] (10), [{Fe2(μ-SCH2)2S(CO)6}{Cr(CO)5}] (11), and [{Fe2(μ-SCH2)2S(CO)6}{W(CO)5}] (12), could be obtained by reaction of complex A with the in situ prepared [{(η5-MeC5H4)(CO)2Fe}(BF4)], [Cr(CO)5(thf)], and [W(CO)5(thf)], respectively. While complex 3 was found to be able to reduce the proton of the weak acid Et3NHCl to give H2, the X-ray crystallographic study confirmed that (i) each P atom of the phosphane ligands in 1 and 8 occupies an apical position at the Fe atoms, (ii) the isocyanide ligand in 3 lies in a basal position of the Fe atom, and (iii) the (η5-MeC5H4)(CO)2Fe, Cr(CO)5, and W(CO)5 units in 10–12 are linked to the central S atom of complex A by an equatorial bond from the two fused six-membered rings of their [2Fe3S]-cluster cores.
Co-reporter:Li-Cheng Song, Shu-Zhen Mei, Cui-Ping Feng, Feng-Hua Gong, Jian-Hua Ge, and Qing-Mei Hu
Organometallics 2010 Volume 29(Issue 21) pp:5050-5056
Publication Date(Web):June 22, 2010
DOI:10.1021/om1002655
We have first studied the reactions of monoanions [(μ-RE)(μ-E)Fe2(CO)6]− (A, E = Se; B, E = S) with electrophile PhC(Cl)═NPh or dianions [(μ-E)2Fe2(CO)6]2− (C, E = Se; D, E = S) with electrophiles PhC(Cl)═NR′ (R′ = Ph, p-MeC6H4). While monoanion A was found to react with PhC(Cl)═NPh to give expected complexes (μ-RSe)[μ-SeC(Ph)═NPh]Fe2(CO)6 (1, R = Me; 2, R = Et; 3, R = m-MeC6H4), monoanion B was found to react with PhC(Cl)═NPh to afford unexpected complexes (μ-RS)[η1-SC(Ph)═NPh-η1]Fe2(CO)6 (4, R = Me; 5, R = Et). Furthermore, while dianion C reacted with PhC(Cl)═NR′ (R′ = Ph, p-MeC6H4) to give expected complexes [μ-SeC(Ph)═NR′]2Fe2(CO)6 (6, R′ = Ph; 7, R′ = p-MeC6H4), dianion D was found to react with PhC(Cl)═NR′ (R′ = Ph, p-MeC6H4) to afford unexpected complexes [μ-SC(Ph)═NR′][η1-C(Ph)═NR′-η1]Fe2(CO)6 (8, R′ = Ph; 9, R′ = p-MeC6H4). All the new complexes 1−9 were characterized by elemental analysis and spectroscopy, and complexes 1 and 4−8 were further structurally confirmed by X-ray crystallography.
Co-reporter:Li-Cheng Song, Pei-Hua Zhao, Zong-Qiang Du, Ming-Yi Tang, and Qing-Mei Hu
Organometallics 2010 Volume 29(Issue 22) pp:5751-5753
Publication Date(Web):October 19, 2010
DOI:10.1021/om100793d
Reactions of butterfly complex (μ-HS)2Fe2(CO)6 (1) with RP(CH2OH)2 under N2 followed by TLC separation in air gave the unexpected tetrahedral complexes (μ-S2)Fe2(CO)5[RP(CH2OH)2] (4a, R = Ph; 4b, η5-C5H4CH2FeCp). While the suggested pathway for production of 4a,b was proved by isolation and characterization of the very air-sensitive intermediate (μ-HS)2Fe2(CO)5[PhP(CH2OH)2] (M1), the in situ reaction of M1 with I(CH2)3I and Et3N allowed us to obtain model complex (μ-PDT)Fe2(CO)5[PhP(CH2OH)2] (5).
Co-reporter:Li-Cheng Song, Jia Cheng, Jing Yan, Chun-Rong Liu and Qing-Mei Hu
Organometallics 2010 Volume 29(Issue 1) pp:205-213
Publication Date(Web):December 8, 2009
DOI:10.1021/om900899h
Trithiol MeC(CH2SH)3 reacted with Fe3(CO)12 and Et3N followed by treatment of the resulting three-μ-CO-containing trianion {[Fe2(μ-CO)(CO)6]3[(μ-SCH2)3CMe]}3− (A) with electrophile Ph2PCl to give triple-butterfly complex [Fe2(μ-Ph2P)(CO)6]3[(μ-SCH2)3CMe] (3), whereas double-butterfly complexes [Fe2(μ-PhC═NPh)(μ-SCH2)(CO)6][Fe2(μ-SCH2)2CMe(CO)6] (4), [Fe2(μ-S═CNHPh)(μ-SCH2)(CO)6][Fe2(μ-SCH2)2CMe(CO)6] (5), and [Fe2(μ-C6H11N═CNHC6H11)(μ-SCH2)(CO)6][Fe2(μ-SCH2)2CMe(CO)6] (6) were produced by reactions of electrophiles PhC(Cl)═NPh, C6H5N═C═S, and C6H11N═C═NC6H11 (DCC) with the one-μ-CO-containing monoanion {[Fe2(μ-CO)(μ-SCH2)(CO)6][Fe2(μ-SCH2)2CMe(CO)6]}− (B) generated in situ from the initially formed trianion A. More interestingly, both triple-butterfly complexes [Fe2(μ-RSC═S)(CO)6]3[(μ-SCH2)3CMe] (8a, R = Me; 8b, PhCH2) and double-butterfly complexes [Fe2(μ-RSC═S)(μ-SCH2)(CO)6][Fe2(μ-SCH2)2CMe(CO)6] (9a, R = Me; 9b, PhCH2) were obtained by reactions of trianion A and monoanion B with CS2/MeI or CS2/Ph2CH2Br, whereas the μ4-S-containing triple-butterfly complexes [Fe2(μ-RS)(CO)6][Fe2(μ4-S)(μ-SCH2)(CO)6][Fe2(μ-SCH2)2CMe(CO)6] (10a, R = Me; 10b, PhCH2) could be obtained by reactions of monoanion B with (μ-S2)Fe2(CO)6/MeI or (μ-S2)Fe2(CO)6/Ph2CH2Br, respectively. All these triple- and double-butterfly complexes were characterized by elemental analysis and spectroscopic techniques, as well as 3, 9a, and 10a,b by X-ray crystallography. In addition, single-butterfly complex Fe2(μ-t-BuS)(μ-C6H11N═CNHC6H11)(CO)6 (7) was prepared by reaction of monoanion [Fe2(μ-CO)(μ-t-BuS)(CO)6]− with DCC in order to confirm the coordination mode of the protonated DCC ligand in double-butterfly complex 6.
Co-reporter:Li-Cheng Song, Xu-Feng Liu, Jiang-Bo Ming, Jian-Hua Ge, Zhao-Jun Xie and Qing-Mei Hu
Organometallics 2010 Volume 29(Issue 3) pp:610-617
Publication Date(Web):January 5, 2010
DOI:10.1021/om9009526
As the active site models of FeFe-hydrogenases, a series of new diiron propanedithiolate compounds (1−7) have been synthesized starting from [(μ-SCH2)2CH(OH)]Fe2(CO)6 (A). Treatment of A with ethyl malonyl chloride or malonyl dichloride in the presence of pyridine gave the malonyl-containing compounds [(μ-SCH2)2CHO2CCH2CO2Et]Fe2(CO)6 (1) and [Fe2(CO)6(μ-SCH2)2CHO2C]2CH2 (2) in 64% and 55% yields, respectively. While A reacted with PPh3 in the presence of Me3NO to give the PPh3-substituted compound [(μ-SCH2)2CH(OH)]Fe2(CO)5(PPh3) (3) in 91% yield, reaction of 3 with malonyl dichloride in the presence of pyridine produced the malonyl-containing compound [Fe2(CO)5(PPh3)(μ-SCH2)2CHO2C]2CH2 (4) in 67% yield. More interestingly, compounds 1, 2, and 4 could react with C60 in the presence of CBr4 and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) via Bingel−Hirsch reaction to afford the [60]fullerene compounds [(μ-SCH2)2CHO2CC(C60)CO2Et]Fe2(CO)6 (5), [Fe2(CO)6(μ-SCH2)2CHO2C]2C(C60) (6), and [Fe2(CO)5(PPh3)(μ-SCH2)2CHO2C]2C(C60) (7) in 36−39% yields. While new compounds 1−7 were characterized by elemental analysis and various spectroscopic methods, 1−4 were further characterized by X-ray crystallography, and the photoinduced H2 evolution catalyzed by 5 was preliminarily investigated.
Co-reporter:Li-Cheng Song ; Jing Yan ; Yu-Long Li ; De-Fu Wang ;Qing-Mei Hu
Inorganic Chemistry 2009 Volume 48(Issue 23) pp:11376-11381
Publication Date(Web):October 27, 2009
DOI:10.1021/ic9006179
Five new l-cysteinyl group-containing diiron/triiron azadithiolate complexes (3−6, 10), which could be regarded as the active site models of [FeFe]-hydrogenases, have been successfully synthesized. Treatment of l-cysteinyl sodium mercaptide CytSNa (1, Cyt = CH2CH(CO2Et)NH(CO2Bu-t) with complex [(μ-SCH2)2NCH2CH2Br]Fe2(CO)6 (2) in THF at room temperature resulted in formation of model complex [(μ-SCH2)2NCH2CH2SCyt]Fe2(CO)6 (3). Further treatment of 3 with decarbonylating agent Me3NO in MeCN at room temperature afforded model complex [(μ-SCH2)2NCH2CH2SCyt]Fe2(CO)5 (4). Similarly, treatment of 3 with an equimolar mixture of Me3NO and Ph3P gave model complex [(μ-SCH2)2NCH2CH2SCyt]Fe2(CO)5(Ph3P) (5) and further treatment of 5 with Me3NO produced model complex [(μ-SCH2)2NCH2CH2SCyt]Fe2(CO)4(Ph3P) (6). More interestingly, model complex [(μ-SCH2)2NCH(CO2Et)CH2SFe(CO)2Cp]Fe2(CO)5 (10) could be synthesized by a “one pot” reaction of the in situ prepared (μ-HS)2Fe2(CO)6 (9) with 37% aqueous formaldehyde followed by treatment with the N-deprotected l-cysteinyl iron mercaptide Cp(CO)2FeSCH2CH(CO2Et)NH2 (8). Complex 8 is new, which was prepared by treatment of complex Cp(CO)2FeSCyt (7) with CF3CO2H followed by 25% aqueous NH3. All the new complexes 3−6, 8, and 10 were characterized by elemental analysis and various spectroscopic techniques, whereas complexes 5 and 10 were further characterized by X-ray crystallography.
Co-reporter:Li-Cheng Song;Guo-Xia Jin;Li-Qun Zhao;Hu-Ting Wang;Wen-Xiong Zhang ;Qing-Mei Hu
European Journal of Inorganic Chemistry 2009 Volume 2009( Issue 3) pp:419-428
Publication Date(Web):
DOI:10.1002/ejic.200800953
Abstract
Reactions of the tetrahedral C2Co2 cluster-bridged bipyridine ligand [(4-C5H4NCO2CH2)2C2Co2(CO)6] (La) with [{M(dppb)(H2O)2}(OTf)2] (M = Pd, Pt; dppb = 1,4-bis(diphenylphosphanyl)butane; OTf = SO3CF3) and AgX (X = NO3, SbF6) gave cationic metallamacrocycles [{M(dppb)La}2(OTf)4] (1, M = Pd; 2, M = Pt) and [(AgLa)2X2] (3, X = NO3; 4, X = SbF6). Furthermore, the isomeric C2Co2 cluster-bridged bipyridine ligand [(3-C5H4NCO2CH2)2C2Co2(CO)6] (Lb) reacted with ZnCl2 and AgX (X = PF6, ClO4) to afford cationic macrocycle [(ZnLbCl2)2] (5) and coordination polymers [(AgLbX)n] (6, X = PF6; 7, X = ClO4), respectively. Whereas flexible ligand La is known, Lb is new and could be prepared by reaction of but-2-yne-1,4-diyl dipyridine-3-carboxylate with [Co2(CO)8]. New self-assembled products 1–7 and ligand Lb were characterized by elemental analysis and spectroscopy, and 1, 3–7, and Lb were additionally characterized by X-ray crystallography. In addition, the electrochemistry of Lb and 5 were studied by cyclic voltammetry. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2009)
Co-reporter:Li-Cheng Song, Bin Gai, Hu-Ting Wang, Qing-Mei Hu
Journal of Inorganic Biochemistry 2009 Volume 103(Issue 5) pp:805-812
Publication Date(Web):May 2009
DOI:10.1016/j.jinorgbio.2009.02.002
As an extension of our study on the H-cluster model compounds, a series of diiron propanediselenolate (PDS)-type models have been successfully synthesized. Reaction of diselenol HSe(CH2)3SeH with Fe3(CO)12 in THF (tetrahydrofuran) at reflux gave the parent model compound [μ-Se(CH2)3Se-μ]Fe2(CO)6 (1) in 48% yield. Further reaction of 1 with PPh3 or PPh2H in the presence of Me3NO in MeCN at room temperature afforded the phosphine-monosubstituted model compounds [μ-Se(CH2)3Se-μ]Fe2(CO)5(L) (2, L = PPh3; 3, L = PPh2H) in 76% and 68% yields, respectively. Similarly, the N-heterocyclic carbene IMes-monosubstituted model compound [μ-Se(CH2)3Se-μ]Fe2(CO)5(IMes) (4) could be prepared in 46% yield by reaction of imidazolium salt IMes · HCl with n-BuLi followed by treatment of the resulting IMes ligand with 1 in THF at room temperature. Compounds 1–4 were fully characterized by elemental analysis and various spectroscopic methods. While the structures of 1–4 were further confirmed by X-ray crystallography, the comparative study of 1 and its analog [μ-S(CH2)3S-μ]Fe2(CO)6 demonstrates that 1 is a better catalyst for TsOH proton reduction to hydrogen under electrochemical conditions.
Co-reporter:Qingshan Li;Chuncheng Luo ;Licheng Song
Chinese Journal of Chemistry 2009 Volume 27( Issue 9) pp:1711-1715
Publication Date(Web):
DOI:10.1002/cjoc.200990288
Abstract
Reactions of one or two equiv. of cyclohexyl isocyanide in THF at room temperature with MoMo triply bonded complexes [Mo(CO)2(η5-C5H4R)]2 (R=COCH3, CO2CH3) gave the isocyanide coordinated MoMo singly bonded complexes with functionally substituted cyclopentadienyl ligands, [Mo(CO)2(η5-C5H4R)]2(μ-η2-CNC6H11) (1a, R=COCH3; 1b, R=CO2CH3) and [Mo(CO)2(η5-C5H4R)(CNC6H11)]2 (2a, R=COCH3; 2b, R=CO2CH3), respectively. Complexes 1a, 1b and 2a, 2b could be more conveniently prepared by thermal decarbonylation of MoMo singly bonded complexes [Mo(CO)3(η5-C5H4R)]2 (R=COCH3, CO2CH3) in toluene at reflux, followed by treatment of the resulting MoMo triply bonded complexes [Mo(CO)2(η5-C5H4R)]2 (R=COCH3, CO2CH3) in situ with cyclohexyl isocyanide. While 1a, 1b and 2a, 2b were characterized by elemental analysis and spectroscopy, 1b was further characterized by X-ray crystallography.
Co-reporter:Li-Cheng Song, Xiang Luo, Yong-Zhen Wang, Bin Gai, Qing-Mei Hu
Journal of Organometallic Chemistry 2009 694(1) pp: 103-112
Publication Date(Web):
DOI:10.1016/j.jorganchem.2008.10.015
Co-reporter:Li-Cheng Song, Liang-Xing Wang, Ming-Yi Tang, Chang-Gong Li, Hai-Bin Song and Qing-Mei Hu
Organometallics 2009 Volume 28(Issue 13) pp:3834-3841
Publication Date(Web):June 11, 2009
DOI:10.1021/om900141x
A series of novel light-driven-type models, which contain a single diiron- ADT(azadithiolate) unit or two and four diiron-ADT units covalently bonded to a photosensitizer porphyrin or metalloporphyrin, have been synthesized and structurally characterized. Reaction of complex [(μ-SCH2)2NC6H4CHO]Fe2(CO)6 (A) with PhCHO, pyrrole, and CF3CO2H in CH2Cl2 followed by treatment with p-chloranil gave light-driven models 5-[(μ-SCH2)2NFe2(CO)6phenyl]-10,15,20-triphenylporphyrin (1), 5,15-[(μ-SCH2)2NFe2(CO)6phenyl]2-10,20-diphenylporphyrin (2), and 5,10-[(μ-SCH2)2NFe2(CO)6phenyl]2-15,20-diphenylporphyrin (3). While light-driven model 5,10,15,20-[(μ-SCH2)2NFe2(CO)6phenyl]4porphyrin (4) could be similarly prepared by reaction of complex A with pyrrole and CF3CO2H followed by treatment with p-chloranil, model 1 could also be prepared by another new method involving a final cyclization step of (μ-HOCH2S)2Fe2(CO)6 with (p-aminophenyl)triphenylporphyrin. In addition, the PPh3-substituted model 5-[(μ-SCH2)2NFe2(CO)5(PPh3)phenyl]-10,15,20-triphenylporphyrin (5) was prepared by reaction of 1 with PPh3 in the presence of Me3NO, whereas treatment of 1 with Zn(OAc)2 afforded the metalloporphyrin-containing model 5-[(μ-SCH2)2NFe2(CO)6phenyl]-10,15,20-triphenylporphyrinozinc (6). X-ray crystallographic studies confirmed that (i) model 3 consists of two diiron-ADT units, which are connected to the two ortho-benzene rings of the porphyrin macrocycle, and (ii) model 6 contains one molecule of MeOH, which is axially coordinated to the Zn atom of the metalloporphyrin macrocycle. Particularly noteworthy is that model 1 was found to be a photoactive catalyst for photoinduced H2 production, and a possible pathway for such H2 production is suggested.
Co-reporter:Li-Cheng Song, Wei Gao, Cui-Ping Feng, De-Fu Wang and Qing-Mei Hu
Organometallics 2009 Volume 28(Issue 20) pp:6121-6130
Publication Date(Web):September 23, 2009
DOI:10.1021/om900572t
As a continuation of our studies on biomimetic chemistry and butterfly cluster chemistry, two series of “closed” and “open” butterfly [2Fe2Se] cluster complexes have been prepared in satisfactory yields. Thus, treatment of Fe3(CO)12 with (HSeCH2)2CHOH in toluene at reflux gave the expected “closed” butterfly [2Fe2Se] cluster complex [(μ-SeCH2)2CH(OH)]Fe2(CO)6 (A), whereas the “open” butterfly cluster complex (μ-EtSe)[(μ-SeCH2CH(OH)(CH2Br)]Fe2(CO)6 (B) was unexpectedly produced along with complex A via a sequential reaction of (μ-Se2)Fe2(CO)6 with Et3BHLi, followed by treatment with (BrCH2)2CHOH. The other “closed” and “open” cluster complexes 1−6 could be further prepared by the hydroxy transformation and CO substitution reactions of complexes A and B. For example, (i) reaction of A with PPh3 and decarbonylating agent Me3NO afforded PPh3-monosubstituted complex [(μ-SeCH2)2CH(OH)]Fe2(CO)5(PPh3) (1), (ii) further reaction of 1 with the acylating agent PhC(O)Cl in the presence of Et3N produced the benzoate-functionalized complex [(μ-SeCH2)2CH(O2CPh)]Fe2(CO)5(PPh3) (2), (iii) treatment of A with the phosphatizing agent Ph2PCl in the presence of Et3N or simply with PhPCl2 yielded the phosphite-functionalized complexes [(μ-SeCH2)2CH(OPPh2-η1)]Fe2(CO)5 (3) and [(μ-SeCH2)2CH(OPPhCl-η1)]Fe2(CO)5 (4), and (iv) treatment of B with 4-pyridinecarboxylic chloride or Ph2PCl in the presence of Et3N resulted in formation of the “open” butterfly cluster complexes (μ-EtSe)[μ-SeCH2CH(CH2Br)(O2CC5H4N-4)]Fe2(CO)6 (5) and (μ-EtSe)[μ-SeCH2CH(CH2Br)(OPPh2-η1)]Fe2(CO)5 (6). All the new complexes have been characterized by elemental analysis and spectroscopy, as well as for A, 1−4, and 6 by X-ray crystallography. Both 1H and 77Se NMR spectral studies demonstrated that complexes B and 5 consist of three isomers of e-Et/a-R, e-Et/e-R, and a-Et/e-R, whereas complex 6 exists only as one isomer of e-Et/a-R. On the basis of an electrochemical study, it was found that the “closed” and “open” complexes A and B can catalyze the proton reduction of TsOH and HOAc to give hydrogen, respectively.
Co-reporter:LiCheng Song
Science China Chemistry 2009 Volume 52( Issue 1) pp:1-14
Publication Date(Web):2009 January
DOI:10.1007/s11426-008-0122-4
This article describes recent developments in chemical study on a series of butterfly-shaped μ-CO-containing Fe/E (E = S, Se, Te) cluster salts. These salts include eleven novel cluster anions, which are the single butterfly one μ-CO-containing [(μ-RE)(μ-CO)Fe2(CO)6]− (A), the double butterfly two μ-CO-containing {[(μ-CO)Fe2(CO)6]2(μ-EZE-μ)}2− (B, E = S; C, E = Se), the triple butterfly three μ-CO-containing {[(μ-CO)Fe2(CO)6]3[(μ-SCH2CH2)3N]}3− (D), {[(μ-CO)Fe2(CO)6]3[1,3,5-(μ-SCH2)3C6H3]}3− (E), {[(μ-CO)Fe2(CO)6]3[(μ-SCH2)3CMe]}3− (F), the double butterfly one μ-CO-containing {[(μ-CO)Fe2(CO)6] [Fe2(CO)6][(μ-SCH2)3CMe]}− (G) derived in situ from F, the quadruple butterfly four μ-CO-containing {[(μ-CO)Fe2(CO)6]4[1,2,4,5-(μ-SCH2)4C6H2]}4− (H), the triple butterfly two μ-CO-containing {[(μ-CO)Fe2(CO)6]2 [Fe2(CO)6][1,2,4,5-(μ-SCH2)4C6H2]}2− (I) derived in situ from H, the quadruple butterfly four μ-CO-containing {[(μ-CO)Fe2(CO)6]4[(μ-SCH2)4C]}4− (J), and the triple butterfly two μ-CO-containing {[(μ-CO)Fe2 (CO)6]2[Fe2(CO)6][(μ-SCH2)4C]}2− (K) derived in situ from J. This article describes not only the synthetic methods for formation of such anionic cluster (A-K) salts, but also their novel reactions leading to various new types of butterfly Fe/E cluster complexes. All these findings described in this article are important both theoretically and practically.
Co-reporter:Li-Cheng Song ; Chang-Gong Li ; Jie Gao ; Bang-Shao Yin ; Xiang Luo ; Xiao-Guang Zhang ; Hai-Lin Bao ;Qing-Mei Hu
Inorganic Chemistry 2008 Volume 47(Issue 11) pp:4545-4553
Publication Date(Web):April 26, 2008
DOI:10.1021/ic701982z
New C-functionalized propanedithiolate-type model complexes (1−8) have been synthesized by functional transformation reactions of the known complex [(μ-SCH2)2CH(OH)]Fe2(CO)6 (A). Treatment of A with the acylating agents PhC(O)Cl, 4-pyridinecarboxylic acid chloride, 2-furancarbonyl chloride, and 2-thiophenecarbonyl chloride in the presence of Et3N affords the expected C-functionalized complexes [(μ-SCH2)2CHO2CPh]Fe2(CO)6 (1), [(μ-SCH2)2CHO2CC5H4N-4]Fe2(CO)6 (2), [(μ-SCH2)2CHO2CC4H3O-2]Fe2(CO)6 (3), and [(μ-SCH2)2CHO2CC4H3S-2]Fe2(CO)6 (4). However, when A is treated with the phosphatizing agents Ph2PCl, PCl3 and PBr3, both C- and Fe-functionalized complexes [(μ-SCH2)2CHOPPh2-η1]Fe2(CO)5 (5), [(μ-SCH2)2CHOPCl2-η1]Fe2(CO)5 (6), and [(μ-SCH2)2CHOPBr2-η1]Fe2(CO)5 (7) are unexpectedly obtained via intramolecular CO substitution by P atoms of the initially formed phosphite complexes. The simplest C-functionalized model complex [(μ-SCH2)2C═O]Fe2(CO)6 (8) can be produced by oxidation of A with Dess−Martin reagent. While 8 is found to be an electrocatalyst for proton reduction to hydrogen, starting complex A can be prepared by another method involving the reaction of HC(OH)(CH2Br)2 with the in situ generated (μ-LiS)2Fe2(CO)6. X-ray crystallographic studies reveal that the bridgehead C atom of 8 is double-bonded to an O atom to form a ketone functionality, whereas the bridgehead C atoms of A, 1, 3, and 4 are equatorially-bonded to their functionalities and those of 5−7 axially-bonded to their functionalities due to formation of the corresponding P−Fe bond-containing heterocycles.
Co-reporter:Li-Cheng Song;Jian-Hua Ge;Jing Yan;Hu-Ting Wang;Xiang Luo ;Qing-Mei Hu
European Journal of Inorganic Chemistry 2008 Volume 2008( Issue 1) pp:164-171
Publication Date(Web):
DOI:10.1002/ejic.200700821
Abstract
We have developed a simple and convenient method for the synthesis of the first H-cluster models in which a L-cysteinyl group is coordinated to one of the two iron atoms of the diiron subsite through its sulfur atom. This synthetic method includes (i) treatment of the Boc-protected L-cysteine ester Boc-NHCH(CH2SH)CO2Et (1, Boc = tert-butoxycarbonyl) with EtONa to give the L-cysteinyl mercaptide NaSCH2CH(NH-Boc)CO2Et (2); (ii) further treatment of 2 with [Cp(CO)2FeI] to produce the metallothioether ligand Cp(CO)2FeSCH2CH(NH-Boc)CO2Et (3); and (iii) treatment of the parent diiron complex [Fe2(μ-SCH2)2CH2(CO)6] (4), [Fe2(μ-SCH2)2N(tBu)(CO)6] (5), or [Fe2(μ-SCH2)2N(C6H4OMe-p)(CO)6] (6) with Me3NO·2H2O followed by ligand 3 to afford the target model compounds [Fe2(μ-SCH2)2CH2(CO)5(ligand 3)] (7), [Fe2(μ-SCH2)2N(tBu)(CO)5(ligand 3)] (8), or [Fe2(μ-SCH2)2N(C6H4OMe-p)(CO)5(ligand 3)] (9), respectively. All the new compounds 2, 3, and 7–9 have been characterized by elemental analysis and various spectroscopic techniques. The X-ray diffraction analysis of 8 has confirmed that these models contain a cysteinyl sulfur atom not only coordinated to one Fe atom of the diiron subsite, but also to the Fe atom of the Cp(CO)2Fe moiety to form the linkage [FeCp-(μ-cysteinyl-S)-Fesubsite], which is similar to [Fecubane-(μ-cysteinyl-S)-Fesubsite] found in natural enzymes. In addition, spectroscopic and electrochemical measurements have further demonstrated that the linkage [FeCp-(μ-cysteinyl-S)-Fesubsite] can provide substantial electronic communication between the diiron subsite and the Cp(CO)2Fe moiety. Under electrochemical conditions, 8 has been shown to be a catalyst for HOAc proton reduction to dihydrogen, and a new type of E*2E2C mechanism for this catalytic reaction is suggested.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2008)
Co-reporter:Li-Cheng Song;Liang-Xing Wang;Bang-Shao Yin;Yu-Long Li;Xiao-Guang Zhang;Yuan-Wei Zhang;Xiang Luo ;Qing-Mei Hu
European Journal of Inorganic Chemistry 2008 Volume 2008( Issue 2) pp:291-297
Publication Date(Web):
DOI:10.1002/ejic.200700845
Abstract
A series of N-acylated diiron azadithiolate complexes as H-cluster models was synthesized and structurally characterized. Treatment of parent complex [{(μ-SCH2)2NH}Fe2(CO)6] (A) with 2-chloroacetic acid in the presence of dicyclohexylcarbodiimide or with 2-chloroacetyl chloride in the presence of Et3N gave N-chloroacetyl complex [{(μ-SCH2)2NC(O)CH2Cl}Fe2(CO)6] (1). Further treatment of 1 with MeC(O)SK afforded N-acetylthioacetyl complex [{(μ-SCH2)2NC(O)CH2SC(O)Me}Fe2(CO)6] (2). N-Ethoxylcarbonylacetyl complex [{(μ-SCH2)2NC(O)CH2CO2Et}Fe2(CO)6] (3) and N-heterocyclic complexes [{(μ-SCH2)2NC(O)C4H3Y-2}Fe2(CO)6] (4, Y = O; 5, Y = S) were produced by reactions of A with EtO2CCH2C(O)Cl, 2-furancarbonyl chloride, and 2-thiophenecarbonyl chloride in the presence of pyridine or Et3N. Similarly, N-malonyl complex [{Fe2(CO)6(μ-SCH2)2NC(O)}2CH2] (6) and N-carbonylbenzaldehyde complex [{(μ-SCH2)2NC(O)C6H4CHO-p}Fe2(CO)6] (7) could be obtained by reaction of A with malonyl dichloride in the presence of pyridine and with p-CHOC6H4C(O)Cl in the presence of Et3N. More interestingly, further reaction of 7 with PhCHO and pyrrole in a 1:3:4 molar ratio in the presence of BF3·OEt2 followed by p-chloranil yielded the first light-driven type of model complex containing an N-carbonylphenylporphyrin moiety [{(μ-SCH2)2NC(O)(TPP)}Fe2(CO)6] (8, TPP = tetraphenylporphyrin group). Whereas the molecular structures of 2, 5, and 7 were established by X-ray crystallography, the electrochemical properties of 2–5 as well as the proton reduction to hydrogen gas catalyzed by 2 and 3 were studied by CV techniques.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2008)
Co-reporter:Li-Cheng Song ; Guang-Huai Zeng ; Shao-Xia Lou ; Hui-Ning Zan ; Jiang-Bo Ming ;Qing-Mei Hu
Organometallics 2008 Volume 27(Issue 15) pp:3714-3721
Publication Date(Web):July 15, 2008
DOI:10.1021/om800077c
The butterfly Fe/S cluster anions (μ-RS)(μ-S−)Fe2(CO)6 (A, R = Et, p-MeC6H4), (μ-S−)2Fe2(CO)6 (C), [(μ-S−)Fe2(CO)6]2(4-μ-SC6H4C6H4S-μ-4′) (D), and [(μ-S−)Fe2(CO)6]2[4-μ-SC6H4OCH2CH2OC6H4S-μ-4′] (E) (generated in situ via reactions of (μ-S2)Fe2(CO)6 with RMgBr, Et3BHLi, 4-LiC6H4C6H4Li-4′, and 4-LiC6H4OCH2CH2OC6H4Li-4′) were found to react with Ph2PCl to give a series of novel butterfly Fe/S/P cluster complexes. Treatment of monoanions A (R = Et, p-MeC6H4) with 1 equiv of Ph2PCl in THF from −78 °C to room temperature gave the single-butterfly Fe2S2P complexes (μ-RS)(η1-Ph2PS-η1)Fe2(CO)6 (7, R = Et; 9, R = p-MeC6H4) and (μ-RS)(η1-Ph2PS-η1)Fe2(CO)5(Ph2PY) (8, R = Et, Y = Cl; 10, R = p-MeC6H4, Y = p-MeC6H4), whereas dianions C, D, and E reacted with 2 equiv of Ph2PCl to give single-butterfly Fe2S2P2 complex (η1-Ph2PS-η1)2Fe2(CO)6 (11) and double-butterfly Fe4S4P2 complexes [(η1-Ph2PS-η1)Fe2(CO)6]2(4-μ-SC6H4C6H4S-μ-4′) (12) and [(η1-Ph2PS-η1)Fe2(CO)6]2[4-μ-SC6H4OCH2CH2OC6H4S-μ-4′] (13), respectively. More interestingly, the novel μ4-S-containing double-butterfly Fe4S2P complexes [(μ-RS)Fe2(CO)6](μ4-S)[(μ-Ph2P)Fe2(CO)6] (14, R = Me; 15, R = Ph; 16, R = Et) could be prepared by reactions of single-butterfly complexes (μ-RS)(η1-Ph2PS-η1)Fe2(CO)6 (1, R = Me; 3, R = Ph; 7 R = Et) with excess Fe2(CO)9 in THF at room temperature, whereas the quadruple-butterfly Fe8S4P2 complexes [(μ-Ph2P)Fe2(CO)6(μ4-S)Fe2(CO)6]2(4-μ-SC6H4C6H4S-μ-4′) (17) and [(μ-Ph2P)Fe2(CO)6(μ4-S)Fe2(CO)6]2[4-μ-SC6H4OCH2CH2OC6H4S-μ-4′] (18) were similarly prepared by reactions of the corresponding double-butterfly complexes 12 and 13 with excess Fe2(CO)9, respectively. All the new complexes 7−18 have been characterized by elemental analysis, by spectroscopy, and for 9, 11, and 14 by X-ray crystallography. In view of the structural similarity of these Fe/S/P complexes to the [FeFe]-hydrogenase active site, they might be regarded as H-cluster models. As a representative, model complex 11 was found to be able to catalyze proton reduction to hydrogen under CV conditions.
Co-reporter:Li-Cheng Song ; Xiao-Niu Fang ; Chang-Gong Li ; Jing Yan ; Hai-Lin Bao ;Qing-Mei Hu
Organometallics 2008 Volume 27(Issue 13) pp:3225-3231
Publication Date(Web):June 11, 2008
DOI:10.1021/om800225y
Tetrathiol 1,2,4,5-(HSCH2)4C6H2 reacted with Fe3(CO)12 and Et3N followed by treatment of the intermediate μ-CO-containing tetraanion {[(μ-CO)Fe2(CO)6]4[1,2,4,5-(μ-SCH2)4C6H2]}4− (7) with 2-furancarbonyl chloride to give quadruple-butterfly complex [(μ-σ,π-C4H3O)Fe2(CO)6]4[1,2,4,5-(μ-SCH2)4C6H2] (9), whereas triple-butterfly complex [(μ-Ph2P)Fe2(CO)6]2[Fe2(CO)6][1,2,4,5-(μ-SCH2)4C6H2] (11) could be produced by reaction of Ph2PCl with the μ-CO-containing dianion {[(μ-CO)Fe2(CO)6]2[Fe2(CO)6][1,2,4,5-(μ-SCH2)4C6H2]}2− (10) generated in situ from the initially formed tetraanion 7. Similarly, the triple-butterfly complexes [(μ-Ph2P)Fe2(CO)6]2[Fe2(CO)6][(μ-SCH2)4C] (14) and [(μ-σ,π-CH2CH═CH2)Fe2(CO)6]2[Fe2(CO)6][(μ-SCH2)4C] (16) were produced by reaction of Ph2PCl or CH2═CHCH2Br with the μ-CO-containing dianion {[(μ-CO)Fe2(CO)6]2[Fe2(CO)6][(μ-SCH2)4C]}2− (13) formed in situ from tetraanion {[(μ-CO)Fe2(CO)6]4[(μ-SCH2)4C]}4− (12) generated initially by reaction of tetrathiol C(CH2SH)4 with Fe3(CO)12 and Et3N. The double-butterfly complex [Fe2(CO)6]2[(μ-SCH2)4C] (15) derived in situ from dianion 13 was also isolated as a minor product along with major products 14 and 15. All the new complexes 9, 11, and 14−16 were characterized by elemental analysis and IR and NMR spectroscopy, as well as by X-ray crystallography for 9, 11, 14, and 15.
Co-reporter:Li-Cheng Song, Hu-Ting Wang, Jian-Hua Ge, Shu-Zhen Mei, Jie Gao, Liang-Xing Wang, Bin Gai, Li-Qun Zhao, Jing Yan and Yong-Zhen Wang
Organometallics 2008 Volume 27(Issue 7) pp:1409-1416
Publication Date(Web):March 14, 2008
DOI:10.1021/om700956e
As the new H-cluster models, a series of N-functionalized azadithiolatodiiron complexes containing mono- and diphosphine ligands 1–7 have been prepared by various methods from complexes [(μ-SCH2N(Fun)]Fe2(CO)6 (A, Fun = C6H4CHO-p; B, Fun = C6H4CO2Me-p; C, Fun = CH2CH2O2CCH2C10H7-1; D, Fun = CH2CH2OH) and [(μ-SCH2)2N(Fun)]Fe2(CO)5(Ph2PH) (E, Fun = C6H4OMe-p). Treatment of A and B with 1 equiv of Me3NO·2H2O followed by 1 equiv of Ph3P or Ph2PH affords the corresponding monophosphine-substituted complexes [(μ-SCH2)2N(C6H4CHO-p)]Fe2(CO)5(Ph3P) (1) and [(μ-SCH2)2N(C6H4CO2Me-p)]Fe2(CO)5L (2, L = Ph3P; 3, Ph2PH). Further treatment of B with ca. 1 equiv of Ph2PC2H4PPh2 (dppe) produced the diphosphine dppe-bridged single model [(μ-SCH2)2N(C6H4CO2Me-p)]Fe2(CO)4(dppe) (4), whereas C reacts with 1 equiv of Me3NO·2H2O followed by 0.5 equiv of (η5-Ph2PC5H4)2Fe (dppf) to give the diphosphine dppf-bridged double model [(μ-SCH2)2N(CH2CH2O2CCH2C10H7-1)Fe2(CO)5]2(dppf) (5). While D reacts with 1 equiv of n-BuLi followed by 1 equiv of Ph2PCl or directly reacts with 1 equiv of Ph2PCl in the presence of Et3N to generate N-alkoxyphosphine-substituted complex [(μ-SCH2)2N(CH2CH2OPPh2-η(1))]Fe2(CO)5 (6), treatment of E with 1 equiv of n-BuLi followed by 1 equiv of CpFe(CO)2I yields organometallic phosphine-substituted complex [(μ-SCH2)2N(C6H4OMe-p)]Fe2(CO)5[Ph2PFe(CO)2Cp] (7). All the new model complexes 1–7 are fully characterized by elemental analysis, spectroscopy, and particularly for 1, 3, 4, 6, and 7 X-ray crystallography. More interestingly, 2 is found to be a catalyst for HOAc proton reduction to hydrogen under CV conditions. In addition, according to electrochemical and spectroelectrochemical studies, an ECEC mechanism is proposed for this electrocatalytic reaction.
Co-reporter:Li-Cheng Song;Jian-Hua Ge;Xiao-Guang Zhang;Yang Liu;Qing-Mei Hu
European Journal of Inorganic Chemistry 2006 Volume 2006(Issue 16) pp:
Publication Date(Web):3 JUL 2006
DOI:10.1002/ejic.200600242
A series of methoxyphenyl-functionalized diiron azadithiolate (ADT) complexes, [{(μ-SCH2)2N(C6H4OMe-p)}Fe2(CO)5L] [L = CO (2); PPh3 (3); PPh2H (4)] and [{(μ-SCH2)2N(C6H4OMe-p)}Fe2(CO)4(CN)2][Et4N]2 (5) as the active site models of Fe-only hydrogenases has been investigated. While model 2 was prepared in 67 % yield by a condensation reaction of N,N-bis(chloromethyl)-p-methoxyaniline (1) with [(μ-LiS)2Fe2(CO)6], models 3–5 were prepared in 49–75 % yields by a CO substitution reaction of 2 with PPh3, PPh2H, or Et4NCN, respectively. The X-ray crystal structures of 2 and 3 revealed that the methoxyphenyl substituent is attached to the N atom by an axial bond and the nitrogen lone electron pair in an equatorial position. On the basis of cyclic voltammetric studies of 2 and 4, it was found that 2 is a catalyst for proton reduction, and an EECC mechanism is proposed for such electrocatalytic H2 production catalyzed by the ADT-type models. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2006)
Co-reporter:Li-Cheng Song;Fu-Hai Su;Qing-Mei Hu;Emanuela Grigiotti;Piero Zanello
European Journal of Inorganic Chemistry 2006 Volume 2006(Issue 2) pp:
Publication Date(Web):9 DEC 2005
DOI:10.1002/ejic.200500745
This paper reports the synthesis, characterization, and some properties of transition metal–fullerene complexes containing trans-1,1′-bis(diphenylphosphanyl)ethylene (dppet, trans-Ph2PCH=CHPPh2) and N,N,N′,N′-tetra(diphenylphosphanylmethyl)ethylene diamine (dppeda, [(Ph2PCH2)2NCH2]2), including [(η2-C60M)(dppet)2] (M = Pt 1, Pd 2), [(η2-C60M)(dppet)]2 (M = Pt 3, Pd 4), [(η2-C60Pd)(η2-C60Pt)(dppet)2](5), [(η2-C60Pd)(η2-C70Pd)(dppet)2] (6), [(η2-C70Pd)(dppet)]2 (7), [(η2-C60M)(dppeda)] (M = Pt 8, Pd 9), and [(η2-C60M)2(dppeda)] (M = Pt 10, Pd 11). Interestingly, while complexes 1, 2, 8, and 9 are the first examples of metallacyclopropafullerene diphosphane ligands containing fullerene cores, complexes 3–7, 10, and 11 are the first group 10 metal-containing, dumbbell-shaped bisfullerenes. All the complexes 1–11 were characterized by elemental analysis, spectroscopy, and, particularly for 4 and 5, by X-ray crystallography and cyclic voltammetry. Pathways for the formation of 1–11 are suggested. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2006)
Co-reporter:Li-Cheng Song ;Ming-Yi Tang Dr.;Fu-Hai Su Dr.;Qing-Mei Hu
Angewandte Chemie 2006 Volume 118(Issue 7) pp:
Publication Date(Web):3 JAN 2006
DOI:10.1002/ange.200503602
Effiziente Kommunikation: An ein biomimetisches Modell des aktiven Zentrums von Fe-Hydrogenasen wurde erstmals ein Porphyrin-Photosensibilisator angehängt (siehe Bild). Das Fluoreszenzspektrum dieses strukturell charakterisierten Systems belegt eine effiziente elektronische Kommunikation zwischen dem Porphyrin und der kovalent angebundenen Dieisenazadithiolat-Einheit.
Co-reporter:Li-Cheng Song, Ming-Yi Tang, Fu-Hai Su, Qing-Mei Hu
Angewandte Chemie International Edition 2006 45(7) pp:1130-1133
Publication Date(Web):
DOI:10.1002/anie.200503602
Co-reporter:Li-Cheng Song,, Fu-Hai Su, Qing-Mei Hu
Journal of Organometallic Chemistry 2005 Volume 690(Issue 5) pp:1121-1124
Publication Date(Web):1 March 2005
DOI:10.1016/j.jorganchem.2004.12.006
The homodinuclear bismetallacyclopropa[60]fullerene complexes (η2-C60)M(μ-η1,η1-trans-Ph2PCHCH PPh2)2M(η2-C60) (1, M = Pt; 2, M = Pd) were prepared by reaction of C60 with M(dba)2 (dba = dibenzylideneacetone) and trans-1,1′-bis(diphenylphosphino)ethylene in 82% and 92% yield, whereas reaction of C60 with Pd(dba)2 and trans-dppet followed by treatment with C60 and Pt2(dba)3 gave rise to the heterodinuclear complex (η2-C60) Pd(μ-η1,η1-trans-Ph2PCHCH PPh2)2Pt(η2-C60) (3) in 65% yield. Mechanistic study showed that these reactions involve the intermediates of monometallacyclopropa[60]fullerene diphosphine ligands (η2-C60)M(η1-trans-Ph2PCHCHPPh2)2 (4, M = Pt; 5, M = Pd). All the mono- and bismetallacyclopropa[60]fullerene complexes 1–5 have been fully characterized by elemental analysis and spectroscopy, as well as for 2 by X-ray crystallography.The homo- and hetero-dinuclear bismetallacyclopropa[60]fullerene complexes (η2-C60)M(μ-η1,η1- trans-Ph2PCHCHPPh2)2M(η2-C60) (1, M = Pt; 2, M = Pd) and (η2-C60)Pd(μ-η1,η1-trans-Ph2PCHCH PPh2)2Pt(η2-C60) (3) were prepared via the intermediate metallacyclopropa[60]fullerene diphosphines (η2-C60)M(η1-trans-Ph2PCHCHPPh2)2 (4, M = Pt; 5, M = Pd). Complexes 1–5 have been fully characterized.
Co-reporter:Li-Cheng Song;Hua-Wei Cheng;Xin Chen;Qing-Mei Hu
European Journal of Inorganic Chemistry 2004 Volume 2004(Issue 15) pp:
Publication Date(Web):1 JUN 2004
DOI:10.1002/ejic.200400088
Treatment of the Cr−Cr singly-bonded dimers [η5-RC5H4Cr(CO)3]2 (1, R = MeCO; 2, R = MeO2C; 3, R = EtO2C) with excess sulfur in refluxing THF gave the cubane Cr4S4 clusters (η5-RC5H4)4Cr4S4 (4, R = MeCO; 5, R = MeO2C; 6, R = EtO2C). The cubane Cr4S4 cluster 4 reacted with excess 2,4-dinitrophenylhydrazine to produce the hydrazone derivative [η5−2,4-(NO2)2C6H3NHN=C(Me)C5H4]4Cr4S4 (7). The singly-bonded dimers of [η5-RC5H4Cr(CO)3]2 (8, R = Me; 9, R = EtO2C), in the presence of excess selenium, reacted similarly to the linear Cr2Se complexes [η5-RC5H4Cr(CO)2]2Se (10, R = Me; 11, R = EtO2C), which reacted with an equimolar quantity of selenium to afford the cubane Cr4Se4 clusters (η5-RC5H4)4Cr4Se4 (12, R = Me; 13, R = EtO2C). A particularly interesting phenomenon is the cross-assembled reaction of the linear Cr2Se complexes [η5-MeC(O)C5H4Cr(CO)2]2Se (14) and [CpCr(CO)2]2Se (15) in the presence of excess selenium in THF that gave rise to a series of cubane Cr4Se4 clusters [η5-MeC(O)C5H4]nCp4−nCr4Se4 (16, n = 0; 17, n = 1; 18, n = 2; 19, n = 3; 20, n = 4). The possible pathway for the cross-assembled reaction is suggested. Furthermore the new clusters were characterized by elemental analysis and spectroscopy, and in the case of 4−6 and 16 also by X-ray diffraction techniques. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2004)
Co-reporter:Li-Cheng Song, Hua-Wei Cheng, Qing-Mei Hu
Journal of Organometallic Chemistry 2004 Volume 689(Issue 10) pp:1849-1855
Publication Date(Web):15 May 2004
DOI:10.1016/j.jorganchem.2004.03.008
The Cr–Cr singly-bonded dimers [{η5-RC5H4Cr(CO)3}2] (1, R=Me; 2, R=CO2Et) reacted with an equivalent of elemental selenium in THF at room temperature to give the linear Cr2Se complexes [{η5-RC5H4Cr(CO)2}2Se] (3, R=Me; 4, R=CO2Et), whereas the linear Cr2Se complex (5, R=MeCO) reacted with excess NaBH4, Ph3PCHPh or 2,4-dinitrophenylhydrazine under respective conditions to afford the linear Cr2Se derivatives [{η5-RC5H4Cr(CO)2}2Se] (6, R=MeCH(OH); 7, R=PhCHCMe; 8, R=2,4-(NO2)2C6H3NHNCMe). Similarly, while the butterfly Cr2Se2 complexes [{η5-RC5H4Cr(CO)2}2Se2] (9, R=Me; 10, R=CO2Et) could be produced either by reaction of dimers 1 and 2 with an excess amount of elemental selenium, or by reaction of the linear complexes 3 and 4 with an equivalent of elemental selenium, the butterfly Cr2Se2 derivatives [{η5-RC5H4Cr(CO)2}2Se2] (12, R=MeCH(OH); 13, R=PhCHCMe; 14, R=2,4-(NO2)2C6H3NHNCMe) were yielded by reaction of the butterfly Cr2Se2 complex (11, R=MeCO) with an excess quantity of NaBH4, Ph3PCHPh and 2,4-dinitrophenylhyazine. Both the linear complexes 3, 4, 6–8 and the butterfly complexes 9, 10, 12–14 are new, which have been fully characterized by elemental analysis, spectroscopy and X-ray crystallography.The linear Cr2Se complexes [{η5-RC5H4Cr(CO)2}2Se] (3, R=Me; 4, R=CO2Et; 6, R=MeCH(OH); 7, R=PhCHCMe; 8, R=2,4-(NO2)2C6H3NHNCMe) and the butterfly Cr2Se2 complexes [{η5-RC5H4Cr(CO)2}2Se2] (9, R=Me; 10, R=CO2Et; 12, R=MeCH(OH); 13, R=PhCHCMe; 14, R=2,4-(NO2)2C6H3NHNCMe) were successfully synthesized and fully characterized by elemental analysis, spectroscopy and X-ray diffraction analysis.
Co-reporter:Li-Cheng Song, Jin-You Wang, Feng-Hua Gong, Jia Cheng, Qing-Mei Hu
Journal of Organometallic Chemistry 2004 Volume 689(Issue 5) pp:930-935
Publication Date(Web):1 March 2004
DOI:10.1016/j.jorganchem.2003.12.023
The [Et3NH]+ salts of dianions {[(μ-CO)Fe2(CO)6]2(μ-SZS-μ)}2−, prepared from Fe3(CO)12, dithiol HSZSH and Et3N, with (μ-S2)Fe2(CO)6 followed by treatment with monohalides ClCH2CO2Et, PhHgCl and PhC(Cl)NPh to give acyclic polyether clusters {(μ-RS)[Fe2(CO)6]2(μ4-S)}2(μ-SZS-μ) [1–5: R=CH2CO2Et, PhHg, PhCNPh; Z=CH2(CH2OCH2)2−4CH2], while they reacted with (μ-S2)Fe2(CO)6 and subsequent treatment with dihalides BrCH2(CH2OCH2)2−4CH2Br under similar conditions to afford macrocyclic crown ether clusters {[Fe2(CO)6]2(μ4-S)}2(μ-SZS-μ)(μ-SYS-μ) [6–11: Z, Y=CH2(CH2OCH2)2−4CH2]. Clusters 1–11 are new, and have been characterized by elemental analysis and spectroscopy, as well as for 6 [Z=Y=CH2(CH2OCH2)2CH2] and 9 [Z=CH2(CH2OCH2)2CH2, Y=CH2(CH2OCH2)3CH2] by X-ray diffraction techniques.Two series of clusters {(μ-RS)[Fe2(CO)6]2(μ4-S)}2(μ-SZS-μ) [1–5: R=CH2CO2Et, PhHg, PhCNPh; Z=CH2(CH2OCH2)2−3CH2] and {[Fe2(CO)6]2(μ4-S)}2(μ-SZS-μ)(μ-SYS-μ) [6–11: Z, Y=CH2(CH2OCH2)2−4CH2] were prepared by “one pot” reactions of the [Et3NH]+ salts of dianions [(μ-CO)Fe2(CO)6]2(μ-SZS-μ) with various halides. Two crystal structures are described.
Co-reporter:Li-Cheng Song, An-Guo Zhu and Yuan-Qiang Guo
Dalton Transactions 2016 - vol. 45(Issue 12) pp:NaN5029-5029
Publication Date(Web):2016/01/04
DOI:10.1039/C5DT04297F
As [FeFe]-hydrogenase models, the first thiodithiolate (TDT) ligand-containing μ-hydride complexes [(μ-TDT)Fe2(CO)4(PMe3)2(μ-H)]+Y− (2–7, Y = Cl, ClO4, PF6, BF4, CF3CO2, CF3SO3) have been prepared by protonation reactions of (μ-TDT)Fe2(CO)4(PMe3)2 (1) with the corresponding HY acids. While the protonation reactions are monitored by in situ1H and 31P{1H} NMR spectroscopy to show the isomer type and stability of 2–7, the structures of the isolated 2–7 are characterized by elemental analysis, spectroscopy and for some of them by X-ray crystallography. Although the H/D exchange of μ-hydride complex 7 (Y = CF3SO3) with D2 or D2O has been proved not to occur under the studied conditions, the H/D exchange of 7 with DCl gives the μ-deuterium complex [(μ-TDT)Fe2(CO)4(PMe3)2(μ-D)]+[CF3SO3]− (8) in a nearly quantitative yield. To our knowledge, 8 is the first crystallographically characterized μ-deuterium-containing butterfly [2Fe2S] complex produced by H/D exchange reaction.
Co-reporter:Li-Cheng Song, Xi-Yue Yang, Meng Cao, Xiu-Yun Gao, Bei-Bei Liu, Liang Zhu and Feng Jiang
Chemical Communications 2017 - vol. 53(Issue 27) pp:NaN3821-3821
Publication Date(Web):2017/03/06
DOI:10.1039/C7CC00149E
The structural and functional modeling of the active site of [NiFe]-hydrogenases has been proved to be challenging to a great extent. Herein, we report the synthesis, structures, and some properties of the NiFe-based dicarbonyl, terminal hydride, and μ-hydroxo models for the active site of [NiFe]-hydrogenases.
Co-reporter:Li-Cheng Song, Fu-Qiang Hu, Miao-Miao Wang, Zhao-Jun Xie, Kai-Kai Xu and Hai-Bin Song
Dalton Transactions 2014 - vol. 43(Issue 21) pp:NaN8071-8071
Publication Date(Web):2014/03/14
DOI:10.1039/C4DT00335G
As biomimetic models for [Fe]-hydrogenase, the 2-acylmethyl-6-ester group-difunctionalized pyridine-containing iron(II) complexes 1–4 have been successfully prepared via the following three separate steps. In the first step, the acylation or esterification of difunctionalized pyridine 2-(p-MeC6H4SO3CH2)-6-HOCH2C5H3N with acetyl chloride or benzoic acid gives the corresponding pyridine derivatives 2-(p-MeC6H4SO3CH2)-6-RCO2CH2C5H3N (A, R = Me; B, R = Ph). The second step involves reaction of A or B with Na2Fe(CO)4 followed by treatment of the intermediate Fe(0) complexes [Na(2-CH2-6-RCO2CH2C5H3N)Fe(CO)4] (M1, R = Me; M2, R = Ph) with iodine to afford 2-acylmethyl-6-acetoxymethyl or 6-benzoyloxymethyl-difunctionalized pyridine-containing Fe(II) iodide complexes [2-C(O)CH2-6-RCO2CH2C5H3N]Fe(CO)2I (1, R = Me; 3, R = Ph). Finally, when 1 or 3 is treated with sodium 2-mercaptopyridinate, the corresponding difunctionalized pyridine-containing Fe(II) mercaptopyridinate complexes [2-C(O)CH2-6-RCO2C5H3N]Fe(CO)2(2-SC5H4N) (2, R = Me; 4, R = Ph) are produced. While the structures of model complexes 1–4 are confirmed by X-ray crystallography, the electrochemical properties of 2 and 4 are compared with those of the two previously reported models. In addition, complexes 2 and 4 have been found to be catalysts for H2 production in the presence of TFA under CV conditions.
Co-reporter:Li-Cheng Song, Xiao-Jing Sun, Pei-Hua Zhao, Jia-Peng Li and Hai-Bin Song
Dalton Transactions 2012 - vol. 41(Issue 29) pp:NaN8950-8950
Publication Date(Web):2012/05/22
DOI:10.1039/C2DT30609C
The [N2S2]-type ligand 1,2-(2-C5H4NCH2S)2C6H4 (L) is prepared in 84% yield by a new method and its structure has been confirmed by X-ray crystallography. The new synthetic method involves sequential reaction of 1,2-phenylenedithiol with EtONa followed by treatment of the resulting disodium salt of 1,2-phenylenedithiol with in situ generated 2-(chloromethyl)pyridine from its HCl salt. Further treatment of ligand L with NiCl2·6H2O or NiI2 affords the expected new mononuclear Ni complexes Ni[1,2-(2-C5H4NCH2S)2C6H4]Cl2 (1) and Ni[1,2-(2-C5H4NCH2S)2C6H4]I2 (3) in 87–88% yields, whereas reaction of L with NiBr2 under similar conditions results in formation of the expected new mononuclear complex Ni[1,2-(2-C5H4NCH2S)2C6H4]Br2 (2) and one unexpected new mononuclear complex Ni[1-(2-C5H4NCH2S)-2-(2-C5H4NCH2SC6H4S)C6H4]Br2 (2*) in 82% and 5% yields, respectively. More interestingly, the ligand L-containing novel trinuclear NiFe2 complex Ni{[1,2-(2-C5H4NCH2S)2C6H4}Fe2(CO)6(μ3-S)2 (4) is found to be prepared by sequential reaction of (μ-S2)Fe2(CO)6 with Et3BHLi, followed by treatment of the resulting (μ-LiS)2Fe2(CO)6 with mononuclear complex 1, 2, or 3 in 12–20% yields. The new complexes 1–4 and 2* are fully characterized by elemental analysis and various spectroscopies, and the crystal structures of 1, 2* and 3 as well as some electrochemical properties of 1–4 are also reported.
Co-reporter:Li-Cheng Song, Zhao-Jun Xie, Xu-Feng Liu, Jiang-Bo Ming, Jian-Hua Ge, Xiao-Guang Zhang, Tian-Ying Yan and Peng Gao
Dalton Transactions 2011 - vol. 40(Issue 4) pp:NaN846-846
Publication Date(Web):2010/12/09
DOI:10.1039/C0DT00909A
A series of new diiron azadithiolate (ADT) complexes (1–8), which could be regarded as the active site models of [FeFe]hydrogenases, have been synthesized starting from parent complex [(μ-SCH2)2NCH2CH2OH]Fe2(CO)6 (A). Treatment of A with ethyl malonyl chloride or malonyl dichloride in the presence of pyridine afforded the malonyl-containing complexes [(μ-SCH2)2NCH2CH2O2CCH2CO2Et]Fe2(CO)6 (1) and [Fe2(CO)6(μ-SCH2)2NCH2CH2O2C]2CH2 (2). Further treatment of 1 and 2 with PPh3 under different conditions produced the PPh3-substituted complexes [(μ-SCH2)2NCH2CH2O2CCH2CO2Et]Fe2(CO)5(PPh3) (3), [(μ-SCH2)2NCH2CH2O2CCH2CO2Et]Fe2(CO)4(PPh3)2 (4), and [Fe2(CO)5(PPh3)(μ-SCH2)2NCH2CH2O2C]2CH2 (5). More interestingly, complexes 1–3 could react with C60 in the presence of CBr4 and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) via Bingel–Hirsch reaction to give the C60-containing complexes [(μ-SCH2)2NCH2CH2O2CC(C60)CO2Et]Fe2(CO)6 (6), [Fe2(CO)6(μ-SCH2)2NCH2CH2O2C]2C(C60) (7), and [(μ-SCH2)2NCH2CH2O2CC(C60)CO2Et]Fe2(CO)5(PPh3) (8). The new ADT-type models 1–8 were characterized by elemental analysis and spectroscopy, whereas 2–4 were further studied by X-ray crystallography and 6–8 investigated in detail by DFT methods.
Co-reporter:Li-Cheng Song, Qian-Li Li, Zhan-Heng Feng, Xiao-Jing Sun, Zhao-Jun Xie and Hai-Bin Song
Dalton Transactions 2013 - vol. 42(Issue 5) pp:NaN1626-1626
Publication Date(Web):2012/10/16
DOI:10.1039/C2DT31976D
Parent complex (μ-PDTe)Fe2(CO)6 (1, PDTe = μ-TeCH2CH2CH2Te-μ) is prepared via a new synthetic route involving the reaction of (μ-Te2)Fe2(CO)6 with Et3BHLi, followed by treatment of (μ-LiTe)2Fe2(CO)6 with Br(CH2)3Br in a 43% yield. Further reactions of 1 with 1 equiv of monophosphines in the presence of the decarbonylating agent Me3NO afford the corresponding monophosphine-substituted complexes (μ-PDTe)Fe2(CO)5(L) (2, L = PPh3; 3, PPh2H; 4, PMe3) in 37%–47% yields, whereas the N-heterocyclic carbene IMes-monosubstituted complex (μ-PDTe)Fe2(CO)5(IMes) (5) can be prepared in a 26% yield by treatment of 1 with the in situ generated IMes from the 1,3-bis(mesityl)imidazolium salt IMes·HCl and n-BuLi. While the diphosphine-bridged single-butterfly complexes (μ-PDTe)Fe2(CO)4(dppm) (6) and (μ-PDTe)Fe2(CO)4(dppn) (7) can be prepared in 28% and 21% yields by treatment of 1 with 1 equiv of the corresponding diphosphines in refluxing xylene, treatment of 1 with 0.5 equiv of diphosphines in the presence of Me3NO results in the formation of the corresponding diphosphine-bridged double-butterfly complexes [(μ-PDTe)Fe2(CO)5]2(dppp) (8), [(μ-PDTe)Fe2(CO)5]2(dppb) (9), and [(μ-PDTe)Fe2(CO)5]2(dppf) (10) in 25–37% yields. All the new substituted model complexes 2–10 are characterized by combustion analysis and spectroscopy, and particularly for 2, 3, 5, and 7–10, by X-ray crystallography. In addition, a comparative study on the electrochemical and electrocatalytic properties of the PDTe-type model complexes 1 and 7 with their corresponding selenium and sulfur analogs are reported.
Co-reporter:Li-Cheng Song, Xiao-Feng Han, Wei Chen, Jia-Peng Li and Xu-Yong Wang
Dalton Transactions 2017 - vol. 46(Issue 30) pp:NaN10013-10013
Publication Date(Web):2017/07/14
DOI:10.1039/C7DT02203D
A new series of the structural and functional models for the active site of [NiFe]-H2ases has been prepared by a simple and convenient synthetic route. Thus, treatment of diphosphines RN(PPh2)2 (1a, R = p-MeC6H4CH2; 1b, R = EtO2CCH2) with an equimolar NiCl2·6H2O, NiBr2·3H2O, and NiI2 in refluxing CH2Cl2/MeOH or EtOH gave the mononuclear Ni complexes RN(PPh2)2NiX2 (2a, R = p-MeC6H4CH2, X = Cl; 2b, R = EtO2CCH2, X = Cl; 3a, R = p-MeC6H4CH2, X = Br; 3b, R = EtO2CCH2, X = Br; 4a, R = p-MeC6H4CH2, X = I; 4b, R = EtO2CCH2, X = I) in 67–97% yields. Further treatment of complexes 2a,b–4a,b with an equimolar mononuclear Fe complex (dppv)(CO)2Fe(pdt) and NaBF4 resulted in formation of the targeted model complexes [RN(PPh2)2Ni(μ-pdt)(μ-X)Fe(CO)(dppv)](BF4) (5a, R = p-MeC6H4CH2, X = Cl; 5b, R = EtO2CCH2, X = Cl; 6a, R = p-MeC6H4CH2, X = Br; 6b, R = EtO2CCH2, X = Br; 7a, R = p-MeC6H4CH2, X = I; 7b, R = EtO2CCH2, X = I) in 60–96% yields. All the new complexes 3a,b–4a,b and 5a,b–7a,b have been characterized by elemental analysis and spectroscopy, and particularly for some of them (3a,b/4a,b and 5b/6b) by X-ray crystallography. More interestingly, the electrochemical and electrocatalytic properties of such halogenido-bridged model complexes are first studied systematically and particularly they have been found to be pre-catalysts for proton reduction to H2 under CV conditions.
Co-reporter:Li-Cheng Song, Meng Cao and Yong-Xiang Wang
Dalton Transactions 2015 - vol. 44(Issue 15) pp:NaN6808-6808
Publication Date(Web):2015/02/23
DOI:10.1039/C5DT00067J
The homodinuclear complexes [Ni(RNPyS4)]2 (1a–1e; RNPyS4 = 2,6-bis(2-mercaptophenylthiomethyl)-4-R-pyridine; R = H, MeO, Cl, Br, i-Pr) were found to be prepared by reactions of the in situ generated Li2[Ni(1,2-S2C6H4)2] with 2,6-bis[(tosyloxy)methyl]pyridine and its substituted derivatives 2,6-bis[(tosyloxy)methyl]-4-R-pyridine. Further reactions of 1a–1e with Fe3(CO)12 gave both heterotrinuclear complexes NiFe2(RNPyS4)(CO)5 (2a–2e) and mononuclear complexes Fe(RNPyS4)(CO) (3a–3e), unexpectedly. Interestingly, complexes 2a–2e and 3a–3e could be regarded as models for the active sites of [NiFe]- and [Fe]-hydrogenases, respectively. All the prepared complexes were characterized by elemental analysis, spectroscopy, and particularly for some of them, by X-ray crystallography. In addition, the electrochemical properties of 2a–2e and 3a–3e as well as the electrocatalytic H2 production catalyzed by 2a–2e and 3a–3e were investigated by CV techniques.

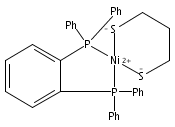





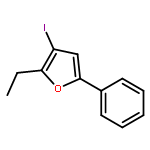
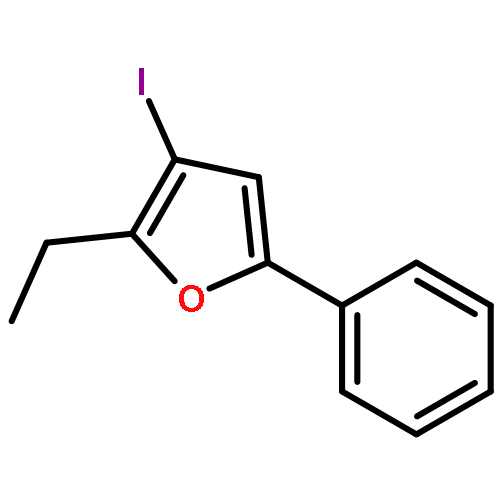


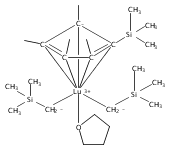


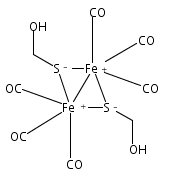




![Benzo[b]thiophene, 3-iodo-2-methyl-](http://img.cochemist.com/ccimg/139700/139625-61-9.png)
![Benzo[b]thiophene, 3-iodo-2-methyl-](http://img.cochemist.com/ccimg/139700/139625-61-9_b.png)

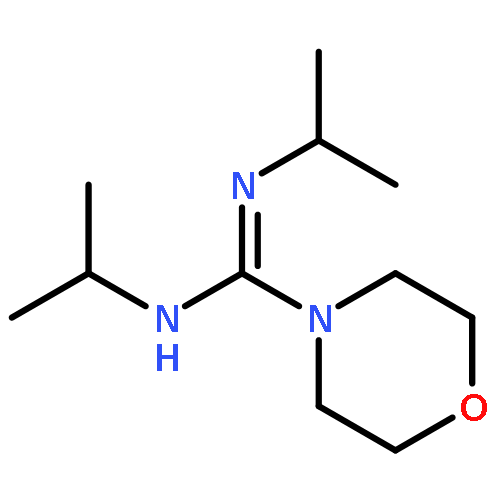




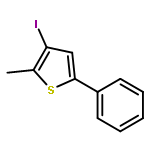
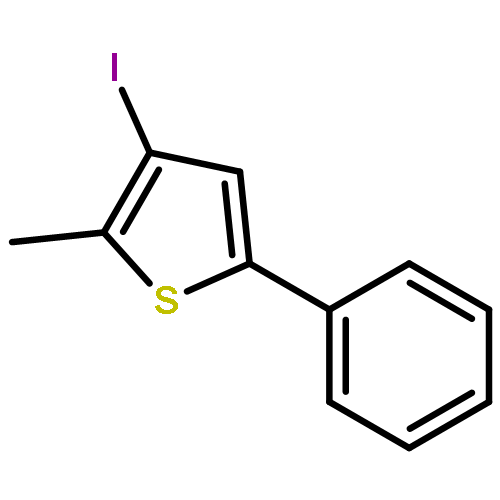
![2-METHYL-5-[(5-METHYLFURAN-2-YL)DISULFANYL]FURAN](http://img.cochemist.com/ccimg/61000/60965-61-9.png)
![2-METHYL-5-[(5-METHYLFURAN-2-YL)DISULFANYL]FURAN](http://img.cochemist.com/ccimg/61000/60965-61-9_b.png)
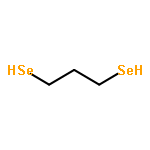
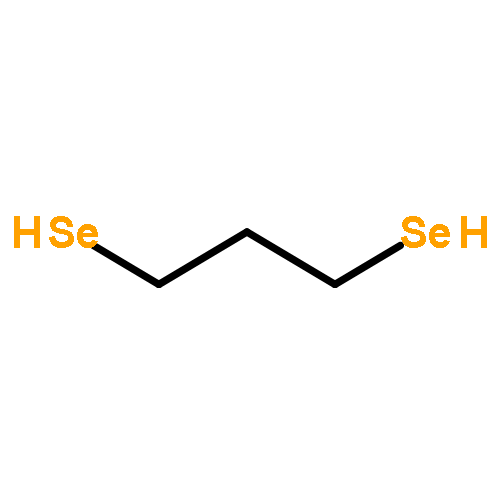




![Platinum,dichloro[1,1'-(1,2-ethanediyl)bis[1,1-diphenylphosphine-kP]]-, (SP-4-2)-](http://img.cochemist.com/ccimg/14700/14647-25-7.png)
![Platinum,dichloro[1,1'-(1,2-ethanediyl)bis[1,1-diphenylphosphine-kP]]-, (SP-4-2)-](http://img.cochemist.com/ccimg/14700/14647-25-7_b.png)