Co-reporter:Brendan Frett, Nick McConnell, Catherine C. Smith, Yuanxiang Wang, Neil P. Shah, Hong-yu Li
European Journal of Medicinal Chemistry 2015 Volume 94() pp:123-131
Publication Date(Web):13 April 2015
DOI:10.1016/j.ejmech.2015.02.052
•A virtual library was screened against kinase crystal structures.•An imidazopyridine-core was found active on the FLT3 tyrosine kinase.•Ligand efficient FLT3 hits were generated with strong biochemical and cell activities.The FLT3 kinase represents an attractive target to effectively treat AML. Unfortunately, no FLT3 targeted therapeutic is currently approved. In line with our continued interests in treating kinase related disease for anti-FLT3 mutant activity, we utilized pioneering synthetic methodology in combination with computer aided drug discovery and identified low molecular weight, highly ligand efficient, FLT3 kinase inhibitors. Compounds were analyzed for biochemical inhibition, their ability to selectively inhibit cell proliferation, for FLT3 mutant activity, and preliminary aqueous solubility. Validated hits were discovered that can serve as starting platforms for lead candidates.
Co-reporter:Brendan Frett ; Robert V. Brown ; Mingliang Ma ; Wenhao Hu ; Haiyong Han
Journal of Medicinal Chemistry 2014 Volume 57(Issue 14) pp:5835-5844
Publication Date(Web):February 11, 2014
DOI:10.1021/jm401719n
The global incidence of cancer is on the rise, and within the next decade, the disease is expected to become the leading cause of death worldwide. Forthcoming strategies used to treat cancers focus on the design and implementation of multidrug therapies to target complementary cancer specific pathways. A more direct means by which this multitargeted approach can be achieved is by identifying and targeting interpathway regulatory factors. Recent advances in understanding Nek2 (NIMA related kinase 2) biology suggest that the kinase potentially represents a multifaceted therapeutic target. In this regard, pharmacologic modulation of Nek2 with a single agent may effect several mechanisms important for tumor growth, survival, progression, and metastasis. We herein review the development of Nek2 as an oncology target and provide a succinct chronology of drug discovery campaigns focused on targeting Nek2.
Co-reporter:Brendan Frett, Marialuisa Moccia, Francesca Carlomagno, Massimo Santoro, Hong-yu Li
European Journal of Medicinal Chemistry 2014 Volume 86() pp:714-723
Publication Date(Web):30 October 2014
DOI:10.1016/j.ejmech.2014.09.023
•MCR based drug discovery led to the identification of two tractable RET leads.•From kinetic binding and modeling studies, data suggests compounds are Type-II inhibitors.•Two lead compounds were found to inhibit RET activity in vivo.From an MCR fragment library, two novel chemical series have been developed as inhibitors of RET, which is a kinase involved in the pathology of medullary thyroid cancer (MTC). Structure activity relationship studies (SAR) identified two sub-micromolar tractable leads, 6g and 13g. 6g was confirmed to be a Type-II RET inhibitor. 13g and 6g inhibited RET in cells transformed by RET/C634. A RET DFG-out homology model was established and utilized to predict Type-II inhibitor binding modes.
Co-reporter:Brendan Frett, Nick McConnell, Yuanxiang Wang, Zhigang Xu, Andrew Ambrose and Hong-yu Li
MedChemComm 2014 vol. 5(Issue 10) pp:1507-1514
Publication Date(Web):11 Aug 2014
DOI:10.1039/C4MD00251B
Trk receptors play a key role in the development and maintenance of neuronal networks. Recent evidence suggests that the Trk family, specifically TrkA, is an important driver for tumour growth, inflammatory and neuropathic pain, and chemoresistance. Through a computational screen, a novel Trk active pharmacophore was identified and a series of pyrazine-based inhibitors were developed, which potently inhibited TrkA. Inhibitors displayed the highest activity on TrkA when screened against a small, tyrosine kinase panel and also exhibited a non-linear SAR. Predicted binding modes of the inhibitors were examined, which identified exploitable regions for future development of more advanced inhibitors.
Co-reporter:Yuanxiang Wang ; Christine E. Kaiser ; Brendan Frett
Journal of Medicinal Chemistry 2013 Volume 56(Issue 13) pp:5219-5230
Publication Date(Web):April 8, 2013
DOI:10.1021/jm3017706
The RAS proteins play a role in cell differentiation, proliferation, and survival. Aberrant RAS signaling has been found to play a role in 30% of all cancers. KRAS, a key member of the RAS protein family, is an attractive cancer target, as frequent point mutations in the KRAS gene render the protein constitutively active. A number of attempts have been made to target aberrant KRAS signaling by identifying small molecule compounds that (1) are synthetic lethal to mutant KRAS, (2) block KRAS/GEF interactions, (3) inhibit downstream KRAS effectors, or (4) inhibit the post-translational processing of RAS proteins. In addition, inhibition of novel targets outside the main KRAS signaling pathway, specifically the cell cycle related kinase PLK1, has been shown have an effect in cells that harbor mutant KRAS. Herein we review the use of various high-throughput screening assays utilized to identify new small-molecule compounds capable of targeting mutant KRAS-driven cancers.
Co-reporter:Biswajit Saha, Brendan Frett, Yuanxiang Wang, Hong-yu Li
Tetrahedron Letters 2013 Volume 54(Issue 19) pp:2340-2343
Publication Date(Web):8 May 2013
DOI:10.1016/j.tetlet.2013.02.055
A mild, cost-effective, and simple three-component Ugi-type reaction using p-toluenesulfinic acid (pTSIA) as the acid catalyst has been developed to synthesize α-amino amides and α-amino amidines. Employing 1 equiv of amine used in the reaction generated α-amino amides exclusively, while 2 equiv of amines, especially with more nucleophilic aniline such as p-anisidine, yielded the α-amino amidines as the major product. This methodology would be suitable for the synthesis of natural or unnatural amino acids and drug-like amidine analogues.
Co-reporter:Brendan Frett;Dr. Yuanxiang Wang;Dr. Hong-yu Li
ChemMedChem 2013 Volume 8( Issue 10) pp:1620-1622
Publication Date(Web):
DOI:10.1002/cmdc.201300311

![3-(4-methylphenyl)-Imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/1338300/1338248-67-1.png)
![3-(4-methylphenyl)-Imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/1338300/1338248-67-1_b.png)
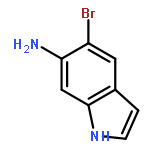
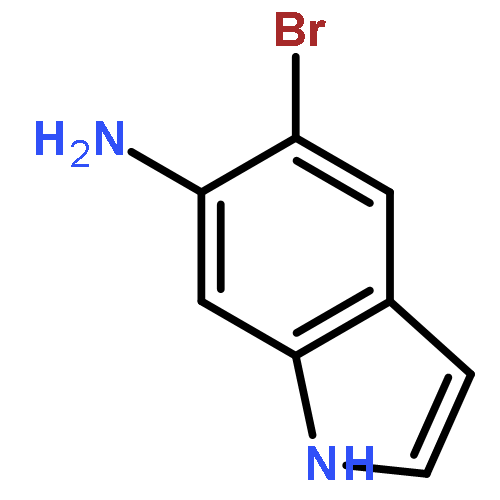








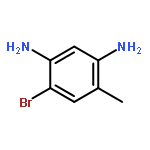
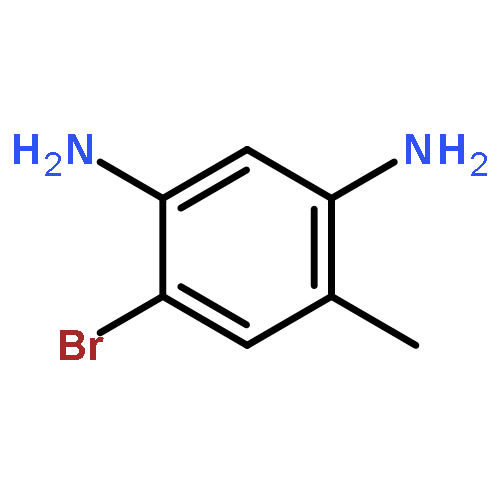
![Imidazo[1,2-a]pyridin-3-ol, 2-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/125700/125634-64-2.png)
![Imidazo[1,2-a]pyridin-3-ol, 2-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/125700/125634-64-2_b.png)
![BENZONITRILE, 4-(6-METHYLIMIDAZO[1,2-A]PYRIDIN-2-YL)-](http://img.cochemist.com/ccimg/118100/118000-52-5.png)
![BENZONITRILE, 4-(6-METHYLIMIDAZO[1,2-A]PYRIDIN-2-YL)-](http://img.cochemist.com/ccimg/118100/118000-52-5_b.png)
![Imidazo[1,2-a]pyridine, 2-(4-chlorophenyl)-6-methyl-](http://img.cochemist.com/ccimg/103900/103844-28-6.png)
![Imidazo[1,2-a]pyridine, 2-(4-chlorophenyl)-6-methyl-](http://img.cochemist.com/ccimg/103900/103844-28-6_b.png)
![7-phenyl-Imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/85200/85102-27-8.png)
![7-phenyl-Imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/85200/85102-27-8_b.png)
![2,3-DIPHENYLIMIDAZO[1,2-A]PYRIDINE](http://img.cochemist.com/ccimg/85200/85102-26-7.png)
![2,3-DIPHENYLIMIDAZO[1,2-A]PYRIDINE](http://img.cochemist.com/ccimg/85200/85102-26-7_b.png)
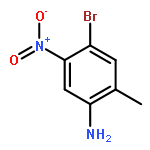
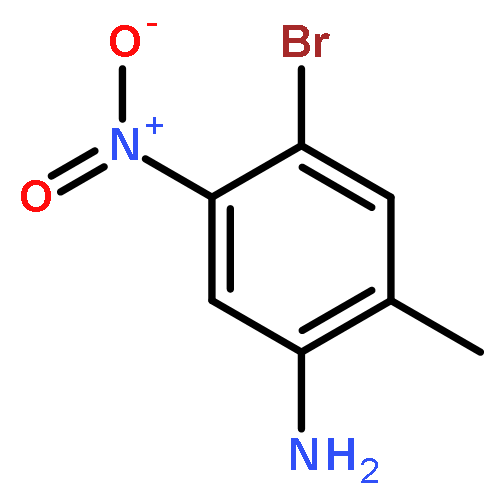

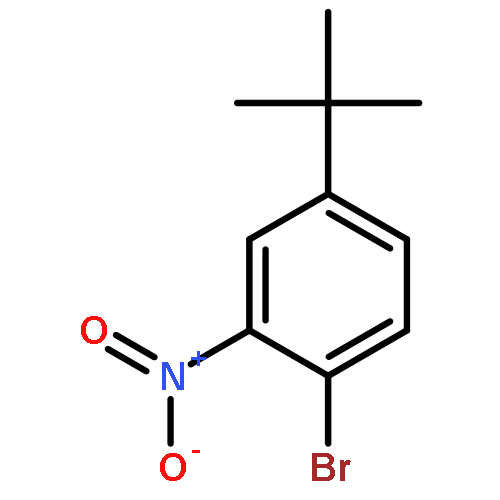
![2-(4-Chloro-phenyl)-7-methyl-1H-imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/66000/65964-62-7.png)
![2-(4-Chloro-phenyl)-7-methyl-1H-imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/66000/65964-62-7_b.png)


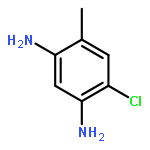
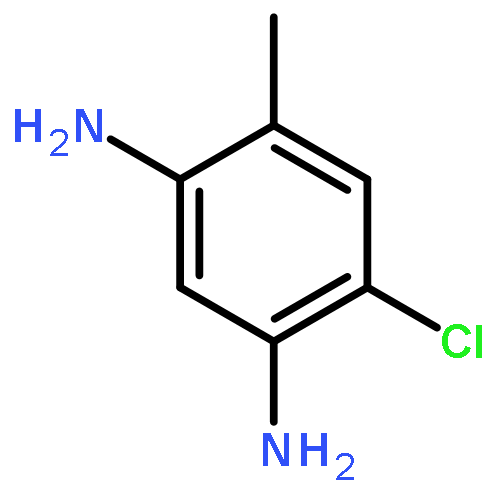





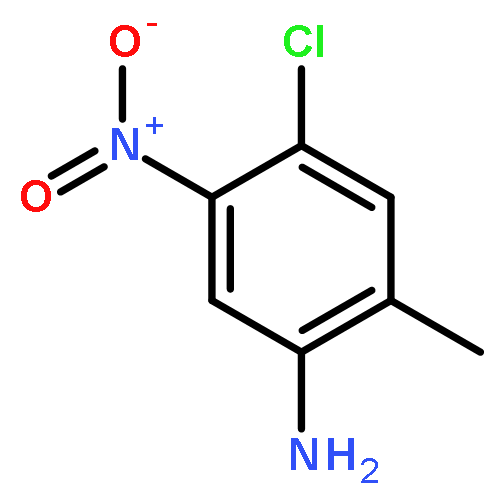
![2-Phenylimidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/4200/4105-21-9.png)
![2-Phenylimidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/4200/4105-21-9_b.png)
![6-methyl-2-phenyl-Imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/1100/1019-89-2.png)
![6-methyl-2-phenyl-Imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/1100/1019-89-2_b.png)
![Methyl 2-phenylimidazo[1,2-a]pyridine-6-carboxylate](http://img.cochemist.com/ccimg/1000/962-24-3.png)
![Methyl 2-phenylimidazo[1,2-a]pyridine-6-carboxylate](http://img.cochemist.com/ccimg/1000/962-24-3_b.png)
![7-Methyl-2-phenyl-1H-imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/900/885-91-6.png)
![7-Methyl-2-phenyl-1H-imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/900/885-91-6_b.png)
![8-methyl-2-phenyl-Imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/900/885-89-2.png)
![8-methyl-2-phenyl-Imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/900/885-89-2_b.png)

