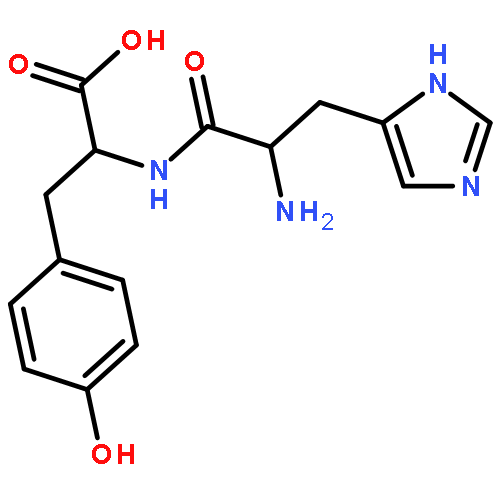
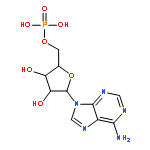
Co-reporter:Aristeidis Tsiamis and Spencer E. Taylor
Energy & Fuels October 19, 2017 Volume 31(Issue 10) pp:10576-10576
Publication Date(Web):September 11, 2017
DOI:10.1021/acs.energyfuels.7b01695
Recent studies have shown that n-C7-precipitated asphaltenes adsorb onto nanoparticles to produce isotherms that are significantly influenced by the dispersed states of both the adsorbate and the adsorbent. In the present work, we investigate this behavior further by determining the adsorption of asphaltene and resin fractions isolated from four different sources onto kaolinite using the depletion method in toluene. Treated conventionally (amount adsorbed, Γ, versus equilibrium bulk concentration, ce), adsorption isotherms for fixed initial concentrations (c0) of C5 and C7 asphaltenes and variable kaolinite mass (ms) are found to be Type I as classified by IUPAC, whereas under the same experimental conditions C5–C7 resins exhibit Type III behavior. By fixing ms and varying c0, however, Type II isotherms are produced by the resins. All of the adsorption results for the same fraction type were found to be very similar, irrespective of the source. The Types I and III isotherms are described very well by the thermodynamic solid–liquid equilibrium (SLE) model of Montoya et al. ( Energy Fuels 2014, 28, 4963−4975) based on the association theory of Talu and Meunier ( AIChE J. 1996, 42, 809−819). Individual isotherms (Γ versus ce) are well-fitted by a shifted Langmuir equation for asphaltenes and by a general Freundlich (power law) relationship for resins. The SLE results verify that in toluene solution the adsorption behavior is complicated by concentration-dependent nanoaggregation of asphaltene species, whereas resin–resin interactions are weaker, but accompanied by adsorbent particle aggregation. On the other hand, when the adsorption data for each fraction type is replotted in terms of the ratio of the experimental parameter c0/ms, as originally done by Wang et al. ( Colloids Surfaces A: Physicochem. Eng. Aspects 2016, 504, 280−286), each set of data merges to a single isotherm which is reasonably well-approximated by a Langmuir-type relationship (we term this a “pseudo-Langmuir equation”), which allows the maximum adsorption to be determined for the different adsorbate/adsorbent systems. The average maximum adsorbed amounts calculated in this way for each of the component types are very similar, being slightly larger for C7A compared with C5A, with the values for the C5–C7R fractions being generally lower and more variable, possibly reflecting some source dependence.
Co-reporter:Weronika M. Swiech, Ian Hamerton, Huang Zeng, David J. Watson, Eleonore Mason, Spencer E. Taylor
Journal of Colloid and Interface Science 2017 Volume 508(Volume 508) pp:
Publication Date(Web):15 December 2017
DOI:10.1016/j.jcis.2017.08.031
Background and hypothesis: Humic acid (HA) is of considerable environmental significance, being a major component of soil, as well as being considered for application in other technological areas. However, its structure and colloidal properties continue to be the subject of debate, largely owing to its molecular complexity and association with other humic substances and mineral matter. As a class, HA is considered to comprise supramolecular assemblies of heterogeneous species, and herein we consider a simple route for the separation of some HA sub-fractions.Experiments: A commercial HA sample from Sigma-Aldrich has been fractionated into two soluble (S1, S2) and two insoluble (I1, I2) fractions by successive dissolution in deionized water at near-neutral pH. These sub-fractions have been characterized by solution and solid-state approaches.Findings: Using this simple approach, the HA has been shown to contain non-covalently bonded species with different polarity and water solubility. The soluble and insoluble fractions have very different chemical structures, as revealed particularly by their solid-state properties (13C NMR and IR spectroscopy, and TGA); in particular, S1 and S2 are characterized by higher carbonyl and aromatic contents, compared with I1 and I2. As shown by solution SAXS measurements and AFM, the soluble fractions behave as hydrophilic colloidal aggregates of at least 50 nm diameter.Download high-res image (122KB)Download full-size image
Co-reporter:Valeska Gonzalez, Marc Jones, and Spencer E. Taylor
Energy & Fuels 2016 Volume 30(Issue 2) pp:844-853
Publication Date(Web):January 6, 2016
DOI:10.1021/acs.energyfuels.5b02352
Improving the efficiency of secondary and tertiary oil recovery is one of the major challenges of the crude oil industry. Arguably the most significant advance made in recent years has been the realization of the importance of water chemistry during oil recovery. This has led to the concept of low-salinity waterflooding for conventional oil reservoirs, for which reducing the total salinity of the injection water has been found to improve oil recovery rates. Although the precise mechanisms responsible for the improvements are not completely understood, it is acknowledged that specific interactions in the crude oil/brine/rock (COBR) systems will modify wettability and interfacial energy. The present paper extends investigations of COBR interactions to higher viscosity oils and considers implications for heavy oil recovery. We have used NMR relaxation time measurements to study interactions in oil/brine/sand (OBS) systems containing either natural bitumen or a polybutene hydrocarbon (Glissopal). Exposing the oil-coated sands to water or aqueous group 1 and group 2 metal chloride solutions enabled the T2 relaxation time spectrum to be determined as a function of time. The observed decreases in the geometric mean T2 values with time obey first-order kinetics; for bitumen-coated sands, the rate constants are consistent with diffusion of water through the bitumen. Opto-digital microscopy verified the formation of ∼1–3 μm diameter water droplets in the initially dry bitumen coating, suggesting that water nucleation and growth also occur. This was not observed for the Glissopal-coated sand samples. No evidence was found for displacement of either viscous oil from the sand grains, although optical microscopy did reveal rearrangement of the bitumen coating, which possibly exposes fresh sand surfaces to the aqueous phase. This behavior is consistent with the finding that the original T2 parameters determined for fresh sand are not fully restored simply by contacting the bitumen-sand surface to water or aqueous salt solutions under the ambient experimental conditions. Glissopal-coated sands exhibited smaller time-dependent T2 changes compared with bitumen-coated sands. While not displacing viscous oils from the sand surface under the experimental conditions used, it is conjectured that water ingress into the surface oil layer could weaken oil/sand interactions which by analogy with recent studies on conventional oil recovery could provide an additional heavy oil recovery mechanism under more dynamic or higher temperature conditions.
Co-reporter:Esther Forte, Spencer E. Taylor
Advances in Colloid and Interface Science 2015 Volume 217() pp:1-12
Publication Date(Web):March 2015
DOI:10.1016/j.cis.2014.12.002
Highlights
- •
A review on advances in modelling asphaltene phase behaviour is provided.
- •
The complexity in modelling these systems is analysed.
- •
A detailed discussion on the application of different fluid theories is offered.
- •
Particularly the application of lattice theories, Cubic Equations of State and SAFT.
Co-reporter:Alisdair Q. Clark , Alastair G. Smith and Steve Threadgold , Spencer E. Taylor
Industrial & Engineering Chemistry Research 2011 Volume 50(Issue 9) pp:5749-5765
Publication Date(Web):March 29, 2011
DOI:10.1021/ie102533e
Jet fuel cleanliness, in terms of dispersed water and dirt, is of paramount importance to ensure aviation safety. In this study, a process image analyzer has been used to determine size distributions of dispersed water droplets and real and standard test dust particulates in jet fuel, both in the laboratory and under representative full-scale operational conditions. The technique is also applied to monitoring water droplet coalescence in a filter-water separator in the presence of a surfactant known to cause coalescer disarming, again under simulated operational conditions. The measured water and dirt count (number) distributions are exclusively log-normal, whereas corresponding volume size distributions show deviations from log-normal behavior as a result of contributions from the relatively small number of larger particles or aggregated droplet clusters, highlighting the importance of volume-based size analyses. Distinguishing between dispersed water droplets and solid particles has been demonstrated quantitatively, using a cosolvent to solubilize the contribution from free water droplets.
Co-reporter:Spencer E. Taylor
Fuel 2011 Volume 90(Issue 10) pp:3028-3039
Publication Date(Web):October 2011
DOI:10.1016/j.fuel.2011.05.028
Nonionic surfactant-stabilised oil-in-water emulsions offer a potentially useful vehicle for transporting heavy crude oils from oilfields to refineries or distribution terminals. Prior to refining, separation of the oil from the emulsion is necessary. Previous studies have suggested that heating the emulsion is sufficient for destabilisation and recovery of the oil. The present work examines this process on a batch laboratory scale and monitors the effect of thermal treatment on the heavy oil/water interfacial tension using spinning drop tensiometry. The present research has confirmed that heating Wolf Lake (Canada) bitumen-in-water emulsions to a temperature close to the cloud point of the surfactant results in efficient bitumen/water resolution, together with separation of a dense surfactant-rich coacervate phase that could recycled in a commercial heavy oil transportation process. The corresponding temperature dependence of the bitumen/water interfacial tension provides further insight into the emulsion resolution process.Highlights► Oil/water emulsions are potentially useful for bitumen transportation from field to refinery. ► We studied thermal destabilisation of nonionic emulsions. ► Surfactant phase behaviour important – potential for recycling? ► Oil/water interfacial tensions determined.
Co-reporter:Spencer E. Taylor
Energy & Fuels 2008 Volume 22(Issue 4) pp:2396
Publication Date(Web):May 21, 2008
DOI:10.1021/ef800090p
In view of its widespread application in aviation turbine fuel, diethyleneglycol monomethylether (DiEGME) and its interactions with water and n-heptane have been characterized using turbidity, interfacial tension, water activity, and water absorption measurements. This additive has been implicated in a number of problems in recent years, which have arguably arisen from its various physicochemical interactions with fuel and fuel system components, for which few data were hitherto available. The present study has therefore addressed the more fundamental aspects underlying such interactions using n-heptane as the hydrocarbon. Turbidity results indicate an increased level of water solubilization, owing to the formation of DiEGME−water clusters (∼1:8 ratio), as the DiEGME concentration exceeds its specification maximum value of 0.15% (w/v) in fuel. Interestingly, this same composition is found in separated water, resulting from additive partitioning from fuel, leading to ∼50% DiEGME/water mixtures. The combined use of interfacial tension, water activity and absorption measurements, and solubility parameters is able to explain this tendency as being due to a reduction in water activity in the presence of DiEGME, with this latter property being reduced significantly above 50% DiEGME, which therefore appears to be the most thermodynamically stable composition. Water activity considerations also provide the basis for understanding the action of DiEGME as a thermodynamic icing inhibitor, consistent with the role that hydrogen bonding plays in reducing water activity and in line with the water-activity-based ice nucleation theory (Koop, T., Luo, B., Tsias, A., and Peter, T. Nature 2000, 406, 611−614). Correspondingly, the thermodynamic activity of DiEGME, derived herein using a Gibbs−Duhem treatment of water activity data, is shown to be reduced in the presence of low levels of water, which would be sufficient to restrict the fuel solubility of this material as observed in practice.