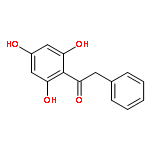
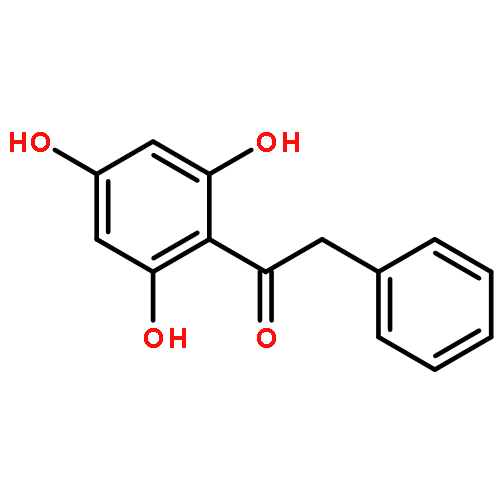
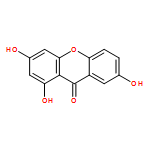
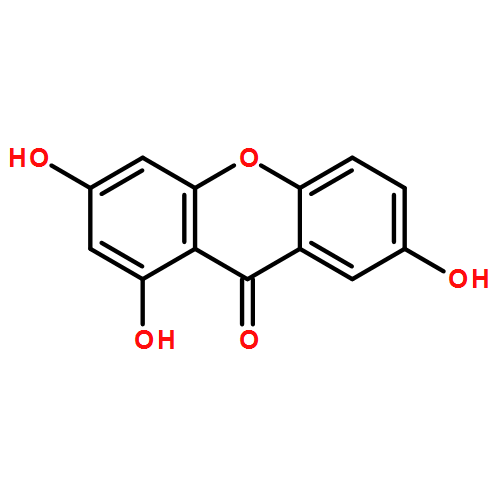
Co-reporter:Zhi Zhang, Nina Xue, Chuancai Bian, Rui Yan, Longlong Jin, Xiaoguang Chen, Xiaoming Yu
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 17) pp:4287-4291
Publication Date(Web):1 September 2016
DOI:10.1016/j.bmcl.2016.07.040
Benzoquinone ansamycins are important leads for the discovery of novel inhibitors of heat shock protein 90 (Hsp90), a promising target of cancer chemotherapeutics. Intrinsic hepatotoxicity caused by the benzoquinone moiety appeared to be a serious limitation to the development of these compounds. To solve this problem by rational structure optimization, a short series of C18-deoxy analogues of herbimycin A were designed based on putative interactions between the compound and the protein. Chemical synthesis of the target molecules were attempted by following the established synthetic route to the natural product, but resulted in the isolation of four serendipitous C15 phenylated final products. In vitro antiproliferative activity and Hsp90 binding affinity of the compounds were determined, suggesting the C18-oxygen of herbimycin A is removable and bulky lipophilic groups can be accommodated at C15 without loss of activity.Benzoquinone ansamycins are important leads for the discovery of novel inhibitors of heat shock protein 90 (Hsp90), a promising target of cancer chemotherapeutics. Intrinsic hepatotoxicity caused by the benzoquinone moiety appeared to be a serious limitation to the development of these compounds. To solve this problem by rational structure optimization, a short series of C18-deoxy analogues of herbimycin A were designed based on putative interactions between the compound and the protein. Chemical synthesis of the target molecules were attempted by following the established synthetic route to the natural product, but resulted in the isolation of four serendipitous C15 phenylated final products. In vitro antiproliferative activity and Hsp90 binding affinity of the compounds were determined, suggesting the C18-oxygen of herbimycin A is removable and bulky lipophilic groups can be accommodated at C15 without loss of activity.
Co-reporter:Meng-Yi Xu, Ni-Na Xue, Di Liu, Yu-Mei Zhou, Wei Li, Yong-Qiang Li, Xiao-Guang Chen, Xiao-Ming Yu
Chinese Chemical Letters 2016 Volume 27(Issue 1) pp:11-15
Publication Date(Web):January 2016
DOI:10.1016/j.cclet.2015.09.024
Heat shock protein 90 (hsp90) is a promising anticancer drug target. A library of 2,4-dihydroxyphenyl (resorcinol) substituted 4,5,6,7-tetrahydro-[1,2,3]triazolo[1,5-a]pyrazine compounds that target this protein were designed and prepared based on our earlier study. The compounds were tested in five cancer cell lines and seven of them showed notable anticancer activity (IC50 2–10 μmol/L). The active subset compounds were further subjected to a polarized fluorescent assay and exhibited high binding affinity toward purified hsp90 (IC50 60–100 nmol/L). These results indicated that the tetrahydro-triazolopyrazine motif of the molecules may represent a novel scaffold for the development of hsp90 inhibitors.2,4-Dihydroxyphenyl (resorcinol) substituted 4,5,6,7-tetrahydro-[1,2,3]triazolo[1,5-a]pyrazine derivatives were found to be potent Hsp90 inhibitors and exhibited remarkable anti-cancer activities in vitro. The results indicated that the title compounds represent a novel class of hsp90 inhibitor.
Co-reporter:Rui Yan, Chuancai Bian, and Xiaoming Yu
Organic Letters 2014 Volume 16(Issue 12) pp:3280-3283
Publication Date(Web):June 9, 2014
DOI:10.1021/ol5012899
Benzoquinone ansamycin antibiotic herbimycin A was synthesized in 19 linear steps and 4.2% yield. Highlighted is the design of a chiral γ-lactone as the C11–C15 synthon that enabled a facile catalytic asymmetric synthesis of the challenging C8–C20 fragment of the target molecule. The easy access to the stereogenic centers and high overall yield made the strategy applicable in the molecular editing of benzoquinone ansamycins.
Co-reporter:Chuancai Bian, Rui Yan, Xiaoming Yu
Tetrahedron 2014 70(18) pp: 2982-2991
Publication Date(Web):
DOI:10.1016/j.tet.2014.03.020
Co-reporter:Di Liu;Liang Huang
Chinese Journal of Chemistry 2013 Volume 31( Issue 3) pp:344-348
Publication Date(Web):
DOI:10.1002/cjoc.201201187
Abstract
A six-step synthesis of (−)-clausenamide is described. Optically pure (R,E)-1,3-diphenylallylic alcohol was acetylated and then subjected to an Ireland-Claisen rearrangement, giving the γ,δ-unsaturated acid, which underwent a substrate-induced stereoselective bromolactonization to afford the expected all-equatorial substituted bromo-δ-lactone. An unusual chemo-selective aminolysis of the lactone resulted in the formation of a γ,δ-epoxy-amide in stereospecific manner. Base-promoted cyclization of this intermediate and the subsequent Davis oxidation furnished the synthesis, delivering the final product in >99% ee and up to 34% overall yield.
Co-reporter:Di Liu, Xiaoming Yu
Tetrahedron Letters 2012 Volume 53(Issue 17) pp:2177-2180
Publication Date(Web):25 April 2012
DOI:10.1016/j.tetlet.2012.02.071
Yield lowering C-silylation was found to be inevitable in all the six tested examples of stoichiometric LDA/TMSCl promoted Ireland-Claisen rearrangement of secondary allyl acetates. In order to circumvent this problem, a higher yielding protocol was devised after the isolated by-products were found to be excellent substrates for further rearrangement. Thus, excessive LDA/TMSCl was applied to achieve complete 3,3′-sigmatropic shift of the substrates, and the resulting mixtures of normal and α-silylated γ,δ-unsaturated carboxylic acids was then desilylated by one-pot application of TBAF.
Co-reporter:Jian Jun Xue, Yu Mei Zhou, Xiao Ming Yu
Chinese Chemical Letters 2011 Volume 22(Issue 11) pp:1261-1264
Publication Date(Web):November 2011
DOI:10.1016/j.cclet.2011.07.002
4- and 6-desphenyl analogues of (−)-clausenamide, 6 and 7, were prepared in optical active form from commercially available d-pyroglutamic acid and the known racemic pyrrolidinone 13, respectively. In order to confirm the absolute stereochemistry of (+)- and (−)-7, intermediate 19b was transferred into the (+)-epi-clausenamide 8.
Co-reporter:Jian Jun Xue, Xiao Ming Yu
Chinese Chemical Letters 2011 Volume 22(Issue 7) pp:761-764
Publication Date(Web):July 2011
DOI:10.1016/j.cclet.2011.01.026
(−)-Clausenamide is a drug candidate under Phase I clinical trial for treatment of Alzheimer's disease (AD). In order to elucidate the substituent related structure–activity relationship, six one-substituent modified (−)-clausenamide analogues were designed, and four of them, namely 3-O-methyl, 6-O-methyl, 3-des-hydroxyl and 6-des-hydroxyl analogues were prepared by selective 3- and 6-OH modification of (−)-clausenamide.
Co-reporter:Jisheng Luo, Xiaoming Yu
Tetrahedron Letters 2011 Volume 52(Issue 19) pp:2450-2453
Publication Date(Web):11 May 2011
DOI:10.1016/j.tetlet.2011.02.092
Generally applicable concise approaches to 3-O-acyl-l-noviose derivatives and their 3-amino bioisosteres, represented by 5 and 6, were described. Chiral aldehyde 7 was thus prepared from dimethyl l-tartrate in five steps, and converted into 5 and 6 by employing substrate induced asymmetric aldehyde or N-sulfinyl aldimine allylation and dihydropyrane epoxidation as key steps, respectively.
Co-reporter:Yiqiu He, Jianjun Xue, Yumei Zhou, Junshan Yang, Xiaoming Yu
Tetrahedron Letters 2009 50(20) pp: 2317-2319
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.02.194

![2-Butenal, 4-[(4-methoxyphenyl)methoxy]-, (2E)-](http://img.cochemist.com/ccimg/842200/842125-04-6.png)
![2-Butenal, 4-[(4-methoxyphenyl)methoxy]-, (2E)-](http://img.cochemist.com/ccimg/842200/842125-04-6_b.png)



























![1-[2,4,6-TRIS(METHOXYMETHOXY)PHENYL]ETHANONE](http://img.cochemist.com/ccimg/36900/36804-11-2.png)
![1-[2,4,6-TRIS(METHOXYMETHOXY)PHENYL]ETHANONE](http://img.cochemist.com/ccimg/36900/36804-11-2_b.png)




![2-[3-(trifluoromethyl)phenyl]-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione](http://img.cochemist.com/ccimg/6100/6052-93-3.png)
![2-[3-(trifluoromethyl)phenyl]-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione](http://img.cochemist.com/ccimg/6100/6052-93-3_b.png)




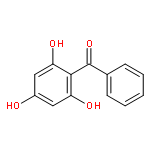
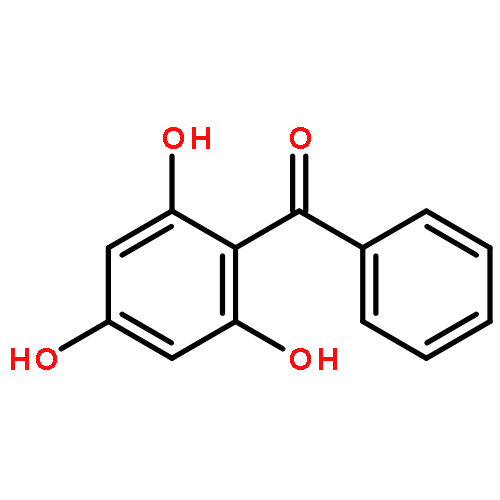
![[(1S,4S)-4-AMINO-1-ISOPROPYL-2-CYCLOPENTEN-1-YL][3-(TRIFLUOROMETH<WBR />YL)-7,8-DIHYDRO-1,6-NAPHTHYRIDIN-6(5H)-YL]METHANONE](http://img.cochemist.com/ccimg/3600/3542-72-1.png)
![[(1S,4S)-4-AMINO-1-ISOPROPYL-2-CYCLOPENTEN-1-YL][3-(TRIFLUOROMETH<WBR />YL)-7,8-DIHYDRO-1,6-NAPHTHYRIDIN-6(5H)-YL]METHANONE](http://img.cochemist.com/ccimg/3600/3542-72-1_b.png)