Co-reporter:P. R. Werkhoven, M. Elwakiel, T. J. Meuleman, H. C. Quarles van Ufford, J. A. W. Kruijtzer and R. M. J. Liskamp
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 2) pp:701-710
Publication Date(Web):05 Nov 2015
DOI:10.1039/C5OB02014J
Mimics of discontinuous epitopes of for example bacterial or viral proteins may have considerable potential for the development of synthetic vaccines, especially if conserved epitopes can be mimicked. However, due to the structural complexity and size of discontinuous epitopes molecular construction of these mimics remains challeging. We present here a convergent route for the assembly of discontinuous epitope mimics by successive azide alkyne cycloaddition on an orthogonal alkyne functionalized scaffold. Here the synthesis of mimics of the HIV gp120 discontinuous epitope that interacts with the CD4 receptor is described. The resulting protein mimics are capable of inhibition of the gp120–CD4 interaction. The route is convergent, robust and should be applicable to other discontinuous epitopes.
Co-reporter:Arwin J. Brouwer, Natalia Herrero Álvarez, Adriano Ciaffoni, Helmus van de Langemheen, Rob M.J. Liskamp
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 16) pp:3429-3435
Publication Date(Web):15 August 2016
DOI:10.1016/j.bmc.2016.05.042
The success of inhibition of the proteasome by formation of covalent bonds is a major victory over the long held-view that this would lead to binding the wrong targets and undoubtedly lead to toxicity. Great challenges are now found in uncovering ensembles of new moieties capable of forming long lasting ties. We have introduced peptido sulfonyl fluorides for this purpose. Tuning the reactivity of this electrophilic trap may be crucial for modulating the biological action. Here we describe incorporation of a vinyl moiety into a peptido sulfonyl fluoride backbone, which should lead to a combined attack of the proteasome active site threonine on the double bond and the sulfonyl fluoride. Although this led to strong proteasome inhibitors, in vitro studies did not unambiguously demonstrate the formation of the proposed seven-membered ring structure. Possibly, formation of a seven-membered covalent adduct with the proteosomal active site threonine can only be achieved within the context of the enzyme. Nevertheless, this dual warhead concept may provide exclusive possibilities for duration and selectivity of proteasome inhibition.
Co-reporter:Helmus van de Langemheen, H. (Linda) C. Quarles van Ufford, John A. W. Kruijtzer, and Rob M. J. Liskamp
Organic Letters 2014 Volume 16(Issue 8) pp:2138-2141
Publication Date(Web):April 7, 2014
DOI:10.1021/ol500604h
Synthetic mimics of protein surfaces have the potential to become inhibitors of protein–protein interactions or even synthetic vaccines. However, the synthesis of these complicated molecular constructs is still difficult. Here we describe an efficient and versatile synthesis of protein mimics containing up to three different cyclic peptides. Using a sequential native chemical ligation strategy, peptide loops containing a thioester handle were introduced onto a triazacyclophane scaffold bearing orthogonal protected cysteine residues.
Co-reporter:Arwin J. Brouwer, Helmus van de Langemheen, Adriano Ciaffoni, Kitty E. Schilder, and Rob M. J. Liskamp
Organic Letters 2014 Volume 16(Issue 11) pp:3106-3109
Publication Date(Web):May 23, 2014
DOI:10.1021/ol5012218
The synthesis of a new triazacyclophane scaffold (TACO scaffold) containing three selectively deprotectable amines is described. The TACO scaffold is conformationally more constrained than our frequently used TAC scaffold, due to introduction of a substituent on the para position of the benzoic acid hinge, which prevents ring flipping and makes it more attractive than the TAC scaffold for preparation of artificial receptor molecules or for mimicking discontinuous epitopes toward protein mimics when more preorganization is required.
Co-reporter:Rik T. C. Cleophas, Martijn Riool, H. (Linda) C. Quarles van Ufford, Sebastian A. J. Zaat, John A. W. Kruijtzer, and Rob M. J. Liskamp
ACS Macro Letters 2014 Volume 3(Issue 5) pp:477
Publication Date(Web):May 7, 2014
DOI:10.1021/mz5001465
This report describes the design and synthesis of a bactericidal poly(ethylene glycol)-based (PEG) hydrogel coating with covalently attached antimicrobial peptides (AMP) stabilized against proteolytic degradation. As such, mimics of the highly active AMP HHC10 (H-KRWWKWIRW-NH2) were designed for optimal stability in human serum while retaining strong antimicrobial activity against Staphylococcus aureus and Staphylococcus epidermidis, the major causative agents of biomaterial associated infection. In order to investigate the selectivity of the AMPs, their hemolytic activity was determined. A N-terminal cysteine facilitated thiol–ene chemistry for a fast, single-step immobilization/photopolymerization strategy. The antimicrobial activity of the resulting thin layer hydrogel coating on a PET surface was established using the Japanese Industrial Standard (JIS) Z2801 assay, showing complete killing (>99.9%) of inocula of S. aureus ATCC 49230, S. epidermidis ATCC 35984, and E. coli ATCC 8739.
Co-reporter:H. van de Langemheen, M. van Hoeke, H. C. Quarles van Ufford, J. A. W. Kruijtzer and R. M. J. Liskamp
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 25) pp:4471-4478
Publication Date(Web):24 Apr 2014
DOI:10.1039/C4OB00190G
The accessibility to collections, libraries and arrays of cyclic peptides is increasingly important since cyclic peptides may provide better mimics of the loop-like structures ubiquitously present in and – especially – on the surface of proteins. The next important step is the preparation of libraries of ensembles of scaffolded cyclic peptides, which upon screening may lead to promising protein mimics. Here we describe the synthesis of a tri-cysteine containing scaffold as well as the simultaneous native chemical ligation of three cyclic peptides thereby affording a clean library of multiple cyclic peptides on this scaffold, representing potential mimics of gp120. Members of this collection of protein mimics showed a decent inhibition of the gp120-CD4 interaction.
Co-reporter:Ronald C. Elgersma, Loes M.J. Kroon-Batenburg, George Posthuma, Johannes D. Meeldijk, Dirk T.S. Rijkers, Rob M.J. Liskamp
European Journal of Medicinal Chemistry 2014 Volume 88() pp:55-65
Publication Date(Web):17 December 2014
DOI:10.1016/j.ejmech.2014.07.089
•pH-controlled aggregation polymorphism of peptides was studied.•Biomaterials based on self-assembling peptides were prepared.•N-terminally functionalized Aβ(16–22) peptides were used as model sequence.•Polarity, amphiphilicity, and overall charge determine aggregation polymorphism.Peptide and protein self-assembly resulting in the formation of amyloidogenic aggregates is generally thought of as a pathological event associated with severe diseases. However, amyloid formation may also provide a basis for advanced bionanomaterials, since amyloid fibrils combine unique material-like properties that make them very useful for design of new types of conducting nanowires, bioactive ligands, and biodegradable coatings as drug-encapsulating materials. The morphology of the supramolecular aggregates determines the properties and application range of these bionanomaterials. An important parameter to control the supramolecular morphology, is the overall charge of the peptide, which is related to the pH of the environment. Herein, we describe the design, synthesis and morphological analysis of a series of N-terminally functionalized Aβ(16–22) peptides (∼Lys-Leu-Val-Phe-Phe-Ala-Glu-OH), that underwent a pH-induced polymorphism, ranging from lamellar sheets, helical tapes, peptide nanotubes, and amyloid fibrils as was observed by transmission electron microscopy. Infrared spectroscopy and wide angle X-ray scattering studies showed that peptide self-assembly was driven by β-sheet formation, and that the supramolecular morphology was directed by subtle variations in electrostatic interactions. Finally, a structural model and hierarchy of self-assembly of a peptide nanotube, assembled at pH 1, is proposed.Tune your tube: the aggregation morphology of the Aβ(16–22) sequence is strongly dependent on the overall charge of the peptide and can be tuned by variations of the N-terminal capping moiety in combination with the pH.
Co-reporter:Jeroen van Ameijde, John Overvoorde, Stefan Knapp, Jeroen den Hertog, Rob Ruijtenbeek, Rob M.J. Liskamp
Analytical Biochemistry 2014 Volume 448() pp:9-13
Publication Date(Web):1 March 2014
DOI:10.1016/j.ab.2013.11.023
Abstract
A versatile assay for protein tyrosine phosphatases (PTP) employing 3-nitrophosphotyrosine containing peptidic substrates is described. These therapeutically important phosphatases feature in signal transduction pathways. The assay involves spectrophotometric detection of 3-nitrotyrosine production from 3-nitrophosphotyrosine containing peptidic substrates, which are accepted by many PTPs. Compared to conventional chromogenic phosphate derivatives, the more realistic peptidic substrates allow evaluating substrate specificity. The assay’s applicability is demonstrated by determining kinetic parameters for several PTP-substrate combinations and inhibitor evaluation, as well as detection of PTP activity in lysates. The convenient new assay may assist further adoption of PTPs in drug development.
Co-reporter:Jeroen van Ameijde, Addy P.R. Zwiebel, Rob Ruijtenbeek, Rob M.J. Liskamp
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 1) pp:113-116
Publication Date(Web):1 January 2014
DOI:10.1016/j.bmcl.2013.11.060
A novel strategy to prepare bisubstrate based inhibitors for histone acetyltransferases is presented. To obtain these, azido peptides derived from histone H3 incorporating either a serine or a phosphoserine residue were connected to a propargyl coenzyme A derivative through copper catalyzed click chemistry. The resulting inhibitors were tested with therapeutically relevant acetyltransferase PCAF. Increased potency of the phosphoserine containing inhibitor was observed. The synthetic strategy presented may be used for developing bisubstrate based inhibitors against any acetyltransferase.
Co-reporter:Steffen van der Wal, Chantelle J. Capicciotti, Stamatia Rontogianni, Robert N. Ben and Rob M. J. Liskamp
MedChemComm 2014 vol. 5(Issue 8) pp:1159-1165
Publication Date(Web):26 Feb 2014
DOI:10.1039/C4MD00013G
Antifreeze glycoproteins (AFGPs) are important naturally occurring biological antifreezes that lower the freezing point of a solution, thereby preventing uncontrolled ice growth. These compounds also inhibit ice recrystallization. Described in this paper is a synthetic antifreeze glycopeptide-based polymer synthesized from an azide/alkyne glycopeptide building block by partial reduction of the azide and subsequent copper catalyzed azide alkyne cycloaddition (CuAAC) polymerization to obtain linear oligomers. To compare the activity with native AFGPs, a linear dodecapeptide (oligomer with four repeating units) was synthesized and isolated which had a comparable length to AFGP-8, the lowest molecular mass glycoprotein AFGP found in nature. In terms of ice recrystallization inhibition (IRI) activity, the triazole-based oligomers displayed only modest IRI activity compared with AFGP-8 and a previously described carbon-linked AFGP analogue. However, CD spectroscopy showed that the triazole-based tetramer possessed a similar secondary structure to the related amide based carbon-linked AFGP tetramer based on AFGP-8.
Co-reporter:Rik T. C. Cleophas, Jelmer Sjollema, Henk J. Busscher, John A. W. Kruijtzer, and Rob M. J. Liskamp
Biomacromolecules 2014 Volume 15(Issue 9) pp:
Publication Date(Web):August 9, 2014
DOI:10.1021/bm500899r
A single step immobilization-polymerization strategy of a highly active antimicrobial peptide into a soft hydrogel network on a poly(ethylene terephthalate) surface using thiol–ene chemistry is described. The bactericidal hydrogel was molecularly characterized via Coomassie and Lowry assay protein staining agents as well as by X-ray photoelectron spectroscopy. The bactericidal activity was established against Staphylococcus aureus and Staphylococcus epidermidis, two bacterial strains commonly associated with biomaterial infections. To gain further insight into the biological stability, the hydrogels were incubated with human serum prior to activity testing without loss of activity. These studies revealed a promising bactericidal hydrogel with good stability under physiological conditions.
Co-reporter:Arwin J. Brouwer, Helmus van de Langemheen, Rob M.J. Liskamp
Tetrahedron 2014 70(26) pp: 4002-4007
Publication Date(Web):
DOI:10.1016/j.tet.2014.04.084
Co-reporter:Jinqiang Zhang, Johan Kemmink, Dirk T. S. Rijkers and Rob M. J. Liskamp
Chemical Communications 2013 vol. 49(Issue 40) pp:4498-4500
Publication Date(Web):28 Mar 2013
DOI:10.1039/C3CC40628H
Herein we report the Ru(II)-catalyzed double-macrocyclization of a hexapeptide to obtain a mimic of the bicyclic CDE-ring of vancomycin, followed by measurement of its binding affinity for small peptide ligands using ITC.
Co-reporter:Loek T. M. van Wandelen, Jeroen van Ameijde, Ahmed F. Ismail-Ali, H. C. (Linda) Quarles van Ufford, Lodewijk A. W. Vijftigschild, Jeffrey M. Beekman, Nathaniel I. Martin, Rob Ruijtenbeek, and Rob M. J. Liskamp
ACS Chemical Biology 2013 Volume 8(Issue 7) pp:1479
Publication Date(Web):April 28, 2013
DOI:10.1021/cb300709g
Although protein kinase inhibitors present excellent pharmaceutical opportunities, lack of selectivity and associated therapeutic side effects are common. Bisubstrate-based inhibitors targeting both the high-selectivity peptide substrate binding groove and the high-affinity ATP pocket address this. However, they are typically large and polar, hampering cellular uptake. This paper describes a modular development approach for bisubstrate-based kinase inhibitors furnished with cell-penetrating moieties and demonstrates their cellular uptake and intracellular activity against protein kinase C (PKC). This enzyme family is a longstanding pharmaceutical target involved in cancer, immunological disorders, and neurodegenerative diseases. However, selectivity is particularly difficult to achieve because of homology among family members and with several related kinases, making PKC an excellent proving ground for bisubstrate-based inhibitors. Besides the pharmacological potential of the novel cell-penetrating constructs, the modular strategy described here may be used for discovering selective, cell-penetrating kinase inhibitors against any kinase and may increase adoption and therapeutic application of this promising inhibitor class.
Co-reporter:Gwenn E. Mulder, H (Linda). C. Quarles van Ufford, Jeroen van Ameijde, Arwin J. Brouwer, John A. W. Kruijtzer and Rob M. J. Liskamp
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 16) pp:2676-2684
Publication Date(Web):22 Feb 2013
DOI:10.1039/C3OB27470E
A diversity of protein surface discontinuous epitope mimics is now rapidly and efficiently accessible. Despite the important role of protein–protein interactions involving discontinuous epitopes in a wide range of diseases, mimicry of discontinuous epitopes using peptide-based molecules remains a major challenge. Using copper(I) catalyzed azide–alkyne cycloaddition (CuAAC), we have developed a general and efficient method for the synthesis of collections of discontinuous epitope mimics. Up to three different cyclic peptides, representing discontinuous epitopes in HIV-gp120, were conjugated to a selection of scaffold molecules. Variation of the scaffold molecule, optimization of the ring size of the cyclic peptides and screening of the resulting libraries for successful protein mimics led to an HIV gp120 mimic with an IC50 value of 1.7 μM. The approach described here provides rapid and highly reproducible access to clean, smart libraries of very complex bio-molecular constructs representing protein mimics for use as synthetic vaccines and beyond.
Co-reporter:Dr. Jeroen vanAmeijde;Dr. John Overvoorde;Dr. Stefan Knapp;Dr. Jeroen denHertog;Dr. Rob Ruijtenbeek;Dr. Rob M. J. Liskamp
ChemPlusChem 2013 Volume 78( Issue 11) pp:1349-1357
Publication Date(Web):
DOI:10.1002/cplu.201300299
Abstract
Phosphatases and kinases regulate the crucial phosphorylation post-translational modification. In spite of their similarly important role in many diseases and therapeutic potential, phosphatases have received arguably less attention. One reason for this is a scarcity of high-throughput phosphatase assays. Herein, a new real-time, dynamic protein tyrosine phosphatase (PTP) substrate microarray assay measuring product formation is described. PTP substrates comprising a novel 3-nitrophosphotyrosine residue are immobilized in discrete spots. After reaction catalyzed by a PTP a 3-nitrotyrosine residue is formed that can be detected by specific, sequence-independent antibodies. The resulting microarray was successfully evaluated with a panel of recombinant PTPs and cell lysates, which afforded results comparable to data from other assays. Its parallel nature, convenience, and low sample requirements facilitate investigation of the therapeutically relevant PTP enzyme family.
Co-reporter:Gwenn E. Mulder, John A. W. Kruijtzer and Rob M. J. Liskamp
Chemical Communications 2012 vol. 48(Issue 80) pp:10007-10009
Publication Date(Web):10 Aug 2012
DOI:10.1039/C2CC35310E
We describe rapid and convenient access to smart libraries of protein surface discontinuous epitope mimics. Up to three different cyclic peptides, representing discontinuous epitopes in HIV-gp120, were conjugated to a triazacyclophane scaffold molecule via CuAAC. In this way protein mimics for use as synthetic vaccines and beyond will become available.
Co-reporter:Arwin J. Brouwer ; Anika Jonker ; Paul Werkhoven ; Ethan Kuo ; Nan Li ; Nerea Gallastegui ; Johan Kemmink ; Bogdan I. Florea ; Michael Groll ; Herman S. Overkleeft ;Rob M. J. Liskamp
Journal of Medicinal Chemistry 2012 Volume 55(Issue 24) pp:10995-11003
Publication Date(Web):November 22, 2012
DOI:10.1021/jm301443r
A new class of potent proteasome inhibitors is described, of which the members contain an amino acid inspired sulfonyl fluoride as the electrophilic trap. In total, 24 peptido sulfonyl fluoride inhibitors have been designed and synthesized, which were inspired by the backbone sequences of the proteasome inhibitors bortezomib, epoxomicin, and Cbz-Leu3-aldehyde. Nine of them were very potent proteasome inhibitors, the best of which had an IC50 of 7 nM. A number of the peptido sulfonyl fluoride inhibitors were found to be highly selective for the β5 proteasome subunit.
Co-reporter:H. Bauke Albada, Fouad Soulimani, Hans J. F. Jacobs, Cees Versluis, Bert M. Weckhuysen and Rob M. J. Liskamp
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 5) pp:1088-1092
Publication Date(Web):16 Nov 2011
DOI:10.1039/C1OB06806G
We describe the synthesis and coordination behaviour to copper(II) of two close structural triazacyclophane-based mimics of two often encountered aspartic acid and histidine containing metalloenzyme active sites. Coordination of these mimics to copper(I) and their reaction with molecular oxygen leads to the formation of dimeric bis(μ-hydroxo) dicopper(II) complexes.
Co-reporter:Monique P. C. Mulder, Peter Fodran, Johan Kemmink, Eefjan J. Breukink, John A. W. Kruijtzer, Adriaan J. Minnaard and Rob M. J. Liskamp
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 37) pp:7491-7502
Publication Date(Web):24 Jul 2012
DOI:10.1039/C2OB25951F
The echinocandins represent the most recent class of antifungal drugs. Previous structure–activity relationship studies on these lipopeptides have relied mainly upon semisynthetic derivatives due to their complex chemical structures. A successful strategy for the rapid enantioselective synthesis of the branched fatty acid chain of caspofungin and analogues was developed to synthesize several simplified analogues of caspofungin. The specific minimum inhibitory activity of each mimic was determined against a panel of Candida strains. This approach gave access to new fully synthetic derived caspofungin mimics with high and selective antifungal activities against Candida strains. In addition, the data suggested an important role of the hydroxy proline residue in the bioactive conformation of the macrocyclic peptide ring structure.
Co-reporter:Dr. Kristina Görmer;Marco Bürger;Dr. John A. W. Kruijtzer;Dr. Ingrid Vetter;Dr. Nachiket Vartak; Dr. Lucas Brunsveld; Dr. Philippe I. H. Bastiaens; Dr. Rob M. J. Liskamp;Dr. Gemma Triola; Dr. Herbert Waldmann
ChemBioChem 2012 Volume 13( Issue 7) pp:1017-1023
Publication Date(Web):
DOI:10.1002/cbic.201200078
Co-reporter:Helmus van de Langemheen, Arwin J. Brouwer, Johan Kemmink, John A. W. Kruijtzer, and Rob M. J. Liskamp
The Journal of Organic Chemistry 2012 Volume 77(Issue 22) pp:10058-10064
Publication Date(Web):October 18, 2012
DOI:10.1021/jo3015566
The synthesis of cyclic peptides containing a thioester handle using a sulfo-click linker is reported. These cyclic peptides can be coupled to N-terminal cysteine-containing constructs via native chemical ligation. A successful application of a cyclic peptide bearing a thioester handle in native chemical ligation is shown by a high yielding ligation.
Co-reporter:Loek T. M. vanWelen;Dr. Jeroen vanAmeijde;Ahmed S. A. Mady;Angelique E. M. Wammes;Alois Bode;Dr. Alex J. Poot;Dr. Rob Ruijtenbeek;Dr. Rob M. J. Liskamp
ChemMedChem 2012 Volume 7( Issue 12) pp:
Publication Date(Web):
DOI:10.1002/cmdc.201290060
Co-reporter:Loek T. M. vanWelen;Dr. Jeroen vanAmeijde;Ahmed S. A. Mady;Angelique E. M. Wammes;Alois Bode;Dr. Alex J. Poot;Dr. Rob Ruijtenbeek;Dr. Rob M. J. Liskamp
ChemMedChem 2012 Volume 7( Issue 12) pp:2113-2121
Publication Date(Web):
DOI:10.1002/cmdc.201200349
Abstract
Kinases present an attractive target for drug development, since they are involved in vital cellular processes and are implicated in a variety of diseases, such as cancer and diabetes. However, obtaining selectivity for a specific kinase over others is difficult since many current kinase inhibitors exclusively target the highly conserved kinase ATP binding domain. Previously, a microarray-based strategy to discover so-called bisubstrate-based inhibitors that target the more specific peptide binding groove in addition to the ATP binding site was described. One attractive feature of this strategy is the opportunity to tune the selectivity of these inhibitors by systematically varying components. In an extension to this previous work, this study explores the potential of this guided selectivity modulation, leading to a series of inhibitors with different selectivity profiles against highly homologous protein kinase C (PKC) isozymes. Of the inhibitors studied, most exhibited improved potency and selectivity compared with their constituent parts. Furthermore, the selectivity was found to be tunable either through modification of the pseudosubstrate peptide (peptide binding groove) or the ATP-competitive part (ATP binding site). In a number of cases, the selectivity of the construct could be predicted from the initial peptide substrate profiling experiment. Since this strategy is applicable to all kinase sets, it could be used to rapidly develop uniquely selective inhibitors.
Co-reporter:Jinqiang Zhang, Johan Kemmink, Dirk T. S. Rijkers, and Rob M. J. Liskamp
Organic Letters 2011 Volume 13(Issue 13) pp:3438-3441
Publication Date(Web):May 26, 2011
DOI:10.1021/ol201184b
Structural mimics comprising 1,4- and 1,5-disubstituted triazole-containing cyclic tripeptides with excellent resemblance toward the DE-ring of vancomycin are conveniently accessible using Cu(I)- or Ru(II)-assisted “click” cyclization.
Co-reporter:Joeri Kuil, Marcel J. E. Fischer, Nico J. de Mol and Rob M. J. Liskamp
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 3) pp:820-833
Publication Date(Web):25 Nov 2010
DOI:10.1039/C0OB00441C
Spleen tyrosine kinase (Syk) is essential for high affinity IgE receptor (FcεRI) mediated mast cell degranulation. Once FcεRI is stimulated, intracellular ITAM motifs of the receptor are diphosphorylated (dpITAM) and Syk is recruited to the receptor by binding of the Syk tandem SH2 domain to dpITAM, resulting in activation of Syk and, eventually, degranulation. To investigate intracellular effects of ITAM mimics, constructs were synthesized with ITAM mimics conjugated to different cell penetrating peptides, i.e. Tat, TP10, octa-Arg and K(Myr)KKK, or a lipophilic C12-chain. In most constructs the cargo and carrier were linked to each other through a disulfide bridge, which is convenient for combining different cargos with different carriers and has the advantage that the cargo and the carrier may be separated by reduction of the disulfide once it is intracellular. The ability of these ITAM constructs to label RBL-2H3 cells was assessed using flow cytometry. Fluorescence microscopy showed that the octa-Arg-SS-Flu-ITAM construct was present in various parts of the cells, although it was not homogeneously distributed. In addition, cell penetrating constructs without fluorescent labels were synthesized to examine degranulation in RBL-2H3 cells. Octa-Arg-SS-ITAM stimulated the mediator release up to 140%, indicating that ITAM mimics may have the ability to activate non-receptor bound Syk.
Co-reporter:H. Bauke Albada;Fiora Rosati;David Coquière;Gerard Roelfes;Rob M. J. Liskamp
European Journal of Organic Chemistry 2011 Volume 2011( Issue 9) pp:1714-1720
Publication Date(Web):
DOI:10.1002/ejoc.201001522
Abstract
A triazacyclophane (TAC) scaffold decorated with three histidine amino acid residues was used as a tridentate ligand in asymmetric copper(II)-catalysed Diels–Alder and Michael addition reactions in water. Enantiomeric excesses up to 55 % were obtained in Diels–Alder reactions using ligands in which the histidine residues were directly attached to the TAC scaffold. Additional amino acid residues on the N-termini of the histidine residues or positioned between the histidine residues and the TAC scaffold, resulted in almost complete loss of enantioselectivity. Modelling studies of the coordination complex of the most specific ligand indicated the presence of a substrate binding pocket in proximity to the catalytically active centre.
Co-reporter:Monique P.C. Mulder, John A.W. Kruijtzer, Eefjan J. Breukink, Johan Kemmink, Roland J. Pieters, Rob M.J. Liskamp
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 21) pp:6505-6517
Publication Date(Web):1 November 2011
DOI:10.1016/j.bmc.2011.08.034
Echinocandins are a novel class of macrocyclic antifungal peptides that act by inhibiting the β-(1,3)-d-glucan synthase complex, which is not present in mammalian cells. Due to the large number of hydroxyl groups present in these complex macrocyclic lipopeptides, most structure–activity relationship studies have relied upon semisynthetic derivatives. In order to probe the influence of the cyclic peptide backbone on the antifungal activity we developed a successful strategy for the synthesis of novel echinocandins analogues by on-resin ring closing metathesis or disulfide formation. The specific minimum inhibitory activity of each mimic was determined against Candida albicans. Our results indicate that ring size is an important factor for antifungal activity.
Co-reporter:Arwin J. Brouwer, Tarik Ceylan, Anika M. Jonker, Tima van der Linden, Rob M.J. Liskamp
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 7) pp:2397-2406
Publication Date(Web):1 April 2011
DOI:10.1016/j.bmc.2011.02.014
We have designed and synthesized novel irreversible serine protease inhibitors containing aliphatic sulfonyl fluorides as an electrophilic trap. These substituted taurine sulfonyl fluorides derived from taurine or protected amino acids were conveniently synthesized from β-aminoethanesulfonyl chlorides using KF/18-crown-6 or from β-aminoethanesulfonates using DAST. Their potency of irreversible inhibition of serine proteases is described in different enzyme assays using chymotrypsin leading to binding affinities up to 22 μM.
Co-reporter: Dr. Rob M. J. Liskamp;Dr. Dirk T. S. Rijkers;Dr. John A. W. Kruijtzer ;Dr. Johan Kemmink
ChemBioChem 2011 Volume 12( Issue 11) pp:1626-1653
Publication Date(Web):
DOI:10.1002/cbic.201000717
Abstract
Despite their enormous diversity in biological function and structure, peptides and proteins are endowed with properties that have induced and stimulated the development of peptidomimetics. Clearly, peptides can be considered as the “stem” of a phylogenetic molecular development tree from which branches of oligomeric peptidomimetics such as peptoids, peptidosulfonamides, urea peptidomimetics, as well as β-peptides have sprouted. It is still a challenge to efficiently synthesize these oligomeric species, and study their structural and biological properties. Combining peptides and peptidomimetics led to the emergence of peptide–peptidomimetic hybrids in which one or more (proteinogenic) amino acid residues have been replaced with these mimetic residues. In scan-like approaches, the influence of these replacements on biological activity can then be studied, to evaluate to what extent a peptide can be transformed into a peptidomimetic structure while maintaining, or even improving, its biological properties. A central issue, especially with the smaller peptides, is the lack of secondary structure. Important approaches to control secondary structure include the introduction of α,α-disubstituted amino acids, or (di)peptidomimetic structures such as the Freidinger lactam. Apart from intra-amino acid constraints, inter-amino acid constraints for formation of a diversity of cyclic peptides have shaped a thick branch. Apart from the classical disulfide bridges, the repertoire has been extended to include sulfide and triazole bridges as well as the single-, double- and even triple-bond replacements, accessible by the extremely versatile ring-closing alkene/alkyne metathesis approaches. The latter approach is now the method of choice for the secondary structure that presents the greatest challenge for structural stabilization: the α-helix.
Co-reporter:Cheng-Bin Yim ;Berend van der Wildt;Dr. Ingrid Dijkgraaf;Lieke Joosten;Annemarie Eek;Cees Versluis;Dr. Dirk T. S. Rijkers; Dr. Otto C. Boerman; Dr. Rob M. J. Liskamp
ChemBioChem 2011 Volume 12( Issue 5) pp:750-760
Publication Date(Web):
DOI:10.1002/cbic.201000639
Abstract
We report on the SSTR2-binding properties of a series of four dimeric [Tyr3]octreotate analogues with different spacer lengths (nine, 19, 41, and 57 atoms) between the peptides. Two analogues (9 and 57 atoms) were selected as precursors for the design, synthesis, and biological evaluation of DOTA-conjugated dimeric [Tyr3]octreotate analogues for tumor targeting. These compounds were synthesized by using a two-stage click ligation procedure: a CuI-catalyzed 1,3-dipolar cycloaddition (“copper-click” reaction) and a thio acid/sulfonyl azide amidation (“sulfo-click” reaction). The IC50 values of these DOTA-conjugated [Tyr3]octreotate analogues were comparable, and internalization studies showed that the nine-atom 111In-DOTA-labeled [Tyr3]octreotate dimer had rapid and high receptor binding. Biodistribution studies with BALB/c nude mice bearing subcutaneous AR42J tumors showed that the 111In-labeled [Tyr3]octreotate dimer (nine atoms) had a high tumor uptake at 1 h p.i. (38.8±8.3 % ID g−1), and excellent tumor retention at 4 h p.i. (40.9±2.5 % ID g−1). However, the introduction of the extended hydrophilic 57 atoms spacer led to rapid clearance from the circulation; this limited tumor accumulation of the radiotracer (21.4±4.9 % ID g−1 at 1 h p.i.). These findings provide important insight on dimerization and spacer effects on the in vivo properties of DOTA-conjugated [Tyr3]octreotate dimers.
Co-reporter:Jeroen van Ameijde, Alex J. Poot, Loek T. M. van Wandelen, Angelique E. M. Wammes, Rob Ruijtenbeek, Dirk T. S. Rijkers and Rob M. J. Liskamp
Organic & Biomolecular Chemistry 2010 vol. 8(Issue 7) pp:1629-1639
Publication Date(Web):04 Feb 2010
DOI:10.1039/B922928K
Efficient strategies for the introduction of arginine residues featuring acetylene or azide moieties in their side chains are described. The substituents are introduced in a way that maintains the basicity of the guanidine moiety. The methodology can be used e.g. for non-invasive labeling of arginine-containing peptides. Its applicability is demonstrated by the introduction of ‘click’ handles into a Protein Kinase C (PKC) pseudosubstrate peptide, and the subsequent preparation and evaluation of a novel bisubstrate-based inhibitor based on such a peptide.
Co-reporter:Maarten van Dijk, Cornelus F. van Nostrum, Wim E. Hennink, Dirk T. S. Rijkers and Rob M. J. Liskamp
Biomacromolecules 2010 Volume 11(Issue 6) pp:
Publication Date(Web):May 24, 2010
DOI:10.1021/bm1002637
Herein we describe the synthesis and rheological characterization of a series of enzymatically sensitive PEG and peptide-based hydrogels by the Cu(I)-catalyzed 1,3-dipolar cycloaddition reaction. The hydrogels were synthesized by a combination of alkyne-functionalized star-shaped PEG molecules (two 4-armed PEGs with Mw 10 and 20 kDa, respectively, and one 8-armed PEG of 20 kDa) and the protease-sensitive bis-azido peptide, Nα-(azido)-d-alanyl-phenylalanyl-lysyl-(2-azidoethyl)-amide (6) in the presence of CuSO4 and sodium ascorbate in aqueous solution. The swelling ratio and the storage modulus (G′) of the hydrogels could be tailored by several parameters, for example, the initial solid content of the hydrogel, the molecular weight of the PEG derivative, and by the architecture of the PEG molecule (4- versus 8-armed PEG derivative). The peptide sequence, d-Ala-Phe-Lys, was sensitive toward the proteases plasmin and trypsin to render the hydrogels biodegradable.
Co-reporter:Jeroen van Ameijde, Thierry K. Slot, Rob M.J. Liskamp
Tetrahedron: Asymmetry 2010 Volume 21(Issue 4) pp:469-475
Publication Date(Web):16 March 2010
DOI:10.1016/j.tetasy.2010.02.005
The scalable, divergent synthesis of all four monomers required for the preparation of sulfonamide-based RNA mimetics is described. Such mimetics may combine excellent mimicry of the parent RNA with enhanced (bio)chemical robustness and convenient oligomerization. As a proof of principle, a dimer resulting from the monomers is described.3-Azido-3-deoxy-1,2-O-isopropylidene-β-d-riboseC8H13N3O4[α]D23=+124.4 (c 0.85, CHCl3)Source of chirality: 1,2:5,6-di-O-isopropylidene-β-d-gluco-furanoseAbsolute configuration: (1R,2R,3R,4S)3-Azido-3,5,6-trideoxy-6-(isobutyloxysulfon)-1,2-O-isopropylidene-β-d-ribo-hexofuranoseC13H23N3O6S[α]D23=+97.1 (c 0.69, CHCl3)Source of chirality: 1,2:5,6-di-O-isopropylidene-β-d-gluco-furanoseAbsolute configuration: (1R,2R,3R,4R)3-Azido-3,5-dideoxy-5-(isobutyloxysulfonmethyl)-N6-benzoyl-adenosineC24H28N8O7S[α]D23=+6.6 (c 2.20, CHCl3)Source of chirality: 1,2:5,6-di-O-isopropylidene-β-d-gluco-furanoseAbsolute configuration: (1R,2R,3R,4R)3-Azido-3,5-dideoxy-5-(isobutyloxysulfonmethyl)-N2-pivaloyl-guanosineC22H32N8O8S[α]D23=+47.7 (c 0.21, CHCl3)Source of chirality: 1,2:5,6-di-O-isopropylidene-β-d-gluco-furanoseAbsolute configuration: (1R,2R,3R,4R)3-Azido-3,5-dideoxy-5-(isobutyloxysulfonmethyl)-N4-benzoyl-cytidineC23H28N6O8S[α]D23=+42.3 (c 0.26, CHCl3)Source of chirality: 1,2:5,6-di-O-isopropylidene-β-d-gluco-furanoseAbsolute configuration: (1R,2R,3R,4R)3-Azido-3,5-dideoxy-5-(isobutyloxysulfonmethyl)-uridineC16H23N5O8S[α]D23=+31.5 (c 1.38, CHCl3)Source of chirality: 1,2:5,6-di-O-isopropylidene-β-d-gluco-furanoseAbsolute configuration: (1R,2R,3R,4R)3-Azido-3,5-dideoxy-5-(chlorosulfonmethyl)-uridineC12H14ClN5O7S[α]D23=+42.1 (c 0.21, CHCl3)Source of chirality: 1,2:5,6-di-O-isopropylidene-β-d-gluco-furanoseAbsolute configuration: (1R,2R,3R,4R)3-Azido-3,5-dideoxy-5-(dimethylaminosulfonmethyl)-N4-benzoyl-cytidineC21H27N7O7S[α]D23=+38.7 (c 0.61, CHCl3)Source of chirality: 1,2:5,6-di-O-isopropylidene-β-d-gluco-furanoseAbsolute configuration: (1R,2R,3R,4R)3-Azido-3,5-dideoxy-5-(isobutylaminosulfonmethyl)-uridineC16H24N6O7S[α]D23=+48.8 (c 0.32, CHCl3)Source of chirality: 1,2:5,6-di-O-isopropylidene-β-d-gluco-furanoseAbsolute configuration: (1R,2R,3R,4R)
Co-reporter:Anton Bunschoten, John A. W. Kruijtzer, Johannes H. Ippel, Carla J. C. de Haas, Jos A. G. van Strijp, Johan Kemmink and Rob M. J. Liskamp
Chemical Communications 2009 (Issue 21) pp:2999-3001
Publication Date(Web):09 Mar 2009
DOI:10.1039/B823425F
In this communication, a new site specific synthesis of highly functionalized and multiple sulfated peptides using convential Fmoc-tBu solid phase peptide synthesis is described.
Co-reporter:Cristina Chamorro, John A. W. Kruijtzer, Marina Farsaraki, Jan Balzarini and Rob M. J. Liskamp
Chemical Communications 2009 (Issue 7) pp:821-823
Publication Date(Web):17 Dec 2008
DOI:10.1039/B817357E
In this Communication, the access to three different peptide loops attached to a small triazacyclophane (TAC) scaffold molecule for the mimicry of discontinuous epitopes present in, for example, antibodies is described for the first time.
Co-reporter:Cheng-Bin Yim, Otto C. Boerman, Monique de Visser, Marion de Jong, Annemarie C. Dechesne, Dirk T. S. Rijkers and Rob M. J. Liskamp
Bioconjugate Chemistry 2009 Volume 20(Issue 7) pp:1323
Publication Date(Web):June 18, 2009
DOI:10.1021/bc900052n
The somatostatin analogue Tyr3-octreotide, which has a high binding affinity for the SSTR2 receptor (somatostatin receptor subtype 2) expressed on tumor cells, is used clinically for the diagnosis and treatment of a variety of neuroendocrine tumors and gastrointestinal disorders. There is growing interest in the development of multivalent peptide systems, because they may have enhanced binding affinity compared to monovalent analogues. In this report, we describe the design and synthesis of a series of Tyr3-octreotide-containing monomeric, dimeric, and tetrameric dendrimeric conjugates. These multivalent dendrimeric cyclic peptides were obtained using Cu(I)-catalyzed 1,3-dipolar cycloaddition between peptidyl azides and dendrimeric alkynes. Their affinities for the SSTR2 receptor were determined by a competitive binding assay on rat brain sections.
Co-reporter:Ronald C. Elgersma, Maarten van Dijk, Annemarie C. Dechesne, Cornelus F. van Nostrum, Wim E. Hennink, Dirk T. S. Rijkers and Rob M. J. Liskamp
Organic & Biomolecular Chemistry 2009 vol. 7(Issue 21) pp:4517-4525
Publication Date(Web):27 Aug 2009
DOI:10.1039/B912851D
We report on the design, synthesis, and structural analysis of cyclic oligomers with an amyloidogenic peptide sequence as the repeating unit to obtain novel self-assembling bionanomaterials. The peptide was derived from the Alzheimer Aβ(16–22) sequence since its strong tendency to form antiparallel β-sheets ensured the formation of intermolecular hydrogen bridges on which the supramolecular assembly of the individual cyclic oligomers was based. The synthesis of the cyclic oligomers was performed via a microwave-assisted Cu(I)-catalyzed 1,3-dipolar cycloaddition reaction of azido-Lys-Leu-Val-Phe-Phe-Ala-Glu-propargyl amide as the monomer. The formation of cyclic oligomers, up to pentamers (35 amino acid residues), was verified by MALDI-TOF analysis and the individual cyclic monomer and dimer could be isolated by HPLC. Gelation behavior and the self-assembly of the linear monomer and the cyclic monomer and dimer were studied by TEM, FTIR and CD. Significant differences were observed in the morphology of the supramolecular aggregates of these three peptides that could be explained by alterations of the hydrogen bond network.
Co-reporter:H. Bauke Albada, Christopher J. Arnusch, Hilbert M. Branderhorst, Anne-Marie Verel, Wouter T.M. Janssen, Eefjan Breukink, Ben de Kruijff, Roland J. Pieters, Rob M.J. Liskamp
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 14) pp:3721-3724
Publication Date(Web):15 July 2009
DOI:10.1016/j.bmcl.2009.05.064
The antibiotic vancomycin—that binds lipid II in the bacterial cell membrane—was conjugated to a mono- and tetravalent mimic of the tris-histidine catalytic triad of metalloenzymes. Targeted hydrolysis by the conjugate was observed using model membranes containing lipid II, and in vitro MIC-values of the targeted mimic constructs could be modulated by Zn-ions.Vancomycin was conjugated to a hydrolysis catalyst (or TACzyme). Targeted hydrolysis by such a conjugate was observed using membranes containing lipid II. MIC-values of targeted hydrolysis catalyst constructs could be modulated by Zn(II).
Co-reporter:Alex J. Poot;Jeroen van Ameijde Dr.;Monique Slijper Dr.;Adriënne van den Berg;Riet Hilhorst Dr.;Rob Ruijtenbeek Dr.;Dirk T. S. Rijkers Dr.;Rob M. J. Liskamp Dr.
ChemBioChem 2009 Volume 10( Issue 12) pp:2042-2051
Publication Date(Web):
DOI:10.1002/cbic.200900199
Abstract
Kinase inhibitors are increasingly important in drug development. Because the majority of current inhibitors target the conserved ATP-binding site, selectivity might become an important issue. This could be particularly problematic for the potential drug target protein kinase C (PKC), of which twelve isoforms with high homology exist in humans. A strategy to increase selectivity is to prepare bisubstrate-based inhibitors that target the more selective peptide-binding site in addition to the ATP-binding site. In this paper a generally applicable, rapid methodology is presented to discover such bisubstrate-based leads. Dynamic peptide microarrays were used to find peptide-binding site inhibitors. These were linked with chemoselective click chemistry to an ATP-binding site inhibitor, and this led to novel bisubstrate structures. The peptide microarrays were used to evaluate the resulting inhibitors. Thus, novel bisubstrate-based inhibitors were obtained that were both more potent and selective compared to their constituent parts. The most promising inhibitor has nanomolar affinity and selectivity towards PKCθ amongst three isozymes.
Co-reporter:Arwin J. Brouwer, Tarik Ceylan, Tima van der Linden, Rob M.J. Liskamp
Tetrahedron Letters 2009 50(26) pp: 3391-3393
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.02.130
Co-reporter:H. Bauke Albada and Rob M. J. Liskamp
ACS Combinatorial Science 2008 Volume 10(Issue 6) pp:814
Publication Date(Web):September 24, 2008
DOI:10.1021/cc800065a
In this report, we present the first library of tripodal synthetic receptor molecules containing three different, temporarily N-terminal protected peptide arms capable of performing hydrolytic reactions. To construct this library, the orthogonally protected triazacyclophane (TAC)-scaffold was used in the preparation of a split−mix library of 19 683 resin bound tripodal receptor molecules. For the construction of the peptide arms, three different sets of amino acids were used, each focused on one part of the catalytic triad as found in several families of hydrolytic enzymes. Therefore, in the sets of amino acids used to assemble these tripeptides, basic (containing His and Lys), nucleophilic (containing Ser and Cys), or acidic (containing Asp and Glu) amino acid residues were present. In addition, nonfunctional hydrophobic amino acid residues were introduced. Possible unfavorable electrostatic interactions of charged N-termini or their acetylation during screening were circumvented by trifluoroacetylation of the N-terminal amines. Screening was performed with a known esterase substrate, 7-acetoxycoumarin, which upon hydrolysis gave the fluorescent 7-hydroxycoumarin, leading to fluorescence of beads containing a hydrolytically active synthetic receptor. Although many synthetic receptors contain catalytic triad combinations, apparently, only a few showed hydrolytic activity. Sequence analysis of the active receptors showed that carboxylate-containing amino acids are frequently found in the acidic arm and that substrate cleavage is mediated by lysine (noncatalytic) or histidine (catalytic) residues. Kinetic analysis of resynthesized receptors showed that catalysis depended on the number of histidine residues and was not assisted by significant substrate binding.
Co-reporter:Maarten van Dijk, Maria L. Nollet, Pascal Weijers, Annemarie C. Dechesne, Cornelus F. van Nostrum, Wim E. Hennink, Dirk T. S. Rijkers and Rob M. J. Liskamp
Biomacromolecules 2008 Volume 9(Issue 10) pp:
Publication Date(Web):September 26, 2008
DOI:10.1021/bm8005984
In this study, the microwave-assisted copper(I)-catalyzed 1,3-dipolar cycloaddition reaction was used to synthesize peptide triazole-based polymers from two novel peptide-based monomers: azido-phenylalanyl-alanyl-lysyl-propargyl amide (1) and azido-phenylalanyl-alanyl-glycolyl-lysyl-propargyl amide (2). The selected monomers have sites for enzymatic degradation as well as for chemical hydrolysis to render the resulting polymer biodegradable. Depending on the monomer concentration in DMF, the molecular mass of the polymers could be tailored between 4.5 and 13.9 kDa (corresponding with 33−100 amino acid residues per polymer chain). As anticipated, both polymers can be enzymatically degraded by trypsin and chymotrypsin, whereas the ester bond in the polymer of 2 undergoes chemical hydrolysis under physiological conditions, as was shown by a ninhydrin-based colorimetric assay and MALDI-TOF analysis. In conclusion, the microwave-assisted copper(I)-catalyzed 1,3-dipolar cycloaddition reaction is an effective tool for synthesizing biodegradable peptide polymers, and it opens up new approaches toward the synthesis of (novel) designed biomedical materials.
Co-reporter:H. Bauke Albada, Fouad Soulimani, Bert M. Weckhuysen and Rob M. J. Liskamp
Chemical Communications 2007 (Issue 46) pp:4895-4897
Publication Date(Web):02 Nov 2007
DOI:10.1039/B709400K
We report the use of triazacyclophane (TAC)-scaffolded amino acids as a structural mimic for 3-histidine metal-binding sites in metalloproteins, especially for the mimicry of type-3 copper binding sites as are present in hemocyanin, tyrosinase and catechol oxidase.
Co-reporter:Dirk-Jan van Zoelen;Maarten R. Egmond Dr.;Rol J. Pieters Dr. and;Rob M. J. Liskamp Dr.
ChemBioChem 2007 Volume 8(Issue 16) pp:
Publication Date(Web):20 SEP 2007
DOI:10.1002/cbic.200700243
An approach for mimicking protein–protein interactions by using a discontinuous epitope to construct a mimic that is about one tenth of the size of a natural inhibitor of papain, namely, cystatin B, is described. The discontinuous epitope of cystatin B, which is involved in the interaction with papain, was mimicked by synthesis of a tripodal molecular construct by using the triazacyclophane (TAC) scaffold. The mimic contains three peptide arms: one that mimics the N terminus, one that mimics the C terminus, and one that mimics the β-hairpin loop structure of cystatin B. These peptide sequences were assembled on the TAC scaffold. The resulting cystatin mimic, CysTACtin 9, showed excellent inhibition of papain with a Ki of 12 nM, which approaches the inhibitory potency of cystatin B (Ki=0.12 nM). Experiments with molecular constructs that contained one or two arms or a mixture of the nonscaffolded peptides showed that both scaffolding and the presence of the three peptide arms are crucial for a successful mimic.
Co-reporter:Cheng-Bin Yim ; Ingrid Dijkgraaf ; Remco Merkx ; Cees Versluis ; Annemarie Eek ; Gwenn E. Mulder ; Dirk T. S. Rijkers ; Otto C. Boerman ;Rob M. J. Liskamp
Journal of Medicinal Chemistry () pp:
Publication Date(Web):April 22, 2010
DOI:10.1021/jm100246m
Herein, we describe the design, synthesis, and biological evaluation of a series of DOTA-conjugated monomeric, dimeric, and tetrameric [Tyr3]octreotide-based analogues as a tool for tumor imaging and/or radionuclide therapy. These compounds were synthesized using a Cu(I)-catalyzed 1,3-dipolar cycloaddition (“click” reaction) between peptidic azides and dendrimer-derived alkynes and a subsequent metal-free introduction of DOTA via the thio acid/sulfonyl azide amidation (“sulfo-click” reaction). In a competitive binding assay using rat pancreatic AR42J tumor cells, the monomeric [Tyr3]octreotide conjugate displayed the highest binding affinity (IC50 = 1.32 nM) followed by dimeric [Tyr3]octreotide (2.45 nM), [DOTA0,Tyr3]octreotide (2.45 nM), and tetrameric [Tyr3]octreotide (14.0 nM). Biodistribution studies with BALB/c nude mice with subcutaneous AR42J tumors showed that the 111In-labeled monomeric [Tyr3]octreotide conjugate had the highest tumor uptake (42.3 ± 2.8 %ID/g) at 2 h p.i., which was better than [111In-DOTA0,Tyr3]octreotide (19.5 ± 4.8 %ID/g). The 111In-labeled dimeric [Tyr3]octreotide conjugate showed a long tumor retention (25.3 ± 5.9 %ID/g at 2 h p.i. and 12.1 ± 1.3 %ID/g at 24 h p.i.). These promising results can be exploited for therapeutic applications.
Co-reporter:P. R. Werkhoven, M. Elwakiel, T. J. Meuleman, H. C. Quarles van Ufford, J. A. W. Kruijtzer and R. M. J. Liskamp
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 2) pp:NaN710-710
Publication Date(Web):2015/11/05
DOI:10.1039/C5OB02014J
Mimics of discontinuous epitopes of for example bacterial or viral proteins may have considerable potential for the development of synthetic vaccines, especially if conserved epitopes can be mimicked. However, due to the structural complexity and size of discontinuous epitopes molecular construction of these mimics remains challeging. We present here a convergent route for the assembly of discontinuous epitope mimics by successive azide alkyne cycloaddition on an orthogonal alkyne functionalized scaffold. Here the synthesis of mimics of the HIV gp120 discontinuous epitope that interacts with the CD4 receptor is described. The resulting protein mimics are capable of inhibition of the gp120–CD4 interaction. The route is convergent, robust and should be applicable to other discontinuous epitopes.
Co-reporter:Gwenn E. Mulder, John A. W. Kruijtzer and Rob M. J. Liskamp
Chemical Communications 2012 - vol. 48(Issue 80) pp:NaN10009-10009
Publication Date(Web):2012/08/10
DOI:10.1039/C2CC35310E
We describe rapid and convenient access to smart libraries of protein surface discontinuous epitope mimics. Up to three different cyclic peptides, representing discontinuous epitopes in HIV-gp120, were conjugated to a triazacyclophane scaffold molecule via CuAAC. In this way protein mimics for use as synthetic vaccines and beyond will become available.
Co-reporter:H. Bauke Albada, Fouad Soulimani, Bert M. Weckhuysen and Rob M. J. Liskamp
Chemical Communications 2007(Issue 46) pp:NaN4897-4897
Publication Date(Web):2007/11/02
DOI:10.1039/B709400K
We report the use of triazacyclophane (TAC)-scaffolded amino acids as a structural mimic for 3-histidine metal-binding sites in metalloproteins, especially for the mimicry of type-3 copper binding sites as are present in hemocyanin, tyrosinase and catechol oxidase.
Co-reporter:Anton Bunschoten, John A. W. Kruijtzer, Johannes H. Ippel, Carla J. C. de Haas, Jos A. G. van Strijp, Johan Kemmink and Rob M. J. Liskamp
Chemical Communications 2009(Issue 21) pp:NaN3001-3001
Publication Date(Web):2009/03/09
DOI:10.1039/B823425F
In this communication, a new site specific synthesis of highly functionalized and multiple sulfated peptides using convential Fmoc-tBu solid phase peptide synthesis is described.
Co-reporter:Jeroen van Ameijde, Alex J. Poot, Loek T. M. van Wandelen, Angelique E. M. Wammes, Rob Ruijtenbeek, Dirk T. S. Rijkers and Rob M. J. Liskamp
Organic & Biomolecular Chemistry 2010 - vol. 8(Issue 7) pp:NaN1639-1639
Publication Date(Web):2010/02/04
DOI:10.1039/B922928K
Efficient strategies for the introduction of arginine residues featuring acetylene or azide moieties in their side chains are described. The substituents are introduced in a way that maintains the basicity of the guanidine moiety. The methodology can be used e.g. for non-invasive labeling of arginine-containing peptides. Its applicability is demonstrated by the introduction of ‘click’ handles into a Protein Kinase C (PKC) pseudosubstrate peptide, and the subsequent preparation and evaluation of a novel bisubstrate-based inhibitor based on such a peptide.
Co-reporter:H. Bauke Albada, Fouad Soulimani, Hans J. F. Jacobs, Cees Versluis, Bert M. Weckhuysen and Rob M. J. Liskamp
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 5) pp:NaN1092-1092
Publication Date(Web):2011/11/16
DOI:10.1039/C1OB06806G
We describe the synthesis and coordination behaviour to copper(II) of two close structural triazacyclophane-based mimics of two often encountered aspartic acid and histidine containing metalloenzyme active sites. Coordination of these mimics to copper(I) and their reaction with molecular oxygen leads to the formation of dimeric bis(μ-hydroxo) dicopper(II) complexes.
Co-reporter:H. van de Langemheen, M. van Hoeke, H. C. Quarles van Ufford, J. A. W. Kruijtzer and R. M. J. Liskamp
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 25) pp:NaN4478-4478
Publication Date(Web):2014/04/24
DOI:10.1039/C4OB00190G
The accessibility to collections, libraries and arrays of cyclic peptides is increasingly important since cyclic peptides may provide better mimics of the loop-like structures ubiquitously present in and – especially – on the surface of proteins. The next important step is the preparation of libraries of ensembles of scaffolded cyclic peptides, which upon screening may lead to promising protein mimics. Here we describe the synthesis of a tri-cysteine containing scaffold as well as the simultaneous native chemical ligation of three cyclic peptides thereby affording a clean library of multiple cyclic peptides on this scaffold, representing potential mimics of gp120. Members of this collection of protein mimics showed a decent inhibition of the gp120-CD4 interaction.
Co-reporter:Jinqiang Zhang, Johan Kemmink, Dirk T. S. Rijkers and Rob M. J. Liskamp
Chemical Communications 2013 - vol. 49(Issue 40) pp:NaN4500-4500
Publication Date(Web):2013/03/28
DOI:10.1039/C3CC40628H
Herein we report the Ru(II)-catalyzed double-macrocyclization of a hexapeptide to obtain a mimic of the bicyclic CDE-ring of vancomycin, followed by measurement of its binding affinity for small peptide ligands using ITC.
Co-reporter:Joeri Kuil, Marcel J. E. Fischer, Nico J. de Mol and Rob M. J. Liskamp
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 3) pp:NaN833-833
Publication Date(Web):2010/11/25
DOI:10.1039/C0OB00441C
Spleen tyrosine kinase (Syk) is essential for high affinity IgE receptor (FcεRI) mediated mast cell degranulation. Once FcεRI is stimulated, intracellular ITAM motifs of the receptor are diphosphorylated (dpITAM) and Syk is recruited to the receptor by binding of the Syk tandem SH2 domain to dpITAM, resulting in activation of Syk and, eventually, degranulation. To investigate intracellular effects of ITAM mimics, constructs were synthesized with ITAM mimics conjugated to different cell penetrating peptides, i.e. Tat, TP10, octa-Arg and K(Myr)KKK, or a lipophilic C12-chain. In most constructs the cargo and carrier were linked to each other through a disulfide bridge, which is convenient for combining different cargos with different carriers and has the advantage that the cargo and the carrier may be separated by reduction of the disulfide once it is intracellular. The ability of these ITAM constructs to label RBL-2H3 cells was assessed using flow cytometry. Fluorescence microscopy showed that the octa-Arg-SS-Flu-ITAM construct was present in various parts of the cells, although it was not homogeneously distributed. In addition, cell penetrating constructs without fluorescent labels were synthesized to examine degranulation in RBL-2H3 cells. Octa-Arg-SS-ITAM stimulated the mediator release up to 140%, indicating that ITAM mimics may have the ability to activate non-receptor bound Syk.
Co-reporter:Ronald C. Elgersma, Maarten van Dijk, Annemarie C. Dechesne, Cornelus F. van Nostrum, Wim E. Hennink, Dirk T. S. Rijkers and Rob M. J. Liskamp
Organic & Biomolecular Chemistry 2009 - vol. 7(Issue 21) pp:NaN4525-4525
Publication Date(Web):2009/08/27
DOI:10.1039/B912851D
We report on the design, synthesis, and structural analysis of cyclic oligomers with an amyloidogenic peptide sequence as the repeating unit to obtain novel self-assembling bionanomaterials. The peptide was derived from the Alzheimer Aβ(16–22) sequence since its strong tendency to form antiparallel β-sheets ensured the formation of intermolecular hydrogen bridges on which the supramolecular assembly of the individual cyclic oligomers was based. The synthesis of the cyclic oligomers was performed via a microwave-assisted Cu(I)-catalyzed 1,3-dipolar cycloaddition reaction of azido-Lys-Leu-Val-Phe-Phe-Ala-Glu-propargyl amide as the monomer. The formation of cyclic oligomers, up to pentamers (35 amino acid residues), was verified by MALDI-TOF analysis and the individual cyclic monomer and dimer could be isolated by HPLC. Gelation behavior and the self-assembly of the linear monomer and the cyclic monomer and dimer were studied by TEM, FTIR and CD. Significant differences were observed in the morphology of the supramolecular aggregates of these three peptides that could be explained by alterations of the hydrogen bond network.
Co-reporter:Monique P. C. Mulder, Peter Fodran, Johan Kemmink, Eefjan J. Breukink, John A. W. Kruijtzer, Adriaan J. Minnaard and Rob M. J. Liskamp
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 37) pp:NaN7502-7502
Publication Date(Web):2012/07/24
DOI:10.1039/C2OB25951F
The echinocandins represent the most recent class of antifungal drugs. Previous structure–activity relationship studies on these lipopeptides have relied mainly upon semisynthetic derivatives due to their complex chemical structures. A successful strategy for the rapid enantioselective synthesis of the branched fatty acid chain of caspofungin and analogues was developed to synthesize several simplified analogues of caspofungin. The specific minimum inhibitory activity of each mimic was determined against a panel of Candida strains. This approach gave access to new fully synthetic derived caspofungin mimics with high and selective antifungal activities against Candida strains. In addition, the data suggested an important role of the hydroxy proline residue in the bioactive conformation of the macrocyclic peptide ring structure.
Co-reporter:Gwenn E. Mulder, H (Linda). C. Quarles van Ufford, Jeroen van Ameijde, Arwin J. Brouwer, John A. W. Kruijtzer and Rob M. J. Liskamp
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 16) pp:NaN2684-2684
Publication Date(Web):2013/02/22
DOI:10.1039/C3OB27470E
A diversity of protein surface discontinuous epitope mimics is now rapidly and efficiently accessible. Despite the important role of protein–protein interactions involving discontinuous epitopes in a wide range of diseases, mimicry of discontinuous epitopes using peptide-based molecules remains a major challenge. Using copper(I) catalyzed azide–alkyne cycloaddition (CuAAC), we have developed a general and efficient method for the synthesis of collections of discontinuous epitope mimics. Up to three different cyclic peptides, representing discontinuous epitopes in HIV-gp120, were conjugated to a selection of scaffold molecules. Variation of the scaffold molecule, optimization of the ring size of the cyclic peptides and screening of the resulting libraries for successful protein mimics led to an HIV gp120 mimic with an IC50 value of 1.7 μM. The approach described here provides rapid and highly reproducible access to clean, smart libraries of very complex bio-molecular constructs representing protein mimics for use as synthetic vaccines and beyond.
Co-reporter:Cristina Chamorro, John A. W. Kruijtzer, Marina Farsaraki, Jan Balzarini and Rob M. J. Liskamp
Chemical Communications 2009(Issue 7) pp:NaN823-823
Publication Date(Web):2008/12/17
DOI:10.1039/B817357E
In this Communication, the access to three different peptide loops attached to a small triazacyclophane (TAC) scaffold molecule for the mimicry of discontinuous epitopes present in, for example, antibodies is described for the first time.







![L-Leucine, N-[(2S)-2-azido-1-oxo-3-phenylpropyl]-, methyl ester](http://img.cochemist.com/ccimg/583900/583827-48-9.png)
![L-Leucine, N-[(2S)-2-azido-1-oxo-3-phenylpropyl]-, methyl ester](http://img.cochemist.com/ccimg/583900/583827-48-9_b.png)
![L-ALANINE, N-[(1,1-DIMETHYLETHOXY)CARBONYL]-L-ISOLEUCYL-, METHYL ESTER](http://img.cochemist.com/ccimg/525000/524953-49-9.png)
![L-ALANINE, N-[(1,1-DIMETHYLETHOXY)CARBONYL]-L-ISOLEUCYL-, METHYL ESTER](http://img.cochemist.com/ccimg/525000/524953-49-9_b.png)








![Carbamic acid,N-[(1S)-1-ethynyl-3-methylbutyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/143400/143327-83-7.png)
![Carbamic acid,N-[(1S)-1-ethynyl-3-methylbutyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/143400/143327-83-7_b.png)


![L-Threonine, N-[(1,1-dimethylethoxy)carbonyl]-L-isoleucyl-](http://img.cochemist.com/ccimg/95400/95364-12-8.png)
![L-Threonine, N-[(1,1-dimethylethoxy)carbonyl]-L-isoleucyl-](http://img.cochemist.com/ccimg/95400/95364-12-8_b.png)




![CARBAMIC ACID, [(1S)-1-FORMYL-3-METHYLBUTYL]-, PHENYLMETHYL ESTER](http://img.cochemist.com/ccimg/71500/71413-15-5.png)
![CARBAMIC ACID, [(1S)-1-FORMYL-3-METHYLBUTYL]-, PHENYLMETHYL ESTER](http://img.cochemist.com/ccimg/71500/71413-15-5_b.png)
![L-Alanine, N-[N-[(1,1-dimethylethoxy)carbonyl]-L-isoleucyl]-](http://img.cochemist.com/ccimg/70400/70396-19-9.png)
![L-Alanine, N-[N-[(1,1-dimethylethoxy)carbonyl]-L-isoleucyl]-](http://img.cochemist.com/ccimg/70400/70396-19-9_b.png)


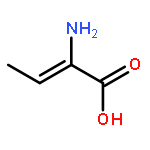
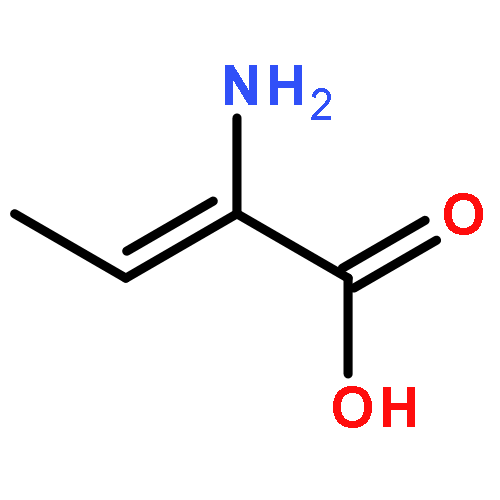
![3,5-bis(acetyloxy)-2-[(acetyloxy)methyl]-6-azidotetrahydro-2H-pyran-4-yl-acetate](http://img.cochemist.com/ccimg/20400/20379-61-7.png)
![3,5-bis(acetyloxy)-2-[(acetyloxy)methyl]-6-azidotetrahydro-2H-pyran-4-yl-acetate](http://img.cochemist.com/ccimg/20400/20379-61-7_b.png)
![L-Leucine, N-[(phenylmethoxy)carbonyl]-L-leucyl-L-leucyl-](http://img.cochemist.com/ccimg/18900/18867-96-4.png)
![L-Leucine, N-[(phenylmethoxy)carbonyl]-L-leucyl-L-leucyl-](http://img.cochemist.com/ccimg/18900/18867-96-4_b.png)










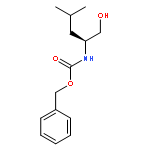
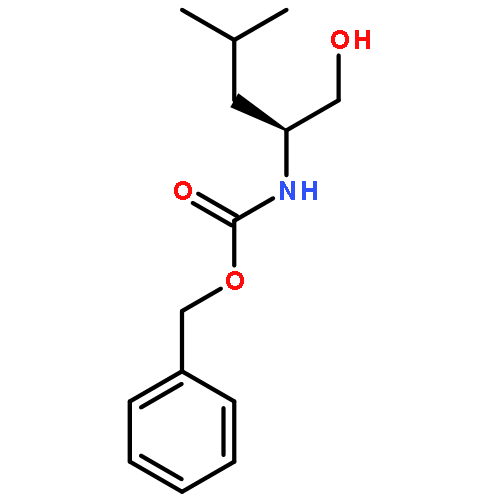







![2-Propyn-1-amine, 3-[tris(1-methylethyl)silyl]-](http://img.cochemist.com/ccimg/918900/918871-51-9.png)
![2-Propyn-1-amine, 3-[tris(1-methylethyl)silyl]-](http://img.cochemist.com/ccimg/918900/918871-51-9_b.png)