Co-reporter:Jerome C. Nwachukwu, Sathish Srinivasan, Nelson E. Bruno, Jason Nowak, ... Kendall W. Nettles
Cell Chemical Biology 2017 Volume 24, Issue 1(Volume 24, Issue 1) pp:
Publication Date(Web):19 January 2017
DOI:10.1016/j.chembiol.2016.11.014
•Environmental estrogens form structurally distinct complexes with ERα•Allosteric signals occur with within the noise of typical ERα structures•Comparison of many structures allows super-resolution analysis of ligand effects•Specific interatomic distances in ERα predict cellular response to diverse ligandsEnvironmental estrogens and anti-hormone therapies for breast cancer have diverse tissue- and signaling-pathway-selective outcomes, but how estrogen receptor alpha (ERα) mediates this phenotypic diversity is poorly understood. We implemented a statistical approach to allow unbiased, parallel analyses of multiple crystal structures, and identified subtle perturbations of ERα structure by different synthetic and environmental estrogens. Many of these perturbations were in the sub-Å range, within the noise of the individual structures, but contributed significantly to the activities of synthetic and environmental estrogens. Combining structural perturbation data from many structures with quantitative cellular activity profiles of the ligands enabled identification of structural rules for ligand-specific allosteric signaling—predicting activity from structure. This approach provides a framework for understanding the diverse effects of environmental estrogens and for guiding iterative medicinal chemistry efforts to generate improved breast cancer therapies, an approach that can be applied to understanding other ligand-regulated allosteric signaling pathways.Download high-res image (195KB)Download full-size image
Co-reporter:Amit Kumar, HaJeung Park, Pengfei Fang, Raman Parkesh, Min Guo, Kendall W. Nettles, and Matthew D. Disney
Biochemistry 2011 Volume 50(Issue 45) pp:
Publication Date(Web):October 11, 2011
DOI:10.1021/bi2013068
RNA internal loops often display a variety of conformations in solution. Herein, we visualize conformational heterogeneity in the context of the 5′CUG/3′GUC repeat motif present in the RNA that causes myotonic dystrophy type 1 (DM1). Specifically, two crystal structures of a model DM1 triplet repeating construct, 5′r[UUGGGC(CUG)3GUCC]2, refined to 2.20 and 1.52 Å resolution are disclosed. Here, differences in the orientation of the 5′ dangling UU end between the two structures induce changes in the backbone groove width, which reveals that noncanonical 1 × 1 nucleotide UU internal loops can display an ensemble of pairing conformations. In the 2.20 Å structure, CUGa, the 5′ UU forms a one hydrogen-bonded pair with a 5′ UU of a neighboring helix in the unit cell to form a pseudoinfinite helix. The central 1 × 1 nucleotide UU internal loop has no hydrogen bonds, while the terminal 1 × 1 nucleotide UU internal loops each form a one-hydrogen bond pair. In the 1.52 Å structure, CUGb, the 5′ UU dangling end is tucked into the major groove of the duplex. While the canonically paired bases show no change in base pairing, in CUGb the terminal 1 × 1 nucleotide UU internal loops now form two hydrogen-bonded pairs. Thus, the shift in the major groove induced by the 5′ UU dangling end alters noncanonical base patterns. Collectively, these structures indicate that 1 × 1 nucleotide UU internal loops in DM1 may sample multiple conformations in vivo. This observation has implications for the recognition of this RNA, and other repeating transcripts, by protein and small molecule ligands.
Co-reporter:Jerome C. Nwachukwu, Mark R. Southern, James R. Kiefer, Pavel V. Afonine, ... Kendall W. Nettles
Structure (5 November 2013) Volume 21(Issue 11) pp:1923-1930
Publication Date(Web):5 November 2013
DOI:10.1016/j.str.2013.07.025
•The ExCoR strategy revealed complex interactions among refinement algorithms•ExCoR can be used to improve both unrefined and refined crystal structures•Structural diversity obtained via ExCoR facilitates automated error correction•ExCoR provides an estimate of uncertainty of refined model parametersIdentifying errors and alternate conformers and modeling multiple main-chain conformers in poorly ordered regions are overarching problems in crystallographic structure determination that have limited automation efforts and structure quality. Here, we show that implementation of a full factorial designed set of standard refinement approaches, termed ExCoR (Extensive Combinatorial Refinement), significantly improves structural models compared to the traditional linear tree approach, in which individual algorithms are tested linearly and are only incorporated if the model improves. ExCoR markedly improved maps and models and reveals building errors and alternate conformations that were masked by traditional refinement approaches. Surprisingly, an individual algorithm that renders a model worse in isolation could still be necessary to produce the best overall model, suggesting that model distortion allows escape from local minima of optimization target function, here shown to be a hallmark limitation of the traditional approach. ExCoR thus provides a simple approach to improving structure determination.Download high-res image (217KB)Download full-size image
Co-reporter:John D. Laughlin, Jerome C. Nwachukwu, Mariana Figuera-Losada, Lisa Cherry, ... Philip V. LoGrasso
Structure (5 December 2012) Volume 20(Issue 12) pp:2174-2184
Publication Date(Web):5 December 2012
DOI:10.1016/j.str.2012.09.021
c-Jun N-terminal (JNK) family kinases have a common peptide-docking site used by upstream activating kinases, substrates, scaffold proteins, and phosphatases, where the ensemble of bound proteins determines signaling output. Although there are many JNK structures, little is known about mechanisms of allosteric regulation between the catalytic and peptide-binding sites, and the activation loop, whose phosphorylation is required for catalytic activity. Here, we compare three structures of unliganded JNK3 bound to different peptides. These were compared as a class to structures that differ in binding of peptide, small molecule ligand, or conformation of the kinase activation loop. Peptide binding induced an inhibitory interlobe conformer that was reversed by alterations in the activation loop. Structure class analysis revealed the subtle structural mechanisms for allosteric signaling between the peptide-binding site and activation loop. Biochemical data from isothermal calorimetry, fluorescence energy transfer, and enzyme inhibition demonstrated affinity differences among the three peptides that were consistent with structural observations.Highlights► Peptide binding induces two distinct autoinhibitory mechanisms in the inactive JNKs ► Activation loop conformer can reverse peptide-induced autoinhibition ► Bidirectional signals are visualized between peptide and activation loop ► Structural class analysis reveals subtle structural features
Co-reporter:John B. Bruning, Michael J. Chalmers, Swati Prasad, Scott A. Busby, ... Patrick R. Griffin
Structure (16 October 2007) Volume 15(Issue 10) pp:1258-1271
Publication Date(Web):16 October 2007
DOI:10.1016/j.str.2007.07.014
Binding to helix 12 of the ligand-binding domain of PPARγ is required for full agonist activity. Previously, the degree of stabilization of the activation function 2 (AF-2) surface was thought to correlate with the degree of agonism and transactivation. To examine this mechanism, we probed structural dynamics of PPARγ with agonists that induced graded transcriptional responses. Here we present crystal structures and amide H/D exchange (HDX) kinetics for six of these complexes. Amide HDX revealed each ligand induced unique changes to the dynamics of the ligand-binding domain (LBD). Full agonists stabilized helix 12, whereas intermediate and partial agonists did not at all, and rather differentially stabilized other regions of the binding pocket. The gradient of PPARγ transactivation cannot be accounted for solely through changes to the dynamics of AF-2. Thus, our understanding of allosteric signaling must be extended beyond the idea of a dynamic helix 12 acting as a molecular switch.
.jpg)
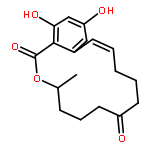
![(7r,11s)-7,15,17-trihydroxy-11-methyl-12-oxabicyclo[12.4.0]octadeca-1(14),15,17-trien-13-one](http://img.cochemist.com/ccimg/26600/26538-44-3.png)
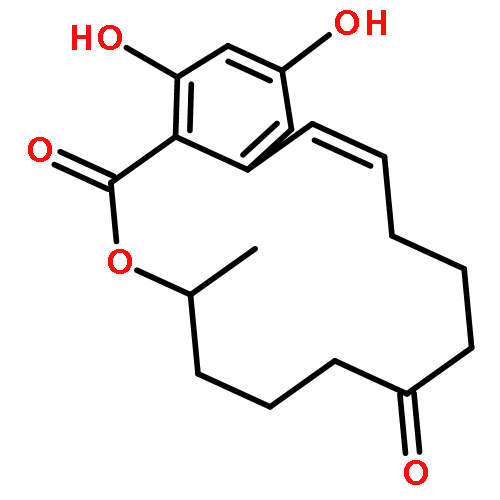
![(7r,11s)-7,15,17-trihydroxy-11-methyl-12-oxabicyclo[12.4.0]octadeca-1(14),15,17-trien-13-one](http://img.cochemist.com/ccimg/26600/26538-44-3_b.png)



![6H-Benzofuro[3,2-c][1]benzopyran-6-one,3,9-dihydroxy-](http://img.cochemist.com/ccimg/500/479-13-0.png)
![6H-Benzofuro[3,2-c][1]benzopyran-6-one,3,9-dihydroxy-](http://img.cochemist.com/ccimg/500/479-13-0_b.png)