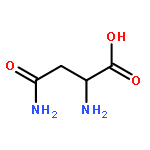
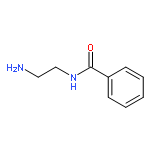
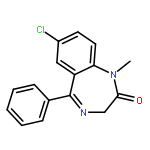
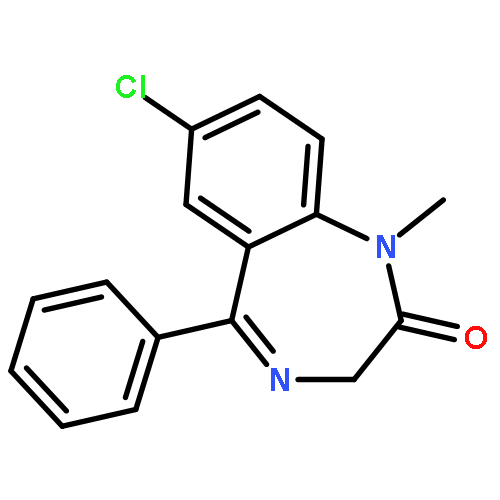
Co-reporter:Bian Li; Jeffrey Mendenhall; Elizabeth Dong Nguyen; Brian E. Weiner; Axel W. Fischer
Journal of Chemical Information and Modeling 2016 Volume 56(Issue 2) pp:423-434
Publication Date(Web):January 24, 2016
DOI:10.1021/acs.jcim.5b00517
Prediction of the three-dimensional (3D) structures of proteins by computational methods is acknowledged as an unsolved problem. Accurate prediction of important structural characteristics such as contact number is expected to accelerate the otherwise slow progress being made in the prediction of 3D structure of proteins. Here, we present a dropout neural network-based method, TMH-Expo, for predicting the contact number of transmembrane helix (TMH) residues from sequence. Neuronal dropout is a strategy where certain neurons of the network are excluded from back-propagation to prevent co-adaptation of hidden-layer neurons. By using neuronal dropout, overfitting was significantly reduced and performance was noticeably improved. For multi-spanning helical membrane proteins, TMH-Expo achieved a remarkable Pearson correlation coefficient of 0.69 between predicted and experimental values and a mean absolute error of only 1.68. In addition, among those membrane protein–membrane protein interface residues, 76.8% were correctly predicted. Mapping of predicted contact numbers onto structures indicates that contact numbers predicted by TMH-Expo reflect the exposure patterns of TMHs and reveal membrane protein–membrane protein interfaces, reinforcing the potential of predicted contact numbers to be used as restraints for 3D structure prediction and protein–protein docking. TMH-Expo can be accessed via a Web server at www.meilerlab.org.
Co-reporter:Brett M. Kroncke, Amanda M. Duran, Jeffrey L. Mendenhall, Jens Meiler, Jeffrey D. Blume, and Charles R. Sanders
Biochemistry 2016 Volume 55(Issue 36) pp:5002
Publication Date(Web):August 26, 2016
DOI:10.1021/acs.biochem.6b00537
There is a compelling and growing need to accurately predict the impact of amino acid mutations on protein stability for problems in personalized medicine and other applications. Here the ability of 10 computational tools to accurately predict mutation-induced perturbation of folding stability (ΔΔG) for membrane proteins of known structure was assessed. All methods for predicting ΔΔG values performed significantly worse when applied to membrane proteins than when applied to soluble proteins, yielding estimated concordance, Pearson, and Spearman correlation coefficients of <0.4 for membrane proteins. Rosetta and PROVEAN showed a modest ability to classify mutations as destabilizing (ΔΔG < −0.5 kcal/mol), with a 7 in 10 chance of correctly discriminating a randomly chosen destabilizing variant from a randomly chosen stabilizing variant. However, even this performance is significantly worse than for soluble proteins. This study highlights the need for further development of reliable and reproducible methods for predicting thermodynamic folding stability in membrane proteins.
Co-reporter:Alexander R. Geanes, Hykeyung P. Cho, Kellie D. Nance, Kevin M. McGowan, P. Jeffrey Conn, Carrie K. Jones, Jens Meiler, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 18) pp:4487-4491
Publication Date(Web):15 September 2016
DOI:10.1016/j.bmcl.2016.07.071
This Letter describes a ligand-based virtual screening campaign utilizing SAR data around the M5 NAMs, ML375 and VU6000181. Both QSAR and shape scores were employed to virtually screen a 98,000-member compound library. Neither approach alone proved productive, but a consensus score of the two models identified a novel scaffold which proved to be a modestly selective, but weak inhibitor (VU0549108) of the M5 mAChR (M5 IC50 = 6.2 μM, M1–4 IC50s >10 μM) based on an unusual 8-((1,3,5-trimethyl-1H-pyrazol-4-yl)sulfonyl)-1-oxa-4-thia-8-azaspiro[4,5]decane scaffold. [3H]-NMS binding studies showed that VU0549108 interacts with the orthosteric site (Ki of 2.7 μM), but it is not clear if this is negative cooperativity or orthosteric binding. Interestingly, analogs synthesized around VU0549108 proved weak, and SAR was very steep. However, this campaign validated the approach and warranted further expansion to identify additional novel chemotypes.
Co-reporter:Jessica A. Finn;Jordan R. Willis;Bryan Briney;Gopal Sapparapu;Vidisha Singh;Hannah King;Celia C. LaBranche;David C. Montefiori;James E. Crowe, Jr.
PNAS 2016 Volume 113 (Issue 16 ) pp:4446-4451
Publication Date(Web):2016-04-19
DOI:10.1073/pnas.1518405113
Development of broadly neutralizing antibodies (bnAbs) against HIV-1 usually requires prolonged infection and induction of
Abs with unusual features, such as long heavy-chain complementarity-determining region 3 (HCDR3) loops. Here we sought to
determine whether the repertoires of HIV-1–naïve individuals contain Abs with long HCDR3 loops that could mediate HIV-1 neutralization.
We interrogated at massive scale the structural properties of long Ab HCDR3 loops in HIV-1–naïve donors, searching for structured
HCDR3s similar to those of the HIV-1 bnAb PG9. We determined the nucleotide sequences encoding 2.3 × 107 unique HCDR3 amino acid regions from 70 different HIV-1–naïve donors. Of the 26,917 HCDR3 loops with 30-amino acid length
identified, we tested 30 for further study that were predicted to have PG9-like structure when chimerized onto PG9. Three
of these 30 PG9 chimeras bound to the HIV-1 gp120 monomer, and two were neutralizing. In addition, we found 14 naturally occurring
HCDR3 sequences that acquired the ability to bind to the HIV-1 gp120 monomer when adding 2- to 7-amino acid mutations via
computational design. Of those 14 designed Abs, 8 neutralized HIV-1, with IC50 values ranging from 0.7 to 98 µg/mL. These data suggest that the repertoire of HIV-1–naïve individuals contains rare B cells
that encode HCDR3 loops that bind or neutralize HIV-1 when presented on a PG9 background with relatively few or no additional
mutations. Long HCDR3 sequences are present in the HIV-naïve B-cell repertoire, suggesting that this class of bnAbs is a favorable
target for rationally designed preventative vaccine efforts.
Co-reporter:Karen J. Gregory, Elizabeth D. Nguyen, Chrysa Malosh, Jeffrey L. Mendenhall, Jessica Z. Zic, Brittney S. Bates, Meredith J. Noetzel, Emma F. Squire, Eric M. Turner, Jerri M. Rook, Kyle A. Emmitte, Shaun R. Stauffer, Craig W. Lindsley, Jens Meiler, and P. Jeffrey Conn
ACS Chemical Neuroscience 2014 Volume 5(Issue 4) pp:282
Publication Date(Web):February 16, 2014
DOI:10.1021/cn400225x
A common metabotropic glutamate receptor 5 (mGlu5) allosteric site is known to accommodate diverse chemotypes. However, the structural relationship between compounds from different scaffolds and mGlu5 is not well understood. In an effort to better understand the molecular determinants that govern allosteric modulator interactions with mGlu5, we employed a combination of site-directed mutagenesis and computational modeling. With few exceptions, six residues (P654, Y658, T780, W784, S808, and A809) were identified as key affinity determinants across all seven allosteric modulator scaffolds. To improve our interpretation of how diverse allosteric modulators occupy the common allosteric site, we sampled the wealth of mGlu5 structure–activity relationship (SAR) data available by docking 60 ligands (actives and inactives) representing seven chemical scaffolds into our mGlu5 comparative model. To spatially and chemically compare binding modes of ligands from diverse scaffolds, the ChargeRMSD measure was developed. We found a common binding mode for the modulators that placed the long axes of the ligands parallel to the transmembrane helices 3 and 7. W784 in TM6 not only was identified as a key NAM cooperativity determinant across multiple scaffolds, but also caused a NAM to PAM switch for two different scaffolds. Moreover, a single point mutation in TM5, G747V, altered the architecture of the common allosteric site such that 4-nitro-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide (VU29) was noncompetitive with the common allosteric site. Our findings highlight the subtleties of allosteric modulator binding to mGlu5 and demonstrate the utility in incorporating SAR information to strengthen the interpretation and analyses of docking and mutational data.Keywords: affinity; cooperativity; metabotropic glutamate receptor 5; Mutagenesis; operational model; structure−activity relationships
Co-reporter:Steffen Lindert, Phoebe L. Stewart, Jens Meiler
Computational Biology and Chemistry 2013 Volume 42() pp:1-4
Publication Date(Web):February 2013
DOI:10.1016/j.compbiolchem.2012.11.001
DNA dependent protein kinase catalytic subunit (DNA-PKcs) is an important regulatory protein in non-homologous end joining a process used to repair DNA double strand breaks. Medium resolution structures both from cryoEM and X-ray crystallography show the general topology of the protein and positions of helices in parts of DNA-PKcs. EM-Fold, an algorithm developed for building protein models into medium resolution density maps has been used to generate models for the heat repeat-like “Ring structure” of the molecule. We were able to computationally corroborate placement of the N-terminus of the domain that supports a previously published hypothesis. Targeted experiments are suggested to test the model.Graphical abstractHighlights► Computational determination of orientation of heat repeat domain of DNA-PKcs. ► Presentation of structural model for heat repeat domain to enable design of experiments. ► Adaption of EM-Fold to work with X-ray density maps.
Co-reporter:Stephanie H. DeLuca;Daniel Rathmann;Annette G. Beck-Sickinger
Biopolymers 2013 Volume 99( Issue 5) pp:314-325
Publication Date(Web):
DOI:10.1002/bip.22162
Abstract
The prolactin releasing peptide (PrRP) is involved in regulating food intake and body weight homeostasis, but molecular details on the activation of the PrRP receptor remain unclear. C-terminal segments of PrRP with 20 (PrRP20) and 13 (PrRP8-20) amino acids, respectively, have been suggested to be fully active. The data presented herein indicate this is true for the wildtype receptor only; a 5-10-fold loss of activity was found for PrRP8-20 compared to PrRP20 at two extracellular loop mutants of the receptor. To gain insight into the secondary structure of PrRP, we used CD spectroscopy performed in TFE and SDS. Additionally, previously reported NMR data, combined with ROSETTANMR, were employed to determine the structure of amidated PrRP20. The structural ensemble agrees with the spectroscopic data for the full-length peptide, which exists in an equilibrium between α- and 310-helix. We demonstrate that PrRP8-20's reduced propensity to form an α-helix correlates with its reduced biological activity on mutant receptors. Further, distinct amino acid replacements in PrRP significantly decrease affinity and activity but have no influence on the secondary structure of the peptide. We conclude that formation of a primarily α-helical C-terminal region of PrRP is critical for receptor activation. © 2012 Wiley Periodicals, Inc. Biopolymers 99: 273–281, 2013.
Co-reporter:Simone Eisenbeis ; William Proffitt ; Murray Coles ; Vincent Truffault ; Sooruban Shanmugaratnam ; Jens Meiler ;Birte Höcker
Journal of the American Chemical Society 2012 Volume 134(Issue 9) pp:4019-4022
Publication Date(Web):February 13, 2012
DOI:10.1021/ja211657k
It is hypothesized that protein domains evolved from smaller intrinsically stable subunits via combinatorial assembly. Illegitimate recombination of fragments that encode protein subunits could have quickly led to diversification of protein folds and their functionality. This evolutionary concept presents an attractive strategy to protein engineering, e.g., to create new scaffolds for enzyme design. We previously combined structurally similar parts from two ancient protein folds, the (βα)8-barrel and the flavodoxin-like fold. The resulting “hopeful monster” differed significantly from the intended (βα)8-barrel fold by an extra β-strand in the core. In this study, we ask what modifications are necessary to form the intended structure and what potential this approach has for the rational design of functional proteins. Guided by computational design, we optimized the interface between the fragments with five targeted mutations yielding a stable, monomeric protein whose predicted structure was verified experimentally. We further tested binding of a phosphorylated compound and detected that some affinity was already present due to an intact phosphate-binding site provided by one fragment. The affinity could be improved quickly to the level of natural proteins by introducing two additional mutations. The study illustrates the potential of recombining protein fragments with unique properties to design new and functional proteins, offering both a possible pathway of protein evolution and a protocol to rapidly engineer proteins for new applications.
Co-reporter:Gordon Lemmon;Kristian Kaufmann
Chemical Biology & Drug Design 2012 Volume 79( Issue 6) pp:888-896
Publication Date(Web):
DOI:10.1111/j.1747-0285.2012.01356.x
Predicting HIV-1 protease/inhibitor binding affinity as the difference between the free energy of the inhibitor bound and unbound state remains difficult as the unbound state exists as an ensemble of conformations with various degrees of flap opening. We improve computational prediction of protease/inhibitor affinity by invoking the hypothesis that the free energy of the unbound state while difficult to predict is less sensitive to mutation. Thereby the HIV-1 protease/inhibitor binding affinity can be approximated with the free energy of the bound state alone. Bound state free energy can be predicted from comparative models of HIV-1 protease mutant/inhibitor complexes. Absolute binding energies are predicted with R = 0.71 and SE = 5.91 kJ/mol. Changes in binding free energy upon mutation can be predicted with R = 0.85 and SE = 4.49 kJ/mol. Resistance mutations that lower inhibitor binding affinity can thereby be recognized early in HIV-1 protease inhibitor development.
Co-reporter:Dr. Ralf Mueller;Dr. Eric S. Dawson; Jens Meiler;Dr. Alice L. Rodriguez;Dr. Brian A. Chauder;Brittney S. Bates;Dr. Andrew S. Felts;Jeffrey P. Lamb;Usha N. Menon;Dr. Sataywan B. Jadhav;Dr. Alexer S. Kane;Dr. Carrie K. Jones;Dr. Karen J. Gregory; Colleen M. Niswender; P. Jeffrey Conn;Dr. Christopher M. Olsen; Danny G. Winder; Kyle A. Emmitte; Craig W. Lindsley
ChemMedChem 2012 Volume 7( Issue 3) pp:406-414
Publication Date(Web):
DOI:10.1002/cmdc.201100510
Co-reporter:Steffen Lindert;Tommy Hofmann;Nils Wötzel;Mert Karaka&x15f;;Phoebe L. Stewart
Biopolymers 2012 Volume 97( Issue 9) pp:669-677
Publication Date(Web):
DOI:10.1002/bip.22027
Abstract
EM-Fold was used to build models for nine proteins in the maps of GroEL (7.7 Å resolution) and ribosome (6.4 Å resolution) in the ab initio modeling category of the 2010 cryo-electron microscopy modeling challenge. EM-Fold assembles predicted secondary structure elements (SSEs) into regions of the density map that were identified to correspond to either α-helices or β-strands. The assembly uses a Monte Carlo algorithm where loop closure, density-SSE length agreement, and strength of connecting density between SSEs are evaluated. Top-scoring models are refined by translating, rotating, and bending SSEs to yield better agreement with the density map. EM-Fold produces models that contain backbone atoms within SSEs only. The RMSD values of the models with respect to native range from 2.4 to 3.5 Å for six of the nine proteins. EM-Fold failed to predict the correct topology in three cases. Subsequently, Rosetta was used to build loops and side chains for the very best scoring models after EM-Fold refinement. The refinement within Rosetta's force field is driven by a density agreement score that calculates a cross-correlation between a density map simulated from the model and the experimental density map. All-atom RMSDs as low as 3.4 Å are achieved in favorable cases. Values above 10.0 Å are observed for two proteins with low overall content of secondary structure and hence particularly complex loop modeling problems. RMSDs over residues in secondary structure elements range from 2.5 to 4.8 Å. © 2012 Wiley Periodicals, Inc. Biopolymers 97: 669–677, 2012.
Co-reporter:Miyeon Kim;Sergey A. Vishnivetskiy;Susan M. Hanson;Oliver P. Ernst;Vsevolod V. Gurevich;Whitney M. Cleghorn;Ned Van Eps;Wayne L. Hubbell;Nathan S. Alexander;Xuanzhi Zhan;Takefumi Morizumi
PNAS 2012 Volume 109 (Issue 45 ) pp:18407-18412
Publication Date(Web):2012-11-06
DOI:10.1073/pnas.1216304109
Arrestin-1 (visual arrestin) binds to light-activated phosphorylated rhodopsin (P-Rh*) to terminate G-protein signaling. To
map conformational changes upon binding to the receptor, pairs of spin labels were introduced in arrestin-1 and double electron–electron
resonance was used to monitor interspin distance changes upon P-Rh* binding. The results indicate that the relative position
of the N and C domains remains largely unchanged, contrary to expectations of a “clam-shell” model. A loop implicated in P-Rh*
binding that connects β-strands V and VI (the “finger loop,” residues 67–79) moves toward the expected location of P-Rh* in
the complex, but does not assume a fully extended conformation. A striking and unexpected movement of a loop containing residue
139 away from the adjacent finger loop is observed, which appears to facilitate P-Rh* binding. This change is accompanied
by smaller movements of distal loops containing residues 157 and 344 at the tips of the N and C domains, which correspond
to “plastic” regions of arrestin-1 that have distinct conformations in monomers of the crystal tetramer. Remarkably, the loops
containing residues 139, 157, and 344 appear to have high flexibility in both free arrestin-1 and the P-Rh*complex.
Co-reporter:Carie Fortenberry ; Elizabeth Anne Bowman ; Will Proffitt ; Brent Dorr ; Steven Combs ; Joel Harp ; Laura Mizoue
Journal of the American Chemical Society 2011 Volume 133(Issue 45) pp:18026-18029
Publication Date(Web):October 6, 2011
DOI:10.1021/ja2051217
It has been demonstrated previously that symmetric, homodimeric proteins are energetically favored, which explains their abundance in nature. It has been proposed that such symmetric homodimers underwent gene duplication and fusion to evolve into protein topologies that have a symmetric arrangement of secondary structure elements—“symmetric superfolds”. Here, the ROSETTA protein design software was used to computationally engineer a perfectly symmetric variant of imidazole glycerol phosphate synthase and its corresponding symmetric homodimer. The new protein, termed FLR, adopts the symmetric (βα)8 TIM-barrel superfold. The protein is soluble and monomeric and exhibits two-fold symmetry not only in the arrangement of secondary structure elements but also in sequence and at atomic detail, as verified by crystallography. When cut in half, FLR dimerizes readily to form the symmetric homodimer. The successful computational design of FLR demonstrates progress in our understanding of the underlying principles of protein stability and presents an attractive strategy for the in silico construction of larger protein domains from smaller pieces.
Co-reporter:Julia Koehler, Jens Meiler
Progress in Nuclear Magnetic Resonance Spectroscopy 2011 Volume 59(Issue 4) pp:360-389
Publication Date(Web):November 2011
DOI:10.1016/j.pnmrs.2011.05.001
Graphical abstractIntroducing a paramagnetic metal ion into a protein allow the measurement of Residual Dipolar Couplings, Paramagnetic Relaxation Enhancements, and Pseudo-Contact Shifts.Highlights► Introduction of a lanthanide ion into a protein leads to paramagnetic effects and partial alignment. ► Paramagnetic Relaxation Enhancements (PREs), Residual Dipolar Couplings (RDCs), and Pseudo-Contact Shifts (PCSs), among others, can be measured. ► Amplitude of paramagnetic effects depends on metal ions, protein size, and magnetic field strength. ► All paramagnetic effects and effects resulting from partial alignment are reviewed. ► Practical aspects include overview of tags used and characteristics of metal ions.
Co-reporter:Steven Combs, Kristian Kaufmann, Julie R. Field, Randy D. Blakely, and Jens Meiler
ACS Chemical Neuroscience 2011 Volume 2(Issue 2) pp:75
Publication Date(Web):October 27, 2010
DOI:10.1021/cn100066p
The human serotonin (5-hydroxytryptamine, 5-HT) transporter (hSERT) is responsible for the reuptake of 5-HT following synaptic release, as well as for import of the biogenic amine into several non-5-HT synthesizing cells including platelets. The antidepressant citalopram blocks SERT and thereby inhibits the transport of 5-HT. To identify key residues establishing high-affinity citalopram binding, we have built a comparative model of hSERT and Drosophila melanogaster SERT (dSERT) based on the Aquifex aeolicus leucine transporter (LeuTAa) crystal structure. In this study, citalopram has been docked into the homology model of hSERT and dSERT using RosettaLigand. Our models reproduce the differential binding affinities for the R- and S-isomers of citalopram in hSERT and the impact of several hSERT mutants. Species-selective binding affinities for hSERT and dSERT also can be reproduced. Interestingly, the model predicts a hydrogen bond between E444 in transmembrane domain 8 (TM8) and Y95 in TM1 that places Y95 in a downward position, thereby removing Y95 from a direct interaction with S-citalopram. Mutation of E444D results in a 10-fold reduced binding affinity for S-citalopram, supporting the hypothesis that Y95 and E444 form a stabilizing interaction in the S-citalopram/hSERT complex.Keywords (keywords): computational docking; homology model; hSERT; ligand; S-citalopram
Co-reporter:Samuel DeLuca, Brent Dorr, and Jens Meiler
Biochemistry 2011 Volume 50(Issue 40) pp:
Publication Date(Web):September 9, 2011
DOI:10.1021/bi200664b
We hypothesize that the degree of surface exposure of amino acid side chains within a globular, soluble protein has been optimized in evolution, not only to minimize the solvation free energy of the monomeric protein but also to prevent protein aggregation. This effect needs to be taken into account when engineering proteins de novo. We test this hypothesis through addition of a knowledge-based, exposure-dependent energy term to the RosettaDesign solvation potential [Lazaridis, T., and Karplus, M. (1999) Proteins 35, 133–152]. Correlation between amino acid type and surface exposure is determined from a representative set of experimental protein structures. The amino acid solvent accessible surface area (SASA) is estimated with a neighbor vector measure that increases in accuracy compared to the neighbor count measure while remaining pairwise decomposable [Durham, E., et al. (2009) J. Mol. Model. 15, 1093–1108]. Benchmarking of this potential in protein design displays a 3.2% improvement in the overall sequence recovery and an 8.5% improvement in recovery of amino acid types tolerated in evolution.
Co-reporter:Kristian Kaufmann;Nicole Shen;Laura Mizoue
Journal of Molecular Modeling 2011 Volume 17( Issue 2) pp:315-324
Publication Date(Web):2011 February
DOI:10.1007/s00894-010-0725-5
The PDZ domain is an interaction motif that recognizes and binds the C-terminal peptides of target proteins. PDZ domains are ubiquitous in nature and help assemble multiprotein complexes that control cellular organization and signaling cascades. We present an optimized energy function to predict the binding free energy (ΔΔG) of PDZ domain/peptide interactions computationally. Geometry-optimized models of PDZ domain/peptide interfaces were built using Rosetta, and protein and peptide side chain and backbone degrees of freedom are minimized simultaneously. Using leave-one-out cross-validation, Rosetta’s energy function is adjusted to reproduce experimentally determined ΔΔG values with a correlation coefficient of 0.66 and a standard deviation of 0.79 kcal mol−1. The energy function places an increased weight on hydrogen bonding interactions when compared to a previously developed method to analyze protein/protein interactions. Binding free enthalpies (ΔΔH) and entropies (ΔS) are predicted with reduced accuracies of R = 0.60 and R = 0.17, respectively. The computational method improves prediction of PDZ domain specificity from sequence and allows design of novel PDZ domain/peptide interactions.
Co-reporter:Anette Schreiber
BIOspektrum 2011 Volume 17( Issue 2) pp:158-161
Publication Date(Web):2011 March
DOI:10.1007/s12268-011-0021-7
Die Verknüpfung funktioneller und struktureller Informationen über G-Protein-gekoppelte Rezeptoren (GPCRs) ist für das Verständnis dieser komplexen Systeme von Vorteil. Wir beschreiben hier die Modellierung von GPCRs mit der Software ROSETTA, wobei auch flexible Regionen abgebildet werden.Combination of functional and structural information of G-protein coupled receptors (GPCRs) aids the understanding of these complex systems. Here we describe modeling of GPCRs using the ROSETTA software suite, which also covers variable regions like loops or termini.
Co-reporter:Ned Van Eps;Anita M. Preininger;Nathan Alexander;Ali I. Kaya;Scott Meier;Wayne L. Hubbell;Heidi E. Hamm
PNAS 2011 Volume 108 (Issue 23 ) pp:9420-9424
Publication Date(Web):2011-06-07
DOI:10.1073/pnas.1105810108
In G-protein signaling, an activated receptor catalyzes GDP/GTP exchange on the Gα subunit of a heterotrimeric G protein. In an initial step, receptor interaction with Gα acts to allosterically trigger GDP release from a binding site located between the nucleotide binding domain and a helical
domain, but the molecular mechanism is unknown. In this study, site-directed spin labeling and double electron–electron resonance
spectroscopy are employed to reveal a large-scale separation of the domains that provides a direct pathway for nucleotide
escape. Cross-linking studies show that the domain separation is required for receptor enhancement of nucleotide exchange
rates. The interdomain opening is coupled to receptor binding via the C-terminal helix of Gα, the extension of which is a high-affinity receptor binding element.
Co-reporter:Ralf Mueller, Alice L. Rodriguez, Eric S. Dawson, Mariusz Butkiewicz, Thuy T. Nguyen, Stephen Oleszkiewicz, Annalen Bleckmann, C. David Weaver, Craig W. Lindsley, P. Jeffrey Conn and Jens Meiler
ACS Chemical Neuroscience 2010 Volume 1(Issue 4) pp:288
Publication Date(Web):January 28, 2010
DOI:10.1021/cn9000389
Selective potentiators of glutamate response at metabotropic glutamate receptor subtype 5 (mGluR5) have exciting potential for the development of novel treatment strategies for schizophrenia. A total of 1,382 compounds with positive allosteric modulation (PAM) of the mGluR5 glutamate response were identified through high-throughput screening (HTS) of a diverse library of 144,475 substances utilizing a functional assay measuring receptor-induced intracellular release of calcium. Primary hits were tested for concentration-dependent activity, and potency data (EC50 values) were used for training artificial neural network (ANN) quantitative structure−activity relationship (QSAR) models that predict biological potency from the chemical structure. While all models were trained to predict EC50, the quality of the models was assessed by using both continuous measures and binary classification. Numerical descriptors of chemical structure were used as input for the machine learning procedure and optimized in an iterative protocol. The ANN models achieved theoretical enrichment ratios of up to 38 for an independent data set not used in training the model. A database of ∼450,000 commercially available drug-like compounds was targeted in a virtual screen. A set of 824 compounds was obtained for testing based on the highest predicted potency values. Biological testing found 28.2% (232/824) of these compounds with various activities at mGluR5 including 177 pure potentiators and 55 partial agonists. These results represent an enrichment factor of 23 for pure potentiation of the mGluR5 glutamate response and 30 for overall mGluR5 modulation activity when compared with those of the original mGluR5 experimental screening data (0.94% hit rate). The active compounds identified contained 72% close derivatives of previously identified PAMs as well as 28% nontrivial derivatives of known active compounds.Keywords: allosteric modulator; artificial neural network (ANN); cheminformatics, virtual screen; high-throughput screen (HTS); Metabotropic glutamate receptor (mGluR); quantitative structure−activity relationship (QSAR)
Co-reporter:Kristian W. Kaufmann, Gordon H. Lemmon, Samuel L. DeLuca, Jonathan H. Sheehan and Jens Meiler
Biochemistry 2010 Volume 49(Issue 14) pp:
Publication Date(Web):March 17, 2010
DOI:10.1021/bi902153g
The objective of this review is to enable researchers to use the software package Rosetta for biochemical and biomedicinal studies. We provide a brief review of the six most frequent research problems tackled with Rosetta. For each of these six tasks, we provide a tutorial that illustrates a basic Rosetta protocol. The Rosetta method was originally developed for de novo protein structure prediction and is regularly one of the best performers in the community-wide biennial Critical Assessment of Structure Prediction. Predictions for protein domains with fewer than 125 amino acids regularly have a backbone root-mean-square deviation of better than 5.0 Å. More impressively, there are several cases in which Rosetta has been used to predict structures with atomic level accuracy better than 2.5 Å. In addition to de novo structure prediction, Rosetta also has methods for molecular docking, homology modeling, determining protein structures from sparse experimental NMR or EPR data, and protein design. Rosetta has been used to accurately design a novel protein structure, predict the structure of protein−protein complexes, design altered specificity protein−protein and protein−DNA interactions, and stabilize proteins and protein complexes. Most recently, Rosetta has been used to solve the X-ray crystallographic phase problem.
Co-reporter:Edward W. Lowe Jr., Alysia Ferrebee, Alice L. Rodriguez, P. Jeffrey Conn, Jens Meiler
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 19) pp:5922-5924
Publication Date(Web):1 October 2010
DOI:10.1016/j.bmcl.2010.07.061
Positive allosteric modulation of the metabotropic glutamate receptor subtype 5 was studied by conducting a comparative molecular field analysis on 118 benzoxazepine derivatives. The model with the best predictive ability retained significant cross-validated correlation coefficients of q2 = 0.58 (r2 = 0.81) yielding a standard error of 0.20 in pEC50 for this class of compounds. The subsequent contour maps highlight the structural features pertinent to the bioactivity values of benzoxazepines.Positive allosteric modulation of the metabotropic glutamate receptor subtype 5 was studied by conducting a comparative molecular field analysis on 118 benzoxazepine derivatives. The model with the best predictive ability retained significant cross-validated correlation coefficients of q2 = 0.58 (r2 = 0.81) yielding a standard error of 0.20 in pEC50 for this class of compounds. The subsequent contour maps highlight the structural features pertinent to the bioactivity values of benzoxazepines.
Co-reporter:Elizabeth Durham;Brent Dorr;Nils Woetzel
Journal of Molecular Modeling 2009 Volume 15( Issue 9) pp:1093-1108
Publication Date(Web):2009 September
DOI:10.1007/s00894-009-0454-9
The burial of hydrophobic amino acids in the protein core is a driving force in protein folding. The extent to which an amino acid interacts with the solvent and the protein core is naturally proportional to the surface area exposed to these environments. However, an accurate calculation of the solvent-accessible surface area (SASA), a geometric measure of this exposure, is numerically demanding as it is not pair-wise decomposable. Furthermore, it depends on a full-atom representation of the molecule. This manuscript introduces a series of four SASA approximations of increasing computational complexity and accuracy as well as knowledge-based environment free energy potentials based on these SASA approximations. Their ability to distinguish correctly from incorrectly folded protein models is assessed to balance speed and accuracy for protein structure prediction. We find the newly developed “Neighbor Vector” algorithm provides the most optimal balance of accurate yet rapid exposure measures.
Co-reporter:Kelli Kazmier, Nathan S. Alexander, Jens Meiler, Hassane S. Mchaourab
Journal of Structural Biology (March 2011) Volume 173(Issue 3) pp:549-557
Publication Date(Web):1 March 2011
DOI:10.1016/j.jsb.2010.11.003
A hybrid protein structure determination approach combining sparse Electron Paramagnetic Resonance (EPR) distance restraints and Rosetta de novo protein folding has been previously demonstrated to yield high quality models (Alexander et al. (2008)). However, widespread application of this methodology to proteins of unknown structures is hindered by the lack of a general strategy to place spin label pairs in the primary sequence. In this work, we report the development of an algorithm that optimally selects spin labeling positions for the purpose of distance measurements by EPR. For the α-helical subdomain of T4 lysozyme (T4L), simulated restraints that maximize sequence separation between the two spin labels while simultaneously ensuring pairwise connectivity of secondary structure elements yielded vastly improved models by Rosetta folding. 54% of all these models have the correct fold compared to only 21% and 8% correctly folded models when randomly placed restraints or no restraints are used, respectively. Moreover, the improvements in model quality require a limited number of optimized restraints, which is determined by the pairwise connectivities of T4L α-helices. The predicted improvement in Rosetta model quality was verified by experimental determination of distances between spin labels pairs selected by the algorithm. Overall, our results reinforce the rationale for the combined use of sparse EPR distance restraints and de novo folding. By alleviating the experimental bottleneck associated with restraint selection, this algorithm sets the stage for extending computational structure determination to larger, traditionally elusive protein topologies of critical structural and biochemical importance.
Co-reporter:Stephanie J. Hirst, Nathan Alexander, Hassane S. Mchaourab, Jens Meiler
Journal of Structural Biology (March 2011) Volume 173(Issue 3) pp:506-514
Publication Date(Web):1 March 2011
DOI:10.1016/j.jsb.2010.10.013
Site-directed spin labeling electron paramagnetic resonance (SDSL-EPR) is often used for the structural characterization of proteins that elude other techniques, such as X-ray crystallography and nuclear magnetic resonance (NMR). However, high-resolution structures are difficult to obtain due to uncertainty in the spin label location and sparseness of experimental data. Here, we introduce RosettaEPR, which has been designed to improve de novo high-resolution protein structure prediction using sparse SDSL-EPR distance data. The “motion-on-a-cone” spin label model is converted into a knowledge-based potential, which was implemented as a scoring term in Rosetta. RosettaEPR increased the fractions of correctly folded models (RMSDCα < 7.5 Å) and models accurate at medium resolution (RMSDCα < 3.5 Å) by 25%. The correlation of score and model quality increased from 0.42 when using no restraints to 0.51 when using bounded restraints and again to 0.62 when using RosettaEPR. This allowed for the selection of accurate models by score. After full-atom refinement, RosettaEPR yielded a 1.7 Å model of T4-lysozyme, thus indicating that atomic detail models can be achieved by combining sparse EPR data with Rosetta. While these results indicate RosettaEPR’s potential utility in high-resolution protein structure prediction, they are based on a single example. In order to affirm the method’s general performance, it must be tested on a larger and more versatile dataset of proteins.
Co-reporter:Sandeepkumar Kothiwale, Corina M. Borza, Edward W. Lowe, Ambra Pozzi, Jens Meiler
Drug Discovery Today (February 2015) Volume 20(Issue 2) pp:255-261
Publication Date(Web):1 February 2015
DOI:10.1016/j.drudis.2014.09.025
•DDR1 is a potential therapeutic target as it is implicated in organ fibrosis and several cancers.•DDR1 has a characteristic kinase domain containing several functionally conserved residues.•Homology modeling and docking studies of DDR1 demonstrates possibilities for computer aided drug discovery.Discoidin domain receptor (DDR) 1 and 2 are transmembrane receptors that belong to the family of receptor tyrosine kinases (RTK). Upon collagen binding, DDRs transduce cellular signaling involved in various cell functions, including cell adhesion, proliferation, differentiation, migration, and matrix homeostasis. Altered DDR function resulting from either mutations or overexpression has been implicated in several types of disease, including atherosclerosis, inflammation, cancer, and tissue fibrosis. Several established inhibitors, such as imatinib, dasatinib, and nilotinib, originally developed as Abelson murine leukemia (Abl) kinase inhibitors, have been found to inhibit DDR kinase activity. As we review here, recent discoveries of novel inhibitors and their co-crystal structure with the DDR1 kinase domain have made structure-based drug discovery for DDR1 amenable.Download high-res image (287KB)Download full-size image
Co-reporter:Steffen Lindert, Nathan Alexander, Nils Wötzel, Mert Karakaş, ... Jens Meiler
Structure (7 March 2012) Volume 20(Issue 3) pp:464-478
Publication Date(Web):7 March 2012
DOI:10.1016/j.str.2012.01.023
Electron density maps of membrane proteins or large macromolecular complexes are frequently only determined at medium resolution between 4 Å and 10 Å, either by cryo-electron microscopy or X-ray crystallography. In these density maps, the general arrangement of secondary structure elements (SSEs) is revealed, whereas their directionality and connectivity remain elusive. We demonstrate that the topology of proteins with up to 250 amino acids can be determined from such density maps when combined with a computational protein folding protocol. Furthermore, we accurately reconstruct atomic detail in loop regions and amino acid side chains not visible in the experimental data. The EM-Fold algorithm assembles the SSEs de novo before atomic detail is added using Rosetta. In a benchmark of 27 proteins, the protocol consistently and reproducibly achieves models with root mean square deviation values <3 Å.Highlights► EM-Fold yields atomic detail not present in the medium resolution density map ► EM-Fold consistently finds the correct topology from medium-resolution density maps
Co-reporter:Ulrike Krug, Nathan S. Alexander, Richard A. Stein, Antje Keim, ... Jens Meiler
Structure (5 January 2016) Volume 24(Issue 1) pp:43-56
Publication Date(Web):5 January 2016
DOI:10.1016/j.str.2015.11.007
•Almost isoenergetic states of a domain motion can be characterized by EPR•X-Ray structures are combined with spectroscopic data by ensemble fitting•A quantitative characterization of the equilibrium can be obtained•Substrate binding changes the equilibrium from open to open and closed statesEscherichia coli 5′-nucleotidase is a two-domain enzyme exhibiting a unique 96° domain motion that is required for catalysis. Here we present an integrated structural biology study that combines DEER distance distributions with structural information from X-ray crystallography and computational biology to describe the population of presumably almost isoenergetic open and closed states in solution. Ensembles of models that best represent the experimental distance distributions are determined by a Monte Carlo search algorithm. As a result, predominantly open conformations are observed in the unliganded state indicating that the majority of enzyme molecules await substrate binding for the catalytic cycle. The addition of a substrate analog yields ensembles with an almost equal mixture of open and closed states. Thus, in the presence of substrate, efficient catalysis is provided by the simultaneous appearance of open conformers (binding substrate or releasing product) and closed conformers (enabling the turnover of the substrate).Download high-res image (169KB)Download full-size image
Co-reporter:Brian E. Weiner, Nils Woetzel, Mert Karakaş, Nathan Alexander, Jens Meiler
Structure (2 July 2013) Volume 21(Issue 7) pp:1107-1117
Publication Date(Web):2 July 2013
DOI:10.1016/j.str.2013.04.022
•BCL::Fold is a de novo membrane protein structure prediction algorithm•Monte Carlo minimization is combined with a knowledge-based energy function•Thirty-four of 40 native membrane protein topologies were sampled in a benchmark testMembrane protein structure determination remains a challenging endeavor. Computational methods that predict membrane protein structure from sequence can potentially aid structure determination for such difficult target proteins. The de novo protein structure prediction method BCL::Fold rapidly assembles secondary structure elements into three-dimensional models. Here, we describe modifications to the algorithm, named BCL::MP-Fold, in order to simulate membrane protein folding. Models are built into a static membrane object and are evaluated using a knowledge-based energy potential, which has been modified to account for the membrane environment. Additionally, a symmetry folding mode allows for the prediction of obligate homomultimers, a common property among membrane proteins. In a benchmark test of 40 proteins of known structure, the method sampled the correct topology in 34 cases. This demonstrates that the algorithm can accurately predict protein topology without the need for large multiple sequence alignments, homologous template structures, or experimental restraints.
Co-reporter:Steffen Lindert, René Staritzbichler, Nils Wötzel, Mert Karakaş, ... Jens Meiler
Structure (15 July 2009) Volume 17(Issue 7) pp:990-1003
Publication Date(Web):15 July 2009
DOI:10.1016/j.str.2009.06.001
In medium-resolution (7–10 Å) cryo-electron microscopy (cryo-EM) density maps, α helices can be identified as density rods whereas β-strand or loop regions are not as easily discerned. We are proposing a computational protein structure prediction algorithm “EM-Fold” that resolves the density rod connectivity ambiguity by placing predicted α helices into the density rods and adding missing backbone coordinates in loop regions. In a benchmark of 11 mainly α-helical proteins of known structure a native-like model is identified in eight cases (rmsd 3.9–7.9 Å). The three failures can be attributed to inaccuracies in the secondary structure prediction step that precedes EM-Fold. EM-Fold has been applied to the ∼6 Å resolution cryo-EM density map of protein IIIa from human adenovirus. We report the first topological model for the α-helical 400 residue N-terminal region of protein IIIa. EM-Fold also has the potential to interpret medium-resolution density maps in X-ray crystallography.
Co-reporter:Soumya Ganguly, Brian E. Weiner, Jens Meiler
Structure (13 April 2011) Volume 19(Issue 4) pp:441-443
Publication Date(Web):13 April 2011
DOI:10.1016/j.str.2011.03.008
The combination of paramagnetic tagging strategies with NMR or EPR spectroscopic techniques can revolutionize de novo structure determination of helical membrane proteins. Leveraging the full potential of this approach requires optimal labeling strategies and prediction of membrane protein topology from sparse and low-resolution distance restraints, as addressed by Chen et al. (2011).
Co-reporter:Nathan Alexander, Ahmad Al-Mestarihi, Marco Bortolus, Hassane Mchaourab, Jens Meiler
Structure (12 February 2008) Volume 16(Issue 2) pp:181-195
Publication Date(Web):12 February 2008
DOI:10.1016/j.str.2007.11.015
As many key proteins evade crystallization and remain too large for nuclear magnetic resonance spectroscopy, electron paramagnetic resonance (EPR) spectroscopy combined with site-directed spin labeling offers an alternative approach for obtaining structural information. Such information must be translated into geometric restraints to be used in computer simulations. Here, distances between spin labels are converted into distance ranges between β carbons by using a “motion-on-a-cone” model, and a linear-correlation model links spin-label accessibility to the number of neighboring residues. This approach was tested on T4-lysozyme and αA-crystallin with the de novo structure prediction algorithm Rosetta. The results demonstrate the feasibility of obtaining highly accurate, atomic-detail models from EPR data by yielding 1.0 Å and 2.6 Å full-atom models, respectively. Distance restraints between amino acids far apart in sequence but close in space are most valuable for structure determination. The approach can be extended to other experimental techniques such as fluorescence spectroscopy, substituted cysteine accessibility method, or mutational studies.
Co-reporter:Susan M. Hanson, Eric S. Dawson, Derek J. Francis, Ned Van Eps, ... Vsevolod V. Gurevich
Structure (11 June 2008) Volume 16(Issue 6) pp:924-934
Publication Date(Web):11 June 2008
DOI:10.1016/j.str.2008.03.006
Visual rod arrestin has the ability to self-associate at physiological concentrations. We previously demonstrated that only monomeric arrestin can bind the receptor and that the arrestin tetramer in solution differs from that in the crystal. We employed the Rosetta docking software to generate molecular models of the physiologically relevant solution tetramer based on the monomeric arrestin crystal structure. The resulting models were filtered using the Rosetta energy function, experimental intersubunit distances measured with DEER spectroscopy, and intersubunit contact sites identified by mutagenesis and site-directed spin labeling. This resulted in a unique model for subsequent evaluation. The validity of the model is strongly supported by model-directed crosslinking and targeted mutagenesis that yields arrestin variants deficient in self-association. The structure of the solution tetramer explains its inability to bind rhodopsin and paves the way for experimental studies of the physiological role of rod arrestin self-association.
.jpg)


















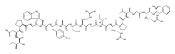
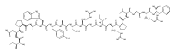
![N-(3-METHYLBUTANOYL)-L-VALYL-N-[(3S,4S)-1-{[(2S)-1-{[(2S,3S)-1-CA<WBR />RBOXY-2-HYDROXY-5-METHYL-3-HEXANYL]AMINO}-1-OXO-2-PROPANYL]AMINO}<WBR />-3-HYDROXY-6-METHYL-1-OXO-4-HEPTANYL]-L-VALINAMIDE](http://img.cochemist.com/ccimg/39400/39324-30-6.png)
![N-(3-METHYLBUTANOYL)-L-VALYL-N-[(3S,4S)-1-{[(2S)-1-{[(2S,3S)-1-CA<WBR />RBOXY-2-HYDROXY-5-METHYL-3-HEXANYL]AMINO}-1-OXO-2-PROPANYL]AMINO}<WBR />-3-HYDROXY-6-METHYL-1-OXO-4-HEPTANYL]-L-VALINAMIDE](http://img.cochemist.com/ccimg/39400/39324-30-6_b.png)






![3-Azaspiro[5.5]undecane](http://img.cochemist.com/ccimg/200/180-44-9.png)
![3-Azaspiro[5.5]undecane](http://img.cochemist.com/ccimg/200/180-44-9_b.png)



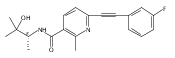
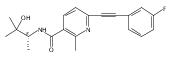
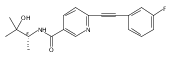
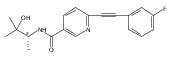



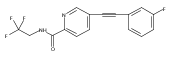
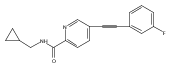
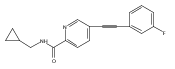

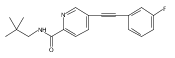
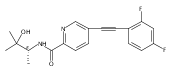
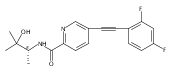

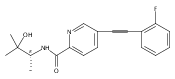
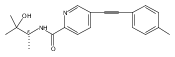

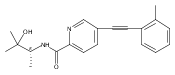
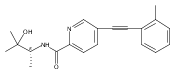
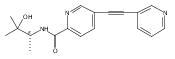
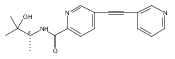









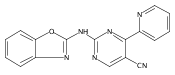
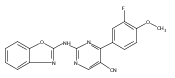
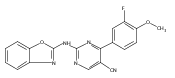
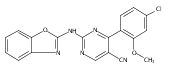



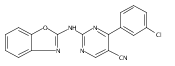
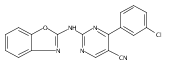
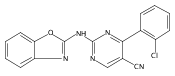
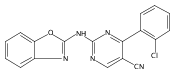


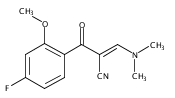
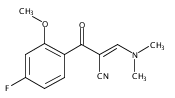
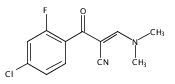
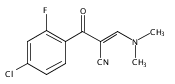

















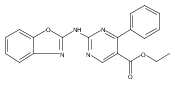
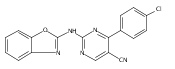
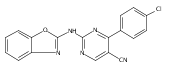
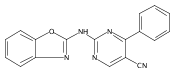
![(3aR,4S,7aR)-Octahydro-4-hydroxy-4-[2-(3-methylphenyl)ethynyl]-1H-indole-1-carboxylic acid methyl ester](/data/chemimg/98800/543906-09-8.png)
![(3aR,4S,7aR)-Octahydro-4-hydroxy-4-[2-(3-methylphenyl)ethynyl]-1H-indole-1-carboxylic acid methyl ester](/data/chemimg/98800/543906-09-8_b.png)
![Benzoic acid, 4-[(1-ethyl-1H-benzimidazol-2-yl)methoxy]-](http://img.cochemist.com/ccimg/502200/502178-66-7.png)
![Benzoic acid, 4-[(1-ethyl-1H-benzimidazol-2-yl)methoxy]-](http://img.cochemist.com/ccimg/502200/502178-66-7_b.png)

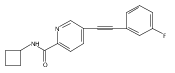
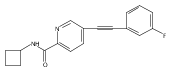
![Benzoic acid, 4-[(3-fluorophenyl)ethynyl]-](http://img.cochemist.com/ccimg/387400/387398-44-9.png)
![Benzoic acid, 4-[(3-fluorophenyl)ethynyl]-](http://img.cochemist.com/ccimg/387400/387398-44-9_b.png)









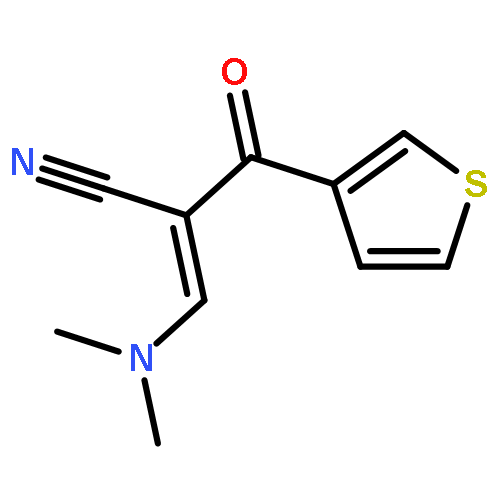
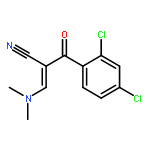
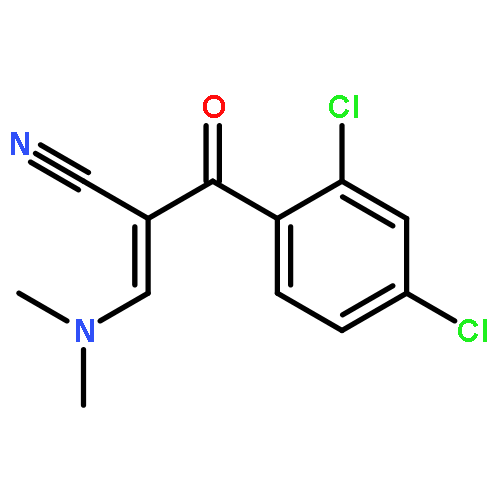
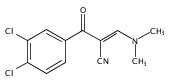







![5-Isobenzofurancarbonitrile,1-[3-(dimethylamino)propyl]-1-(4-fluorophenyl)-1,3-dihydro-, (1R)-](http://img.cochemist.com/ccimg/128200/128196-02-1.png)
![5-Isobenzofurancarbonitrile,1-[3-(dimethylamino)propyl]-1-(4-fluorophenyl)-1,3-dihydro-, (1R)-](http://img.cochemist.com/ccimg/128200/128196-02-1_b.png)



![N-[4-(4-chlorophenyl)butyl]-N,N-diethylheptan-1-aminium](http://img.cochemist.com/ccimg/68400/68379-02-2.png)
![N-[4-(4-chlorophenyl)butyl]-N,N-diethylheptan-1-aminium](http://img.cochemist.com/ccimg/68400/68379-02-2_b.png)
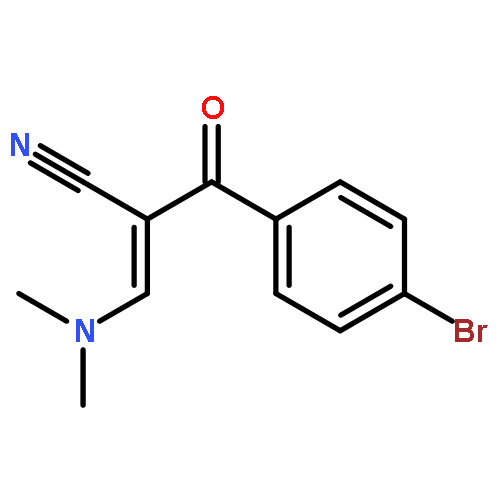
![Benzenepropanenitrile,2-chloro-a-[(dimethylamino)methylene]-b-oxo-](http://img.cochemist.com/ccimg/52300/52200-17-6.png)
![Benzenepropanenitrile,2-chloro-a-[(dimethylamino)methylene]-b-oxo-](http://img.cochemist.com/ccimg/52300/52200-17-6_b.png)
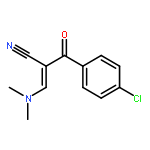
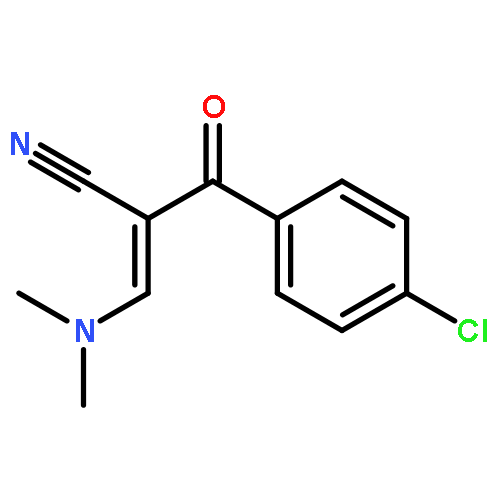
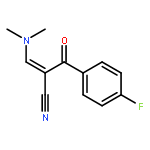
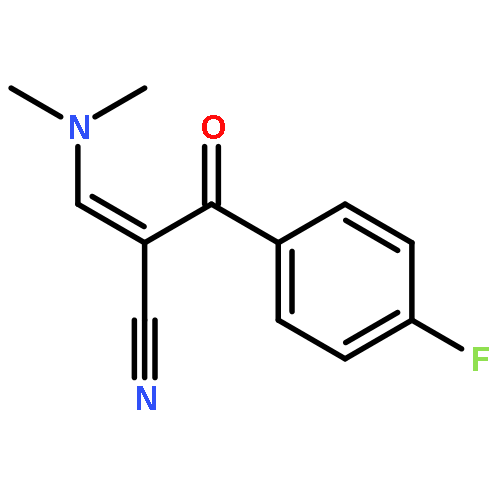
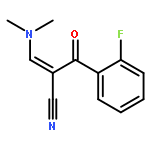
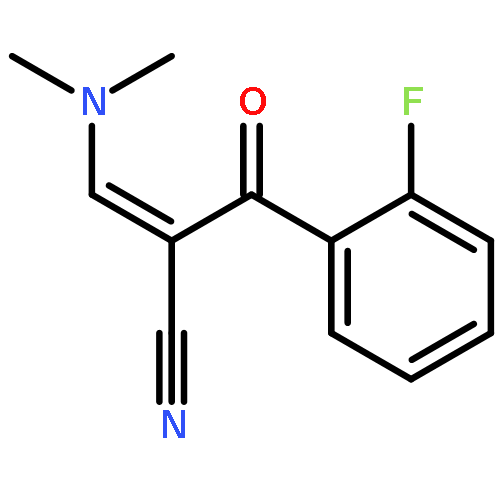
![Benzenepropanenitrile, 3-chloro-a-[(dimethylamino)methylene]-b-oxo-](http://img.cochemist.com/ccimg/52300/52200-08-5.png)
![Benzenepropanenitrile, 3-chloro-a-[(dimethylamino)methylene]-b-oxo-](http://img.cochemist.com/ccimg/52300/52200-08-5_b.png)