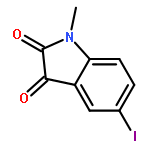
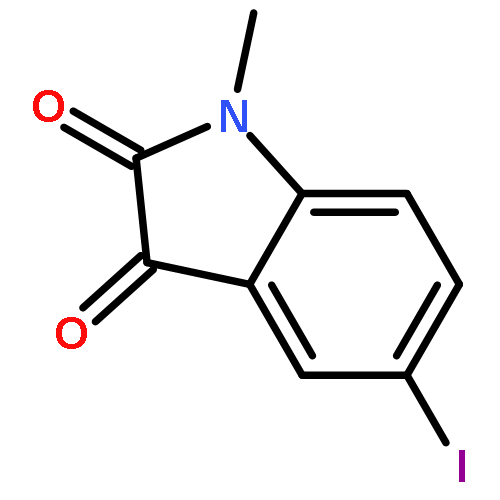
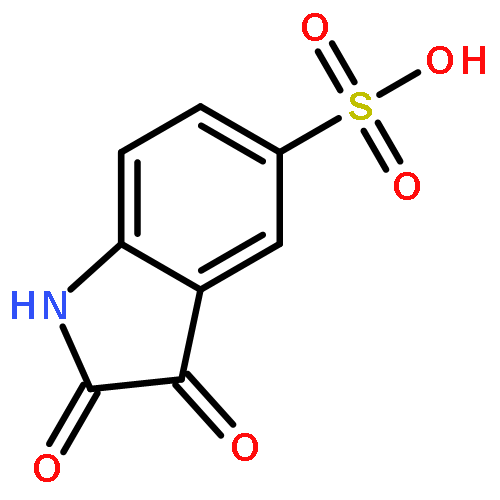
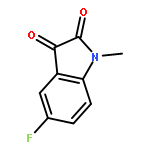
Co-reporter:Li Wang, Bo-Bo Bao, Guo-Qing Song, Cheng Chen, Xu-Meng Zhang, Wei Lu, Zefang Wang, Yan Cai, Shuang Li, Sheng Fu, Fu-Hang Song, Haitao Yang, Jian-Guo Wang
European Journal of Medicinal Chemistry 2017 Volume 137(Volume 137) pp:
Publication Date(Web):8 September 2017
DOI:10.1016/j.ejmech.2017.05.045
•40 novel unsymmetrical aromatic disulfides were synthesized.•The synthesized disulfide compounds are potent inhibitors of SARS main protease.•Possible binding mode and structure-activity relationships of the compounds were established.The worldwide outbreak of severe acute respiratory syndrome (SARS) in 2003 had caused a high rate of mortality. Main protease (Mpro) of SARS-associated coronavirus (SARS-CoV) is an important target to discover pharmaceutical compounds for the therapy of this life-threatening disease. During the course of screening new anti-SARS agents, we have identified that a series of unsymmetrical aromatic disulfides inhibited SARS-CoV Mpro significantly for the first time. Herein, 40 novel unsymmetrical aromatic disulfides were synthesized chemically and their biological activities were evaluated in vitro against SARS-CoV Mpro. These novel compounds displayed excellent IC50 data in the range of 0.516–5.954 μM. Preliminary studies indicated that these disulfides are reversible and mpetitive inhibitors. A possible binding mode was generated via molecular docking simulation and a comparative field analysis (CoMFA) model was constructed to understand the structure-activity relationships. The present research therefore has provided some meaningful guidance to design and identify anti-SARS drugs with totally new chemical structures.Download high-res image (295KB)Download full-size image
Co-reporter:Wei Lu, Irshad Ahmed Baig, Hui-Jie Sun, Chang-Jun Cui, Rui Guo, In-Pil Jung, Di Wang, Mei Dong, Moon-Young Yoon, Jian-Guo Wang
European Journal of Medicinal Chemistry 2015 Volume 94() pp:298-305
Publication Date(Web):13 April 2015
DOI:10.1016/j.ejmech.2015.03.014
•We synthesized 24 quinazolinone benzoate compounds as novel inhibitors of Mycobacterium tuberculosis acetohydroxyacid synthase.•Five compounds exhibit significant inhibition on both MTB-AHAS and various strains of MTB.•Selected compound 5h shows desirable intracellular anti-MTB activity against MDR or XDR strains.Acetohydroxyacid synthase (AHAS) catalyzes the first essential biosynthetic step of branched-chain amino acids and is a biologically safe target against Mycobacterium tuberculosis (MTB). In our previous research, we used virtual screening to identify some novel AHAS inhibitors as potent antituberculosis agents. In this study, we synthesized twenty-four additional quinazolinone benzoates and explored their antitubercular activity. Five of these compounds displayed significant MTB-AHAS inhibition and their IC50 values were determined to be in the range of 6.50 μM–12.08 μM. Importantly, these compounds also exhibited strong in vitro activity (MICs in the range of 2.5–10 mg/L) and intracellular activity against clinically isolated extensively drug-resistant strains of M. tuberculosis. Taken together, these results indicated that the quinazolinone benzoate compounds should be regarded as promising lead compounds for the development of potent antituberculosis drugs with a novel mode of action.IC50 = 6.50 μM MTB-AHAS. MIC = 2.5 mg/L for MDR and XDR MTB.
Co-reporter:Xu-Meng Zhang, Hui Guo, Zai-Shun Li, Fu-Hang Song, Wei-Min Wang, Huan-Qin Dai, Li-Xin Zhang, Jian-Guo Wang
European Journal of Medicinal Chemistry 2015 Volume 101() pp:419-430
Publication Date(Web):28 August 2015
DOI:10.1016/j.ejmech.2015.06.047
•51 isatin-β-thiosemicarbazone (IBT) compounds were synthesized.•The synthesized IBTs are potent inhibitors of MRSA and VRE.•Structure–activity relationships of the anti-MRSA compounds were established.Methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Enterococcus (VRE) have caused an increasing mortality rate, which means that antibiotic resistance is becoming an important health issue. In the course to screen new agents for resistant bacteria, we identified that a series of isatin-β-thiosemicarbazones (IBTs) could inhibit the growth of MRSA and VRE. This was the first time that the “familiar” IBT compounds exhibited significant anti Gram-positive pathogen activity. Against a clinical isolated MRSA strain, 20 of the 51 synthesized compounds showed minimum inhibitory concentration (MIC) data of 0.78 mg/L and another 12 novel compounds had MICs of 0.39 mg/L. Moreover, these compounds also inhibited Enterococcus faecalis and VRE at similar levels, indicating that IBTs might have different mode of action compared with vancomycin. For these IBTs, comparative field analysis (CoMFA) models were further established to understand the structure–activity relationships in order to design new compounds from steric and electrostatic contributions. This work has suggested that IBTs can be considered as potential lead compounds to discover antibacterial inhibitors to combat drug resistance.
Co-reporter:Huifang M. Zhang, Huanqin Dai, Paul J. Hanson, Huidong Li, Hui Guo, Xin Ye, Maged G. Hemida, Luoqiang Wang, Yaojun Tong, Ye Qiu, Selina Liu, Fengping Wang, Fuhang Song, Buchang Zhang, Jian-Guo Wang, Li-Xin Zhang, and Decheng Yang
ACS Chemical Biology 2014 Volume 9(Issue 4) pp:1015
Publication Date(Web):January 31, 2014
DOI:10.1021/cb400775z
We report here an isatin derivative 45 (ID45) against coxsackievirus B3 (CVB3) replication, which was synthesized based on a high-throughput screen of a unique natural product library. ID45 showed the most potent anti-CVB3 activity among the four synthesized compounds. Treatment of cells with ID45 before or after infection significantly reduced viral particle formation, resulting in protection of cells from virus-induced apoptosis. In addition, ID45 treatment caused remarkable up-regulation of glucose-regulated protein 78 (GRP78), a hallmark of endoplasmic reticulum (ER) stress and an indicator of enhanced cell viability. In identifying the ER stress response pathway induced by ID45, we found that ID45 activated PKR-like ER protein kinase (PERK) but failed to up-regulate eIF2α phosphorylation. Instead ID45 activated transcription factor Nrf2 (NF-E2-related factor-2), which is evidenced by its nuclear translocation and upregulation of its downstream target genes NQO1 (NAD(P)H quinone-oxidoreductase 1) and GCLM (glutamate-cysteine ligase, modifier subunit). This observation was further verified by using siRNAs of GRP78 or Nrf2, which blocked both the translocation of Nrf2 and up-regulation of its target genes, leading to aggressive viral replication and enhanced cell apoptosis. Finally, we found that ID45-induced up-regulation of NQO1 protected eIF4GI, a eukaryotic cap-dependent translation initiation factor, from cleavage by CVB3 protease and degradation by proteasomes. Taken together, our findings established that a novel antiviral mechanism of isatin derivative ID45 inhibits CVB3 replication by promoting cell survival through a PERK/Nrf2-dependent ER stress pathway, which benefits host cap-dependent translation but suppresses CVB3 cap-independent translation.
Co-reporter:Yu-Ting Lee ; Chang-Jun Cui ; Eve W. L. Chow ; Nason Pue ; Thierry Lonhienne ; Jian-Guo Wang ; James A. Fraser ;Luke W. Guddat
Journal of Medicinal Chemistry 2013 Volume 56(Issue 1) pp:210-219
Publication Date(Web):December 13, 2012
DOI:10.1021/jm301501k
The sulfonylurea herbicides exert their activity by inhibiting plant acetohydroxyacid synthase (AHAS), the first enzyme in the branched-chain amino acid biosynthesis pathway. It has previously been shown that if the gene for AHAS is deleted in Candida albicans, attenuation of virulence is achieved, suggesting AHAS as an antifungal drug target. Herein, we have cloned, expressed, and purified C. albicans AHAS and shown that several sulfonylureas are inhibitors of this enzyme and possess antifungal activity. The most potent of these compounds is ethyl 2-(N-((4-iodo-6-methoxypyrimidin-2-yl)carbamoyl)sulfamoyl)benzoate (10c), which has a Ki value of 3.8 nM for C. albicans AHAS and an MIC90 of 0.7 μg/mL for this fungus in cell-based assays. For the sulfonylureas tested there was a strong correlation between inhibitory activity toward C. albicans AHAS and fungicidal activity, supporting the hypothesis that AHAS is the target for their inhibitory activity within the cell.
Co-reporter:Di Wang, Xuelian Zhu, Changjun Cui, Mei Dong, Hualiang Jiang, Zhengming Li, Zhen Liu, Weiliang Zhu, and Jian-Guo Wang
Journal of Chemical Information and Modeling 2013 Volume 53(Issue 2) pp:343-353
Publication Date(Web):January 14, 2013
DOI:10.1021/ci3004545
Acetohydroxyacid synthase (AHAS) has been regarded as a promising drug target against Mycobacterium tuberculosis (MTB) as it catalyzes the biosynthesis of branched-chain amino acids. In this study, 23 novel AHAS inhibitors were identified through molecular docking followed by similarity search. The determined IC50 values range from 0.385 ± 0.026 μM to >200 μM against bacterium AHAS. Five of the identified compounds show significant in vitro activity against H37Rv strains (MICs in the range of 2.5–80 mg/L) and clinical MTB strains, including MDR and XDR isolates. More impressively, compounds 5 and 7 can enhance the killing ability against macrophages infected pathogen remarkably. This study suggests our discovered inhibitors can be further developed as novel anti-MTB therapeutics targeting AHAS.
Co-reporter:Zai-Shun Li, Wei-Min Wang, Wei Lu, Cong-Wei Niu, Yong-Hong Li, Zheng-Ming Li, Jian-Guo Wang
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 13) pp:3723-3727
Publication Date(Web):1 July 2013
DOI:10.1016/j.bmcl.2013.05.013
46 Novel nonsymmetrical aromatic disulfides containing [1,3,4]thiadiazole or [1,3,4]oxadiazole groups were synthesized and their biological activities were evaluated as inhibitors of acetohydroxyacid synthase (AHAS, EC 2.2.1.6). Besides their strong in vitro inhibition against plant AHAS, compounds 3e and 3f also display 80–100% post-emergence herbicidal activities in greenhouse bioassay at 1500 g/ha dosage. The assay of exogenous branched-chain amino acids supplementation on rape root growth of 3e suggests that the herbicidal activity has relationship with AHAS inhibition.
Co-reporter:Jian-Li Shang, Hui Guo, Zai-Shun Li, Biao Ren, Zheng-Ming Li, Huan-qin Dai, Li-Xin Zhang, Jian-Guo Wang
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 3) pp:724-727
Publication Date(Web):1 February 2013
DOI:10.1016/j.bmcl.2012.11.103
A total of 29 novel sulfenamide compounds were synthesized, spectroscopically characterized and evaluated in vitro for antimicrobial activity against various infectious pathogens. Compounds 1b and 2c exhibited potent inhibition against clinical Methicillin-resistant Staphylococcus aureus (MRSA) strains with minimum inhibitory concentration (MIC) values of 1.56 μg/mL.Twenty nine novel sulfenamide compounds were synthesized and their antimicrobial activities were evaluated. Compounds 1b and 2c exhibited strong anti-MRSA activity.
Co-reporter:Jun Shang, Wei-Min Wang, Yong-Hong Li, Hai-Bin Song, Zheng-Ming Li, and Jian-Guo Wang
Journal of Agricultural and Food Chemistry 2012 Volume 60(Issue 34) pp:8286-8293
Publication Date(Web):August 20, 2012
DOI:10.1021/jf302206x
Acetohydroxyacid synthase (AHAS; EC 2.2.1.6) is an important bioactive target for the design of environmentally benign herbicides. On the basis of previous virtual screening, 50 asymmetric aryl disulfides containing [1,2,4]triazole groups were synthesized and characterized by 1H NMR, HRMS, and crystal structure. Compounds I-a, I-b, and I-p show Ki values of 1.70, 4.69, and 5.57 μM, respectively, for wild type Arabidopsis thaliana AHAS (AtAHAS) and low resistance against mutant type AtAHAS W574L. At 100 mg L–1 concentration, compounds I-a, II-a, and II-b exhibit 86.6, 81.7, and 87.5% in vivo rape root growth inhibition. CoMFA steric and electrostatic contour maps were established, and a possible binding mode was suggested from molecular docking, which provide valuable information to understand the key structural features of these disulfide compounds. To the authors' knowledge, this is the first comprehensive case suggesting that asymmetric aryl disulfides are novel AHAS inhibitors.
Co-reporter:Jianguo Wang, Haizhong Tan, Yonghong Li, Yi Ma, Zhengming Li, and Luke W. Guddat
Journal of Agricultural and Food Chemistry 2011 Volume 59(Issue 18) pp:9892-9900
Publication Date(Web):August 13, 2011
DOI:10.1021/jf2021607
Acetohydroxyacid synthase (AHAS) catalyzes the first common step in the biosynthesis of the branched-chain amino acids. As a result of its metabolic importance in plants, it is a target for many commercial herbicides. Virtual screening analysis inspired the evaluation of 19 commercially available isatin analogues and 13 newly synthesized isatin derivatives as novel AHAS inhibitors and for their herbicidal activity. The best compound demonstrated 95% inhibition of the activity of Arabidopsis thaliana AHAS at a concentration of 100 mg L–1, whereas the herbicidal activities of three compounds reached 50% inhibition at a concentration of 10 mg L–1 using the rape root growth test. CoMFA contour models were established to understand the structure–activity relationships for this class of AHAS inhibitor. The compounds were docked to the active site cavity of A. thaliana AHAS using FlexX, and the dominant binding mode was consistent with frontier molecular orbital from DFT calculations. This is the first comprehensive study of isatin derivatives as AHAS inhibitors and provides a valuable starting point for the design of new herbicides.
Co-reporter:Yi Ma;Bin Wang;Zheng-Ming Li
Journal of Molecular Modeling 2011 Volume 17( Issue 8) pp:1899-1909
Publication Date(Web):2011 August
DOI:10.1007/s00894-010-0898-y
Since they are potential topoisomerase II (Topo II) inhibitors, naphthoquinone fused cyclic α-aminophosphonates display anticancer activity. In order to explore the inhibitory mechanisms of these compounds, they were docked into the active site of Topo II structure, which allowed their probable binding modes to be predicted. Some meaningful results concerning their structure–activity relationships were obtained from density functional theory calculations. Models based on quantitative comparative molecular field analysis and comparative molecular similarity index analysis were derived for the steric, electrostatic, hydrophobic and H-bonding features of the compounds. The present study provides valuable results that enhance our understanding of the anticancer activities of these inhibitors and will aid the rational drug design of novel Topo II inhibitors in the future.

![Benzoic acid, (2Z)-2-[1-[[4-(1,1-dimethylethyl)phenyl]methyl]-1,2-dihydro-2-oxo-3H-indol-3-ylidene]hydrazide](/data/chemimg/142000/1332882-59-3.png)
![Benzoic acid, (2Z)-2-[1-[[4-(1,1-dimethylethyl)phenyl]methyl]-1,2-dihydro-2-oxo-3H-indol-3-ylidene]hydrazide](/data/chemimg/142000/1332882-59-3_b.png)
![Benzoic acid, (2Z)-2-[1-[(2,4-dichlorophenyl)methyl]-1,2-dihydro-2-oxo-3H-indol-3-ylidene]hydrazide](/data/chemimg/142000/1332882-58-2.png)
![Benzoic acid, (2Z)-2-[1-[(2,4-dichlorophenyl)methyl]-1,2-dihydro-2-oxo-3H-indol-3-ylidene]hydrazide](/data/chemimg/142000/1332882-58-2_b.png)
![Benzoic acid, (2Z)-2-[1,2-dihydro-1-[(2-methylphenyl)methyl]-2-oxo-3H-indol-3-ylidene]hydrazide](/data/chemimg/142000/1332882-57-1.png)
![Benzoic acid, (2Z)-2-[1,2-dihydro-1-[(2-methylphenyl)methyl]-2-oxo-3H-indol-3-ylidene]hydrazide](/data/chemimg/142000/1332882-57-1_b.png)
![Benzoic acid, (2Z)-2-[1-[(4-fluorophenyl)methyl]-1,2-dihydro-2-oxo-3H-indol-3-ylidene]hydrazide](/data/chemimg/142000/1332882-56-0.png)
![Benzoic acid, (2Z)-2-[1-[(4-fluorophenyl)methyl]-1,2-dihydro-2-oxo-3H-indol-3-ylidene]hydrazide](/data/chemimg/142000/1332882-56-0_b.png)
![Benzoic acid, (2Z)-2-[1-[(3-fluorophenyl)methyl]-1,2-dihydro-2-oxo-3H-indol-3-ylidene]hydrazide](/data/chemimg/142000/1332882-55-9.png)
![Benzoic acid, (2Z)-2-[1-[(3-fluorophenyl)methyl]-1,2-dihydro-2-oxo-3H-indol-3-ylidene]hydrazide](/data/chemimg/142000/1332882-55-9_b.png)
![Benzoic acid, (2Z)-2-[1-[(4-cyanophenyl)methyl]-1,2-dihydro-2-oxo-3H-indol-3-ylidene]hydrazide](/data/chemimg/142000/1332882-54-8.png)
![Benzoic acid, (2Z)-2-[1-[(4-cyanophenyl)methyl]-1,2-dihydro-2-oxo-3H-indol-3-ylidene]hydrazide](/data/chemimg/142000/1332882-54-8_b.png)






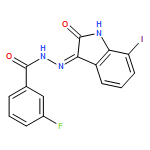

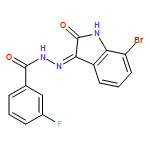

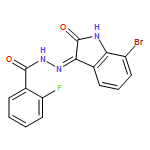







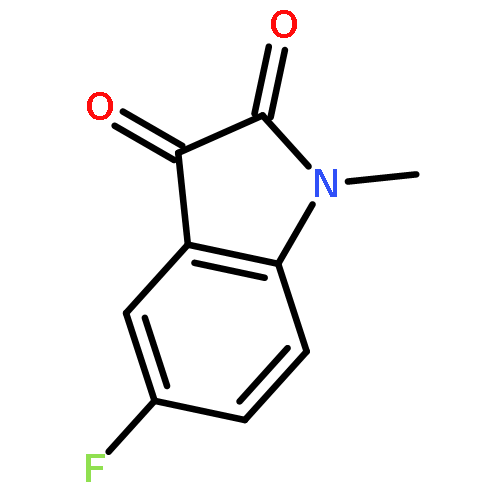
![1H-Indole-2,3-dione, 1-[[4-(1,1-dimethylethyl)phenyl]methyl]-](/data/chemimg/103000/314031-55-5.png)
![1H-Indole-2,3-dione, 1-[[4-(1,1-dimethylethyl)phenyl]methyl]-](/data/chemimg/103000/314031-55-5_b.png)